
TOMM40 Genetic Variants Cause Neuroinflammation in Alzheimer’s Disease
 , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Results
2.1. AD Patients Exhibit Genetic Variants within TOMM40 Gene
2.2. Exonic SNP of TOMM40 Are Linked to Increased AD Susceptibility
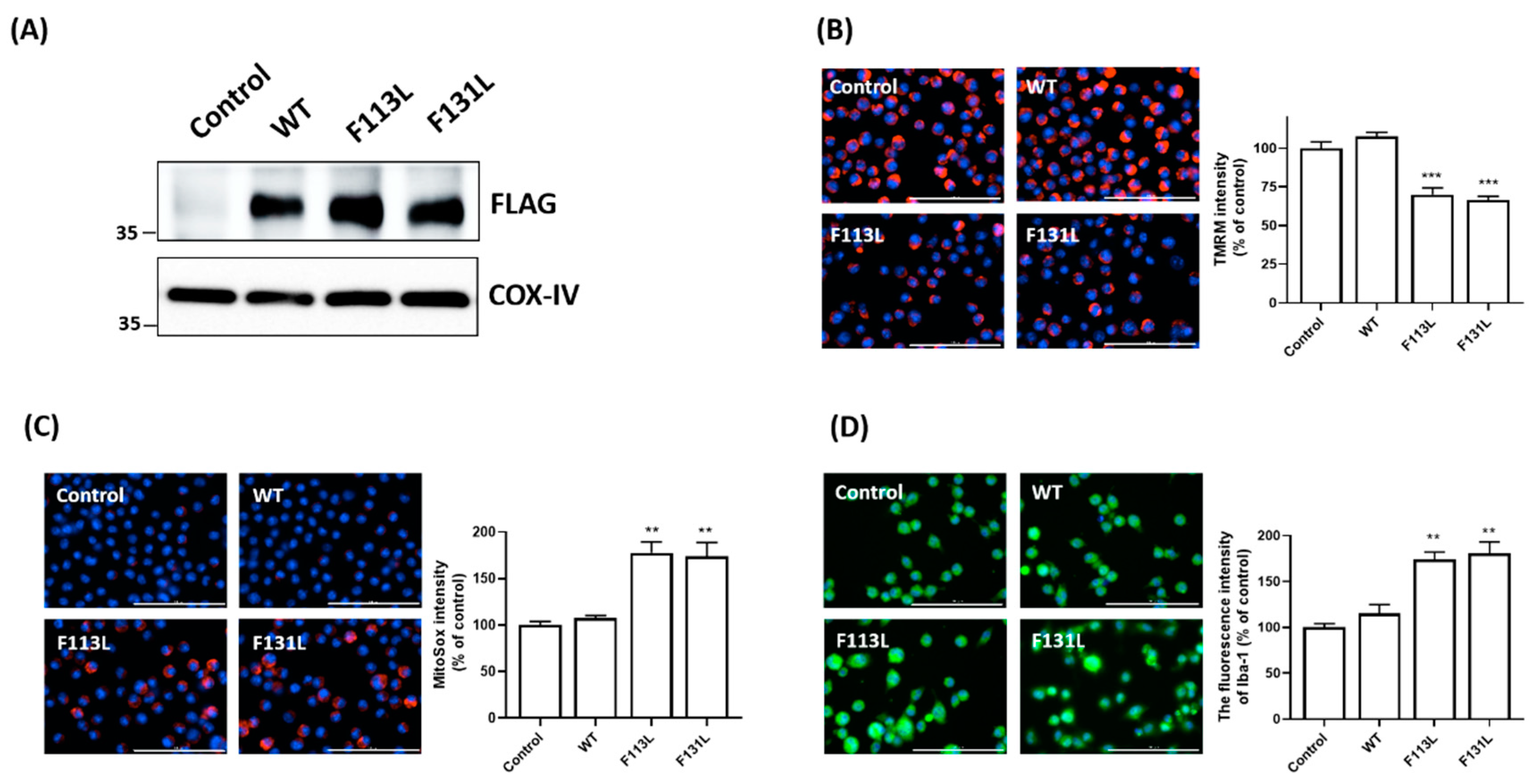
2.3. AD-Associated TOMM40 Genetic Variants, but Not Wild-Type TOMM40, Causes Mitochondrial Dysfunction and Oxidative Stress of Microglial Cells
2.4. AD-Associated TOMM40 Genetic Variants Cause Microglial Activation
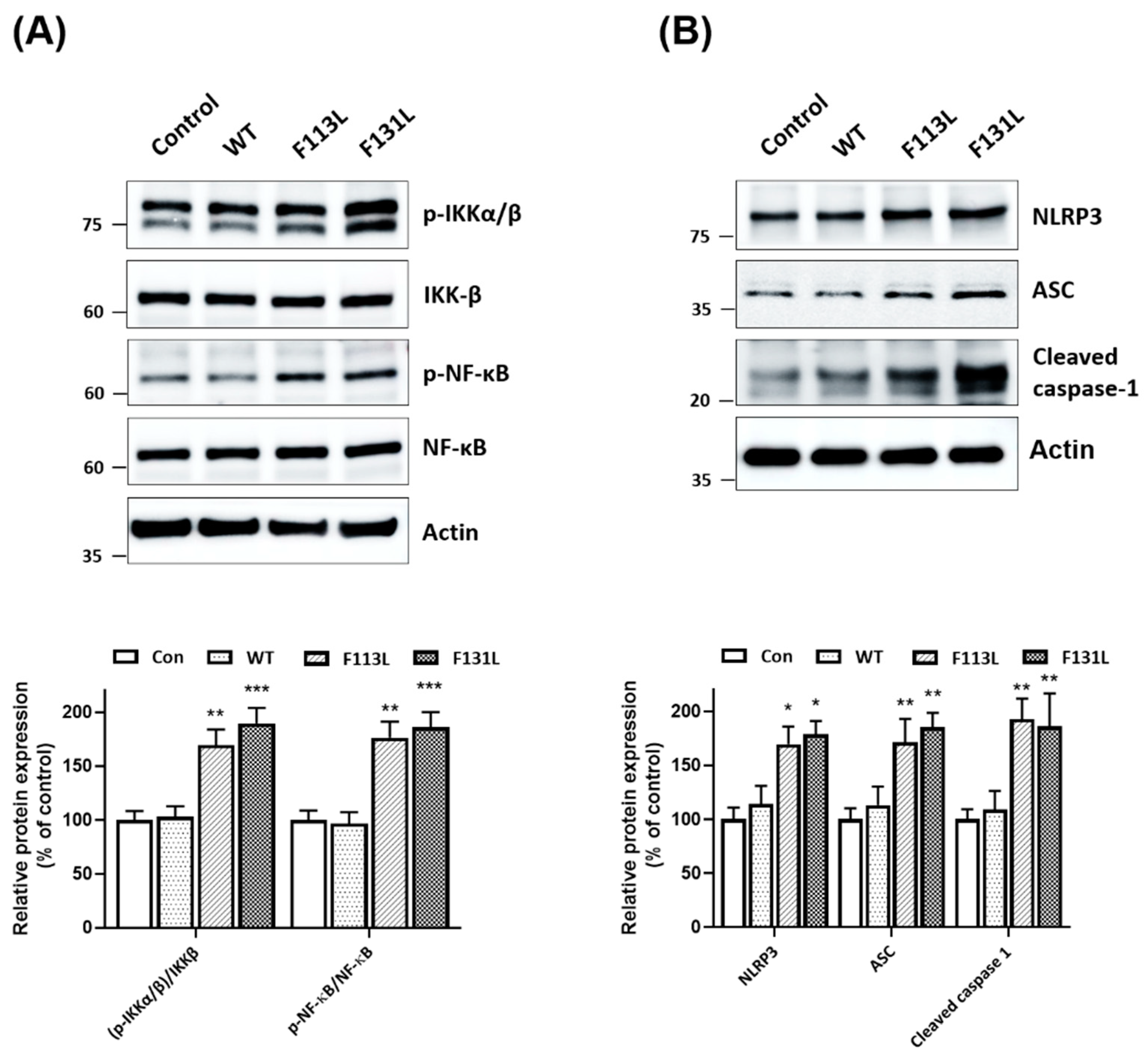
2.5. Mutant (F113L) or (F131L) TOMM40 Activates NF-κB Cascade and NLRP3 Inflammasome in Microglial Cells
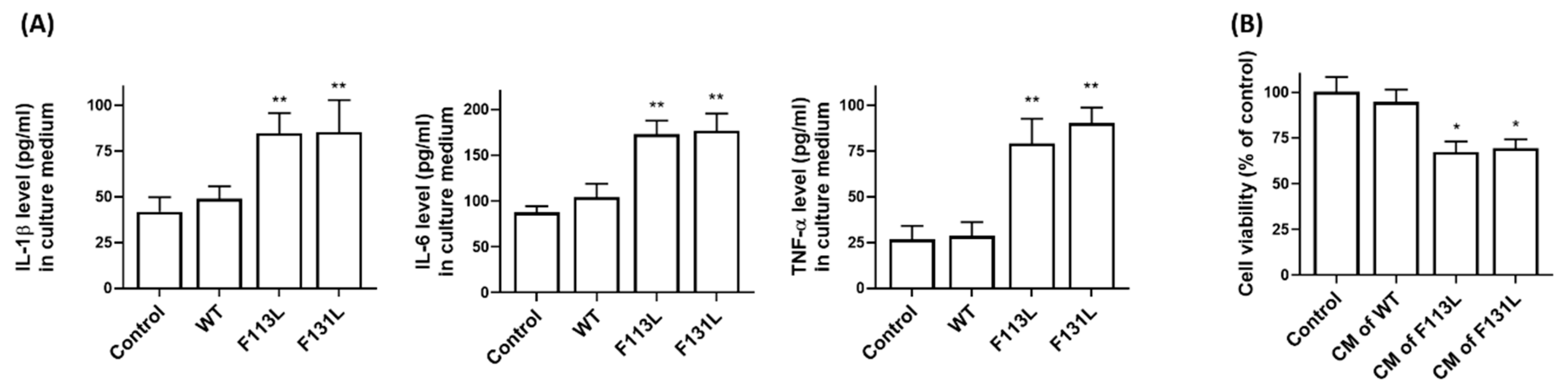
2.6. AD-Associated TOMM40 Genetic Variants Cause the Release of Pro-Inflammatory Cytokines from Microglia Cells, Leading to Cell Death of Hippocampal Neurons
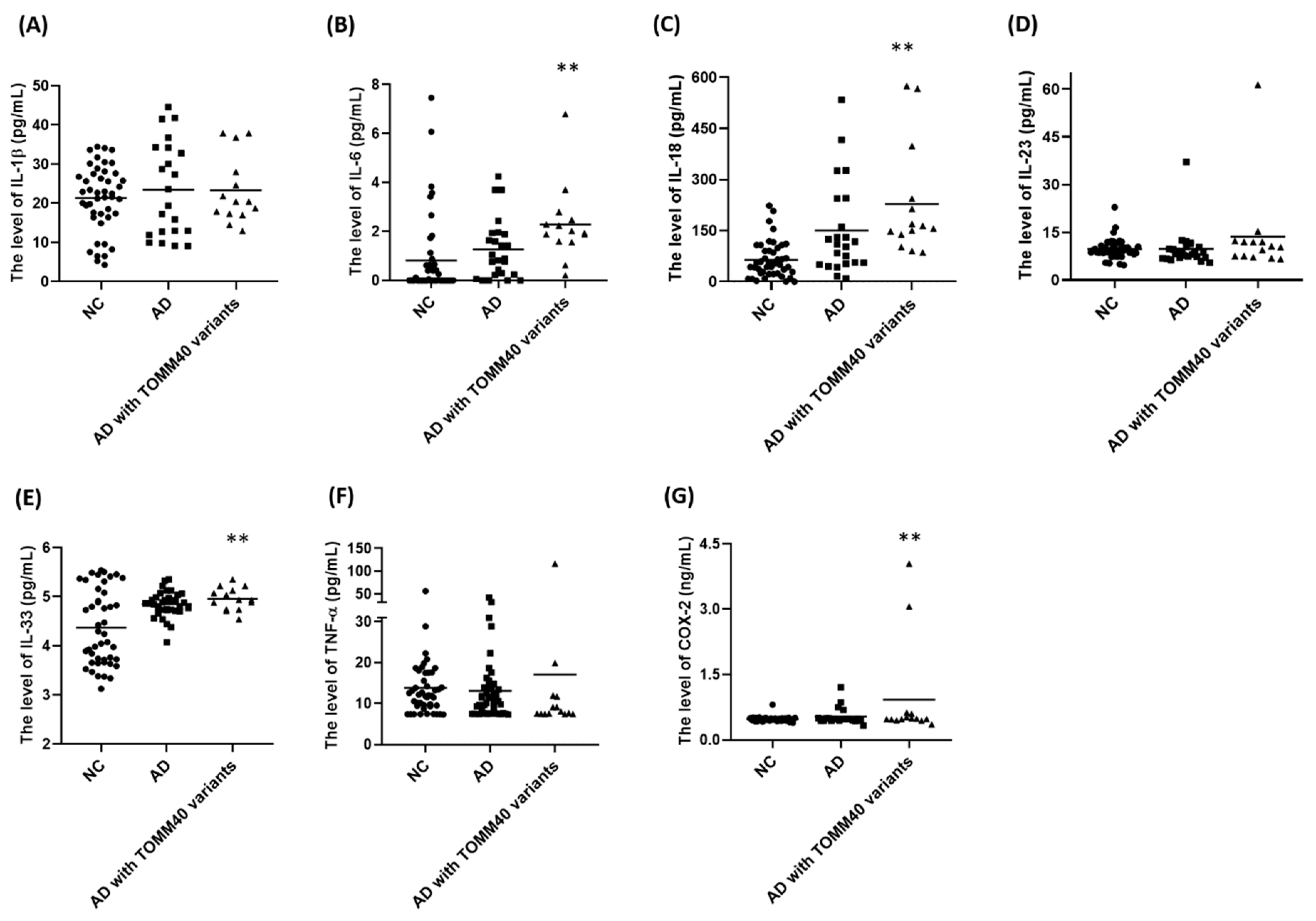
2.7. Plasma Levels of IL-6, IL-18, IL-33, and COX-2 Are Upregulated in AD Patients Carrying TOMM40 Genetic Variants
3. Discussion
4. Materials and Methods
4.1. Patients and Control Subjects
4.2. DNA Extraction, WGS and Data Processing
4.3. Sequencing and Genotyping of TOMM40 Genetic Variants
4.4. Cell Culture
4.5. Transfection of TOMM40 Genetic Variants
4.6. Determination of Mitochondrial Membrane Potential (ΔΨm) and Mitochondrial Superoxide
4.7. Immunofluorescence Staining of Iba-1
4.8. Immunoblotting Assays
4.9. Measurement of Pro-Inflammatory Cytokines in Culture Medium
4.10. Determination of Cell Viability of HT22 Hippocampal Neurons
4.11. Measurement of Plasma Levels of Cytokines or COX-2
4.12. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chetelat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Li, F.; Wei, C.; Zhu, M.; Qu, Q.; Qin, W.; Tang, Y.; Shen, L.; Wang, Y.; Shen, L.; et al. Prediction of Alzheimer’s disease using multi-variants from a Chinese genome-wide association study. Brain 2021, 144, 924–937. [Google Scholar] [CrossRef] [PubMed]
- Sierksma, A.; Escott-Price, V.; De Strooper, B. Translating genetic risk of Alzheimer’s disease into mechanistic insight and drug targets. Science 2020, 370, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Beecham, G.W.; Hamilton, K.; Naj, A.C.; Martin, E.R.; Huentelman, M.; Myers, A.J.; Corneveaux, J.J.; Hardy, J.; Vonsattel, J.P.; Younkin, S.G.; et al. Genome-Wide Association Meta-analysis of Neuropathologic Features of Alzheimer’s Disease and Related Dementias. PLoS Genet. 2014, 10, e1004606. [Google Scholar] [CrossRef] [PubMed]
- Chuang, W.L.; Hsieh, Y.C.; Wang, C.Y.; Kuo, H.C.; Huang, C.C. Association of apolipoproteins e4 and c1 with onset age and memory: A study of sporadic Alzheimer disease in Taiwan. J. Geriatr. Psychiatry Neurol. 2010, 23, 42–48. [Google Scholar] [CrossRef]
- Genin, E.; Hannequin, D.; Wallon, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Bullido, M.J.; Engelborghs, S.; De Deyn, P.; Berr, C.; et al. APOE and Alzheimer disease: A major gene with semi-dominant inheritance. Mol. Psychiatry 2011, 16, 903–907. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s disease: Advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef]
- Martens, Y.A.; Zhao, N.; Liu, C.C.; Kanekiyo, T.; Yang, A.J.; Goate, A.M.; Holtzman, D.M.; Bu, G. ApoE Cascade Hypothesis in the pathogenesis of Alzheimer’s disease and related dementias. Neuron 2022, 110, 1304–1317. [Google Scholar] [CrossRef]
- Raulin, A.C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.C. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. Mol. Neurodegener. 2022, 17, 72. [Google Scholar] [CrossRef]
- Coon, K.D.; Myers, A.J.; Craig, D.W.; Webster, J.A.; Pearson, J.V.; Lince, D.H.; Zismann, V.L.; Beach, T.G.; Leung, D.; Bryden, L.; et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J. Clin. Psychiatry 2007, 68, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.J.; Fulton-Howard, B.; Goate, A. Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet Neurol. 2020, 19, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.J.; Hardy, J. TOMM40 association with Alzheimer disease: Tales of APOE and linkage disequilibrium. Arch. Neurol. 2012, 69, 1243–1244. [Google Scholar] [CrossRef] [PubMed]
- Yashin, A.I.; Fang, F.; Kovtun, M.; Wu, D.; Duan, M.; Arbeev, K.; Akushevich, I.; Kulminski, A.; Culminskaya, I.; Zhbannikov, I.; et al. Hidden heterogeneity in Alzheimer’s disease: Insights from genetic association studies and other analyses. Exp. Gerontol. 2018, 107, 148–160. [Google Scholar] [CrossRef]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Humphries, A.D.; Streimann, I.C.; Stojanovski, D.; Johnston, A.J.; Yano, M.; Hoogenraad, N.J.; Ryan, M.T. Dissection of the mitochondrial import and assembly pathway for human Tom40. J. Biol. Chem. 2005, 280, 11535–11543. [Google Scholar] [CrossRef]
- Lee, E.G.; Chen, S.; Leong, L.; Tulloch, J.; Yu, C.E. TOMM40 RNA Transcription in Alzheimer’s Disease Brain and Its Implication in Mitochondrial Dysfunction. Genes 2021, 12, 871. [Google Scholar] [CrossRef]
- Bezuch, N.; Bradburn, S.; Robinson, A.C.; Pendleton, N.; Payton, A.; Murgatroyd, C. Superior Frontal Gyrus TOMM40-APOE Locus DNA Methylation in Alzheimer’s Disease. J. Alzheimers Dis. Rep. 2021, 5, 275–282. [Google Scholar] [CrossRef]
- Valant, V.; Keenan, B.T.; Anderson, C.D.; Shulman, J.M.; Devan, W.J.; Ayres, A.M.; Schwab, K.; Goldstein, J.N.; Viswanathan, A.; Alzheimer’s Disease Neuroimaging Initiative; et al. TOMM40 in Cerebral Amyloid Angiopathy Related Intracerebral Hemorrhage: Comparative Genetic Analysis with Alzheimer’s Disease. Transl. Stroke Res. 2012, 3 (Suppl. 1), 102–112. [Google Scholar] [CrossRef]
- Siddarth, P.; Burggren, A.C.; Merrill, D.A.; Ercoli, L.M.; Mahmood, Z.; Barrio, J.R.; Small, G.W. Longer TOMM40 poly-T variants associated with higher FDDNP-PET medial temporal tau and amyloid binding. PLoS ONE 2018, 13, e0208358. [Google Scholar] [CrossRef]
- Ferencz, B.; Karlsson, S.; Kalpouzos, G. Promising Genetic Biomarkers of Preclinical Alzheimer’s Disease: The Influence of APOE and TOMM40 on Brain Integrity. Int. J. Alzheimers Dis. 2012, 2012, 421452. [Google Scholar] [PubMed]
- Roses, A.D.; Lutz, M.W.; Amrine-Madsen, H.; Saunders, A.M.; Crenshaw, D.G.; Sundseth, S.S.; Huentelman, M.J.; Welsh-Bohmer, K.A.; Reiman, E.M. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharm. J. 2010, 10, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Linnertz, C.; Anderson, L.; Gottschalk, W.; Crenshaw, D.; Lutz, M.W.; Allen, J.; Saith, S.; Mihovilovic, M.; Burke, J.R.; Welsh-Bohmer, K.A.; et al. The cis-regulatory effect of an Alzheimer’s disease-associated poly-T locus on expression of TOMM40 and apolipoprotein E genes. Alzheimers Dement. 2014, 10, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Takei, N.; Miyashita, A.; Tsukie, T.; Arai, H.; Asada, T.; Imagawa, M.; Shoji, M.; Higuchi, S.; Urakami, K.; Kimura, H.; et al. Genetic association study on in and around the APOE in late-onset Alzheimer disease in Japanese. Genomics 2009, 93, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Lutz, F.; Yu, C.E. Functional analysis of APOE locus genetic variation implicates regional enhancers in the regulation of both TOMM40 and APOE. J. Hum. Genet. 2012, 57, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Mangalmurti, A.; Lukens, J.R. How neurons die in Alzheimer’s disease: Implications for neuroinflammation. Curr. Opin. Neurobiol. 2022, 75, 102575. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Muzio, L.; Viotti, A.; Martino, G. Microglia in Neuroinflammation and Neurodegeneration: From Understanding to Therapy. Front. Neurosci. 2021, 15, 742065. [Google Scholar] [CrossRef]
- Haque, M.E.; Akther, M.; Jakaria, M.; Kim, I.S.; Azam, S.; Choi, D.K. Targeting the microglial NLRP3 inflammasome and its role in Parkinson’s disease. Mov. Disord. 2020, 35, 20–33. [Google Scholar] [CrossRef]
- Lunemann, J.D.; Malhotra, S.; Shinohara, M.L.; Montalban, X.; Comabella, M. Targeting Inflammasomes to Treat Neurological Diseases. Ann. Neurol. 2021, 90, 177–188. [Google Scholar] [CrossRef] [PubMed]
- de Araujo, F.M.; Cuenca-Bermejo, L.; Fernandez-Villalba, E.; Costa, S.L.; Silva, V.D.A.; Herrero, M.T. Role of Microgliosis and NLRP3 Inflammasome in Parkinson’s Disease Pathogenesis and Therapy. Cell. Mol. Neurobiol. 2022, 42, 1283–1300. [Google Scholar] [CrossRef] [PubMed]
- Brahadeeswaran, S.; Sivagurunathan, N.; Calivarathan, L. Inflammasome Signaling in the Aging Brain and Age-Related Neurodegenerative Diseases. Mol. Neurobiol. 2022, 59, 2288–2304. [Google Scholar] [CrossRef] [PubMed]
- Milner, M.T.; Maddugoda, M.; Gotz, J.; Burgener, S.S.; Schroder, K. The NLRP3 inflammasome triggers sterile neuroinflammation and Alzheimer’s disease. Curr. Opin. Immunol. 2021, 68, 116–124. [Google Scholar] [CrossRef]
- Hanslik, K.L.; Ulland, T.K. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front. Neurol. 2020, 11, 570711. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Santos, M.S.; Oliveira, C.R. Alzheimer’s disease: A lesson from mitochondrial dysfunction. Antioxid. Redox Signal. 2007, 9, 1621–1630. [Google Scholar] [CrossRef]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Rehling, P.; Pfanner, N.; Meisinger, C. Insertion of hydrophobic membrane proteins into the inner mitochondrial membrane--a guided tour. J. Mol. Biol. 2003, 326, 639–657. [Google Scholar] [CrossRef]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [PubMed]
- Abyadeh, M.; Gupta, V.; Chitranshi, N.; Gupta, V.; Wu, Y.; Saks, D.; Wander Wall, R.; Fitzhenry, M.J.; Basavarajappa, D.; You, Y.; et al. Mitochondrial dysfunction in Alzheimer’s disease—A proteomics perspective. Expert Rev. Proteom. 2021, 18, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- von Bernhardi, R.; Eugenin-von Bernhardi, L.; Eugenin, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, Y.; Mok, K.Y.; Kwok, T.C.Y.; Mok, V.C.T.; Guo, Q.; Ip, F.C.; Chen, Y.; Mullapudi, N.; Alzheimer’s Disease Neuroimaging, I.; et al. Non-coding variability at the APOE locus contributes to the Alzheimer’s risk. Nat. Commun. 2019, 10, 3310. [Google Scholar] [CrossRef]
- Zhu, Z.; Yang, Y.; Xiao, Z.; Zhao, Q.; Wu, W.; Liang, X.; Luo, J.; Cao, Y.; Shao, M.; Guo, Q.; et al. TOMM40 and APOE variants synergistically increase the risk of Alzheimer’s disease in a Chinese population. Aging Clin. Exp. Res. 2021, 33, 1667–1675. [Google Scholar] [CrossRef]
- Michaelson, D.M. APOE epsilon4: The most prevalent yet understudied risk factor for Alzheimer’s disease. Alzheimers Dement. 2014, 10, 861–868. [Google Scholar] [CrossRef]
- Rebeck, G.W. The role of APOE on lipid homeostasis and inflammation in normal brains. J. Lipid Res. 2017, 58, 1493–1499. [Google Scholar] [CrossRef]
- Babenko, V.N.; Afonnikov, D.A.; Ignatieva, E.V.; Klimov, A.V.; Gusev, F.E.; Rogaev, E.I. Haplotype analysis of APOE intragenic SNPs. BMC Neurosci. 2018, 19 (Suppl. 1), 16. [Google Scholar] [CrossRef]
- Jun, G.; Naj, A.C.; Beecham, G.W.; Wang, L.S.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Ertekin-Taner, N.; Fallin, M.D.; Friedland, R.; et al. Meta-analysis confirms CR1, CLU, and PICALM as alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch. Neurol. 2010, 67, 1473–1484. [Google Scholar] [CrossRef]
- Hu, P.; Qin, Y.H.; Jing, C.X.; Lu, L.; Hu, B.; Du, P.F. Does the geographical gradient of ApoE4 allele exist in China? A systemic comparison among multiple Chinese populations. Mol. Biol. Rep. 2011, 38, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Lobo, A.; Launer, L.J.; Fratiglioni, L.; Andersen, K.; Di Carlo, A.; Breteler, M.M.; Copeland, J.R.; Dartigues, J.F.; Jagger, C.; Martinez-Lage, J.; et al. Prevalence of dementia and major subtypes in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000, 54 (Suppl. 5), S4–S9. [Google Scholar] [PubMed]
- Chiba-Falek, O.; Gottschalk, W.K.; Lutz, M.W. The effects of the TOMM40 poly-T alleles on Alzheimer’s disease phenotypes. Alzheimers Dement. 2018, 14, 692–698. [Google Scholar] [CrossRef]
- Lyall, D.M.; Harris, S.E.; Bastin, M.E.; Munoz Maniega, S.; Murray, C.; Lutz, M.W.; Saunders, A.M.; Roses, A.D.; Valdes Hernandez Mdel, C.; Royle, N.A.; et al. Alzheimer’s disease susceptibility genes APOE and TOMM40, and brain white matter integrity in the Lothian Birth Cohort 1936. Neurobiol. Aging 2014, 35, 1513.e25–1513.e33. [Google Scholar] [CrossRef] [PubMed]
- Ridge, P.G.; Hoyt, K.B.; Boehme, K.; Mukherjee, S.; Crane, P.K.; Haines, J.L.; Mayeux, R.; Farrer, L.A.; Pericak-Vance, M.A.; Alzheimer’s Disease Genetics Consortium; et al. Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobiol. Aging 2016, 41, 200.e13–200.e20. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, S.; Samaranch, L.; Vidal-Taboada, J.M.; Lamet, I.; Bullido, M.J.; Frank-Garcia, A.; Coria, F.; Lleo, A.; Clarimon, J.; Lorenzo, E.; et al. Genetic variation in APOE cluster region and Alzheimer’s disease risk. Neurobiol. Aging 2011, 32, 2107.e7–2107.e17. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Lutz, M.W.; Wilson, R.S.; Burns, D.K.; Roses, A.D.; Saunders, A.M.; Gaiteri, C.; De Jager, P.L.; Barnes, L.L.; Bennett, D.A. TOMM40’523 variant and cognitive decline in older persons with APOE epsilon3/3 genotype. Neurology 2017, 88, 661–668. [Google Scholar] [CrossRef]
- Ortega-Rojas, J.; Arboleda-Bustos, C.E.; Guerrero, E.; Neira, J.; Arboleda, H. Genetic Variants and Haplotypes of TOMM40, APOE, and APOC1 are Related to the Age of Onset of Late-onset Alzheimer Disease in a Colombian Population. Alzheimer Dis. Assoc. Disord. 2022, 36, 29–35. [Google Scholar] [CrossRef]
- Bagnoli, S.; Piaceri, I.; Tedde, A.; Bessi, V.; Bracco, L.; Sorbi, S.; Nacmias, B. TOMM40 polymorphisms in Italian Alzheimer’s disease and frontotemporal dementia patients. Neurol. Sci. 2013, 34, 995–998. [Google Scholar] [CrossRef]
- Lin, R.; Zhang, Y.; Yan, D.; Liao, X.; Gong, G.; Hu, J.; Fu, Y.; Cai, W. Association of common variants in TOMM40/APOE/APOC1 region with human longevity in a Chinese population. J. Hum. Genet. 2016, 61, 323–328. [Google Scholar] [CrossRef]
- Omoumi, A.; Fok, A.; Greenwood, T.; Sadovnick, A.D.; Feldman, H.H.; Hsiung, G.Y. Evaluation of late-onset Alzheimer disease genetic susceptibility risks in a Canadian population. Neurobiol. Aging 2014, 35, 936.e5–936.e12. [Google Scholar] [CrossRef] [PubMed]
- Zeitlow, K.; Charlambous, L.; Ng, I.; Gagrani, S.; Mihovilovic, M.; Luo, S.; Rock, D.L.; Saunders, A.; Roses, A.D.; Gottschalk, W.K. The biological foundation of the genetic association of TOMM40 with late-onset Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2973–2986. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; La Rue, A.; Hermann, B.P.; Xu, G.; Koscik, R.L.; Jonaitis, E.M.; Bendlin, B.B.; Hogan, K.J.; Roses, A.D.; Saunders, A.M.; et al. The effect of TOMM40 poly-T length on gray matter volume and cognition in middle-aged persons with APOE epsilon3/epsilon3 genotype. Alzheimers Dement. 2011, 7, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sarasua, S.M.; Davis, N.J.; DeLuca, J.M.; Boccuto, L.; Thielke, S.M.; Yu, C.E. TOMM40 genetic variants associated with healthy aging and longevity: A systematic review. BMC Geriatr. 2022, 22, 667. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Pappas, C.; Le, S.T.; Wang, Q.; Klinedinst, B.S.; Larsen, B.A.; Pollpeter, A.; Lee, L.Y.; Lutz, M.W.; Gottschalk, W.K.; et al. APOE, TOMM40, and sex interactions on neural network connectivity. Neurobiol. Aging 2022, 109, 158–165. [Google Scholar] [CrossRef]
- Li, Y.; Xia, X.; Wang, Y.; Zheng, J.C. Mitochondrial dysfunction in microglia: A novel perspective for pathogenesis of Alzheimer’s disease. J. Neuroinflamm. 2022, 19, 248. [Google Scholar] [CrossRef]
- Lin, M.M.; Liu, N.; Qin, Z.H.; Wang, Y. Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol. Sin. 2022, 43, 2439–2447. [Google Scholar] [CrossRef]
- Lee, S.; Cho, H.J.; Ryu, J.H. Innate Immunity and Cell Death in Alzheimer’s Disease. ASN Neuro. 2021, 13, 17590914211051908. [Google Scholar] [CrossRef]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar]
- Agrawal, I.; Jha, S. Mitochondrial Dysfunction and Alzheimer’s Disease: Role of Microglia. Front. Aging Neurosci. 2020, 12, 252. [Google Scholar] [CrossRef]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Pinho, C.M.; Teixeira, P.F.; Glaser, E. Mitochondrial import and degradation of amyloid-beta peptide. Biochim. Biophys. Acta 2014, 1837, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Roman, G.C.; Tatemichi, T.K.; Erkinjuntti, T.; Cummings, J.L.; Masdeu, J.C.; Garcia, J.H.; Amaducci, L.; Orgogozo, J.M.; Brun, A.; Hofman, A.; et al. Vascular dementia: Diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology 1993, 43, 250–260. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | SNP | Position | MAF (Cases/NC) | MAF * dbSNP | OR (95% CI) | p Value |
|---|---|---|---|---|---|---|
| PVRL2: Intron variant | rs394221 | 45368424 | 0.51/0.38 | 0.45 | 1.7 (1.2~2.4) | 0.001 |
| TOMM40:Synonymous, p.Ser66= | rs772262361 | 45394870 | 0.013/0.0 | 0.00004 | - | - |
| TOMM40: Intron | rs184017 | 45394969 | 0.34/0.16 | 0.20 | 2.8 (2.0~4.0) | 4.2 × 10−8 |
| TOMM40: Intron | rs2075650 | 45395619 | 0.25/0.07 | 0.13 | 4.2 (2.8~6.1) | 1.1 × 10−10 |
| TOMM40: Missense, p.Phe113Leu | rs157581 * | 45395714 | 0.38/0.23 | 0.23 | 2.1 (1.5~2.9) | 4.4 × 10−5 |
| TOMM40: Missense, p.Phe131Leu | rs11556505 * | 45396144 | 0.26/0.10 | 0.11 | 3.3 (2.2~4.8) | 2.5 × 10−8 |
| TOMM40: Intron | rs157582 | 45396219 | 0.34/0.18 | 0.22 | 2.4 (1.7~3.3) | 3.3 × 10−6 |
| APOE: Missense, p.Asn14Lys | rs440446 * | 45409167 | 0.56/0.33 | 0.38 | 2.6 (1.9~3.6) | 1.4 × 10−8 |
| APOE: Intron | rs769449 | 45412079 | 0.25/0.08 | 0.11 | 3.6 (2.4~5.3) | 2.7 × 10−9 |
| NC | AD Patients | Controls * | p Value 1 | p Value 2 | |
|---|---|---|---|---|---|
| Number | 213 | 393 | 1025 | ||
| Age (years) | 67.4 ± 10.3 | 74.0 ± 8.7 | 58.7 ± 5.4 | <0.0001 | <0.0001 |
| Men, N (%) | 111 (52.1) | 142 (36.1) | 524 (51.1) | 0.0001 | <0.0001 |
| Education (years) | 8.1 ± 4.5 | 6.2 ± 4.7 | 5.3 ± 1.1 | <0.0001 | <0.0001 |
| Hypertension, N (%) | 99 (46.5) | 191 (48.6) | 207 (20.2) | 0.62 | <0.0001 |
| Diabetes, N (%) | 153 (71.8) | 271 (69.0) | 81 (7.9) | 0.46 | <0.0001 |
| Global cognition, MMSE | 24.3 ± 5.4 | 16.5 ± 6.5 | 27.1 ± 2.6 | <0.0001 | <0.0001 |
| APOE ε4 carrier, N (%) | 12 (5.63) | 201 (51.2) | 166 (16.2) | <0.0001 | <0.0001 |
| TOMM40 | |||||
| rs772262361, p.Ser66 = AA/AG (%) | 100/0 | 99.5/0.5 | - | 0.24 ** | - |
| rs157581, p.Phe113Leu TT/TC/CC (%) | 67.3/30.3/2.4 | 40/49.9/10.1 | 59.9/35/5.1 | <0.0001 | <0.0001 |
| rs11556505, p.Phe131Leu CC/CT/TT (%) | 92.6/7.4/0 | 54.4/40/5.6 | 81.9/17.3/0.9 | <0.0001 | <0.0001 |
| NC (n = 45) | AD (n = 37) | AD with TOMM40 Genetic Variants (n = 14) | p Value | |
|---|---|---|---|---|
| Age (years) | 72.1 ± 9.0 | 72.9 ± 6.0 | 71.29 ± 6.3 | 0.6740 |
| Men/Female | 21/24 | 14/23 | 4/10 | 0.4275 |
| APOE ε4 carrier (%) | 0 | 32.4 | 85.7 | <0.0001 |
| IL-1β (pg/mL) | 21.4 ± 8.4 | 23.1 ± 10.8 | 23.3 ± 8.6 | 0.3884 |
| IL-6 (pg/mL) | 0.8 ± 1.6 | 1.6 ± 1.5 | 2.3 ± 1.6 | 0.0237 |
| IL-18 (pg/mL) | 64.4 ± 53.0 | 193.5 ± 200.6 | 228.66 ± 164.7 | <0.0001 |
| IL-23 (pg/mL) | 9.7 ± 3.2 | 10.5 ± 8.9 | 13.7 ± 13.9 | 0.5867 |
| IL-33 (pg/mL) | 4.4 ± 0.8 | 4.9 ± 0.3 | 5.0 ± 0.2 | 0.0005 |
| TNF-α (pg/mL) | 13.8 ± 8.1 | 14.5 ± 18.9 | 17.1 ± 28.8 | 0.9342 |
| COX-2 (ng/mL) | 0.5 ± 0.06 | 0.7 ± 0.7 | 0.9 ± 1.1 | 0.0453 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-C.; Chang, S.-C.; Lee, Y.-S.; Ho, W.-M.; Huang, Y.-H.; Wu, Y.-Y.; Chu, Y.-C.; Wu, K.-H.; Wei, L.-S.; Wang, H.-L.; et al. TOMM40 Genetic Variants Cause Neuroinflammation in Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 4085. https://doi.org/10.3390/ijms24044085
Chen Y-C, Chang S-C, Lee Y-S, Ho W-M, Huang Y-H, Wu Y-Y, Chu Y-C, Wu K-H, Wei L-S, Wang H-L, et al. TOMM40 Genetic Variants Cause Neuroinflammation in Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(4):4085. https://doi.org/10.3390/ijms24044085
Chicago/Turabian StyleChen, Yi-Chun, Shih-Cheng Chang, Yun-Shien Lee, Wei-Min Ho, Yu-Hua Huang, Yah-Yuan Wu, Yi-Chuan Chu, Kuan-Hsuan Wu, Li-Shan Wei, Hung-Li Wang, and et al. 2023. "TOMM40 Genetic Variants Cause Neuroinflammation in Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 4: 4085. https://doi.org/10.3390/ijms24044085
APA StyleChen, Y.-C., Chang, S.-C., Lee, Y.-S., Ho, W.-M., Huang, Y.-H., Wu, Y.-Y., Chu, Y.-C., Wu, K.-H., Wei, L.-S., Wang, H.-L., & Chiu, C.-C. (2023). TOMM40 Genetic Variants Cause Neuroinflammation in Alzheimer’s Disease. International Journal of Molecular Sciences, 24(4), 4085. https://doi.org/10.3390/ijms24044085