
DIS3: The Enigmatic Gene in Multiple Myeloma

Abstract
1. Introduction
2. DIS3: A Catalytic Subunit of the RNA Exosome
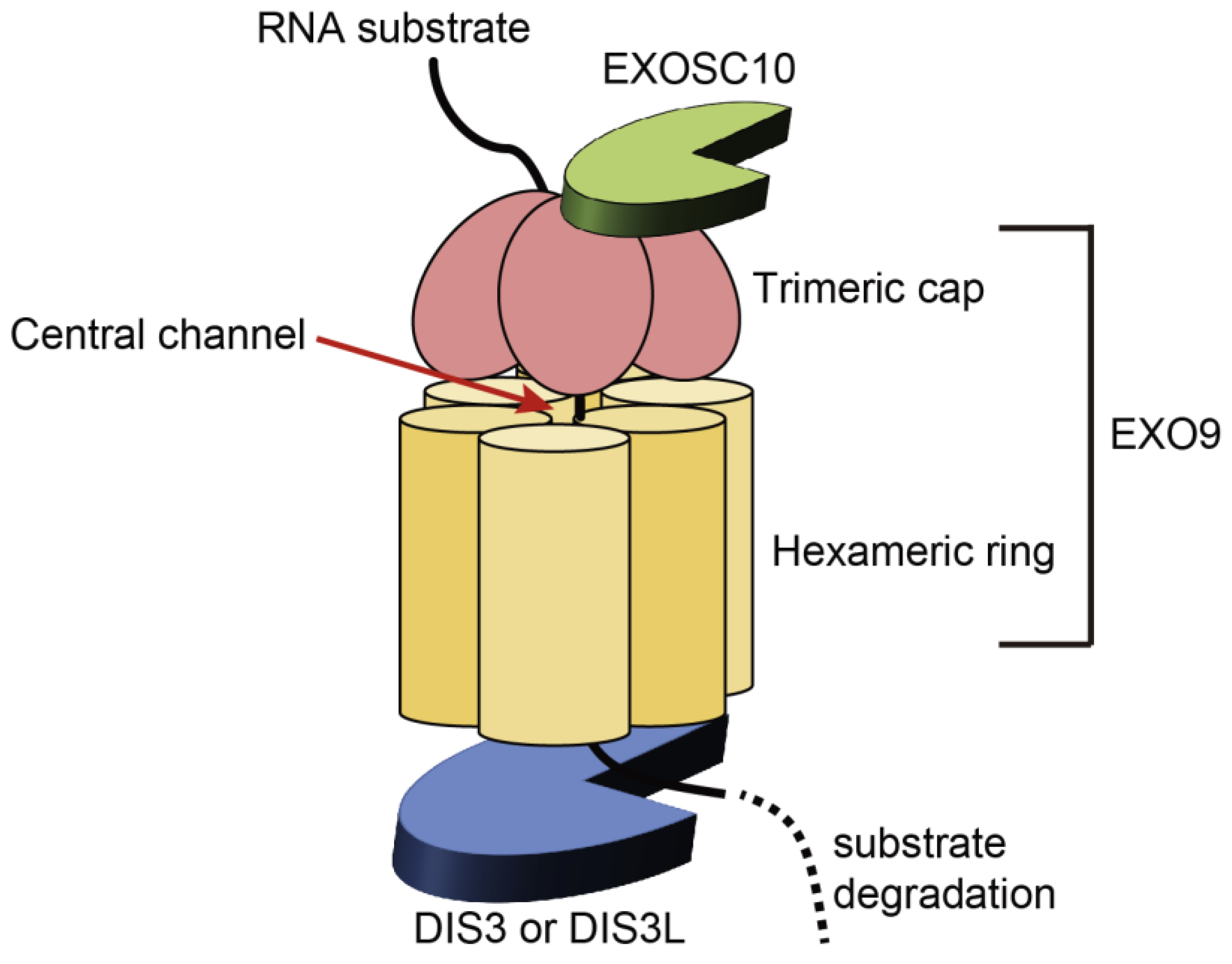
2.1. Structure and Functions of the RNA Exosome
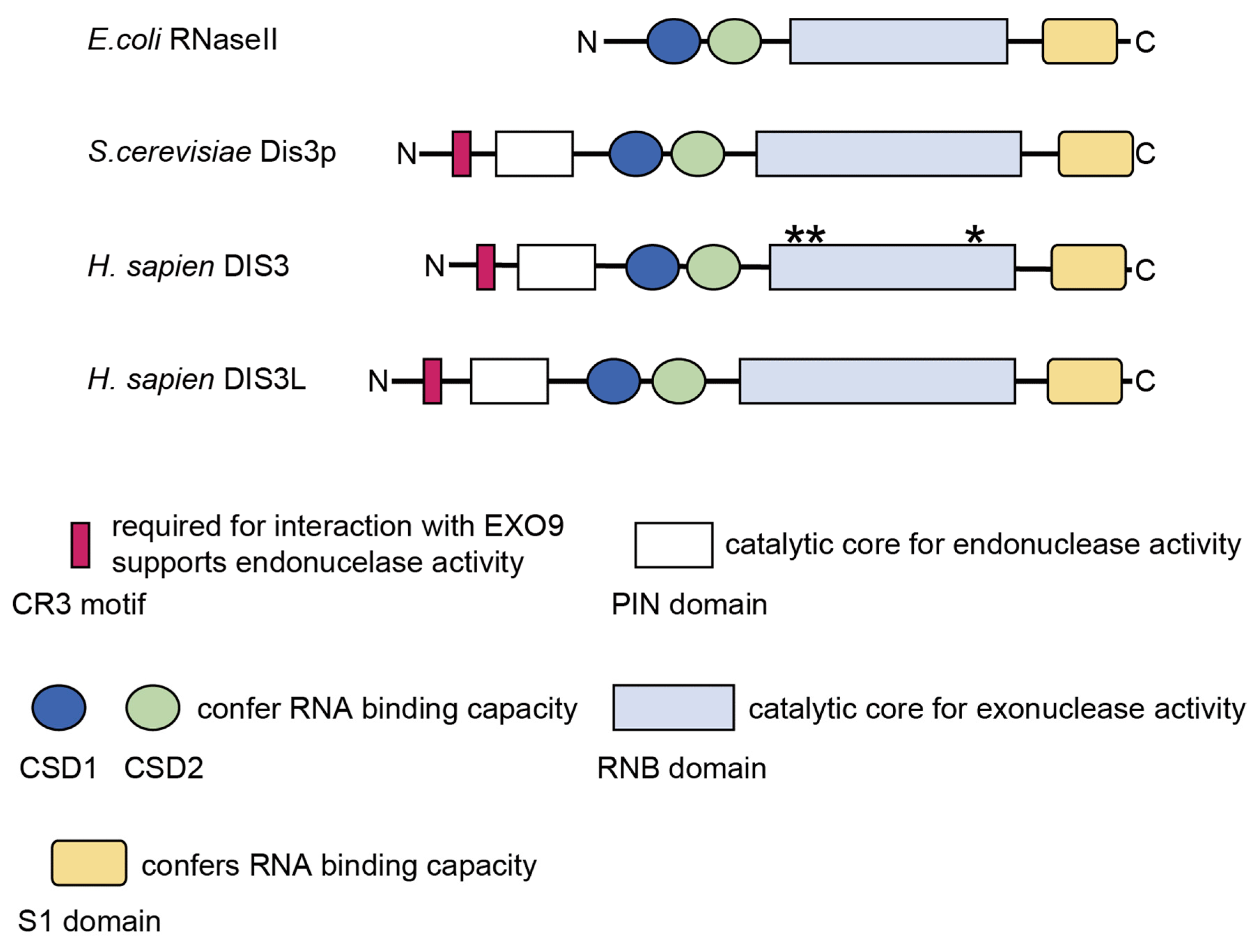
2.2. Structure and Molecular Functions of DIS3
3. Physiological Functions of DIS3 in Hematopoiesis
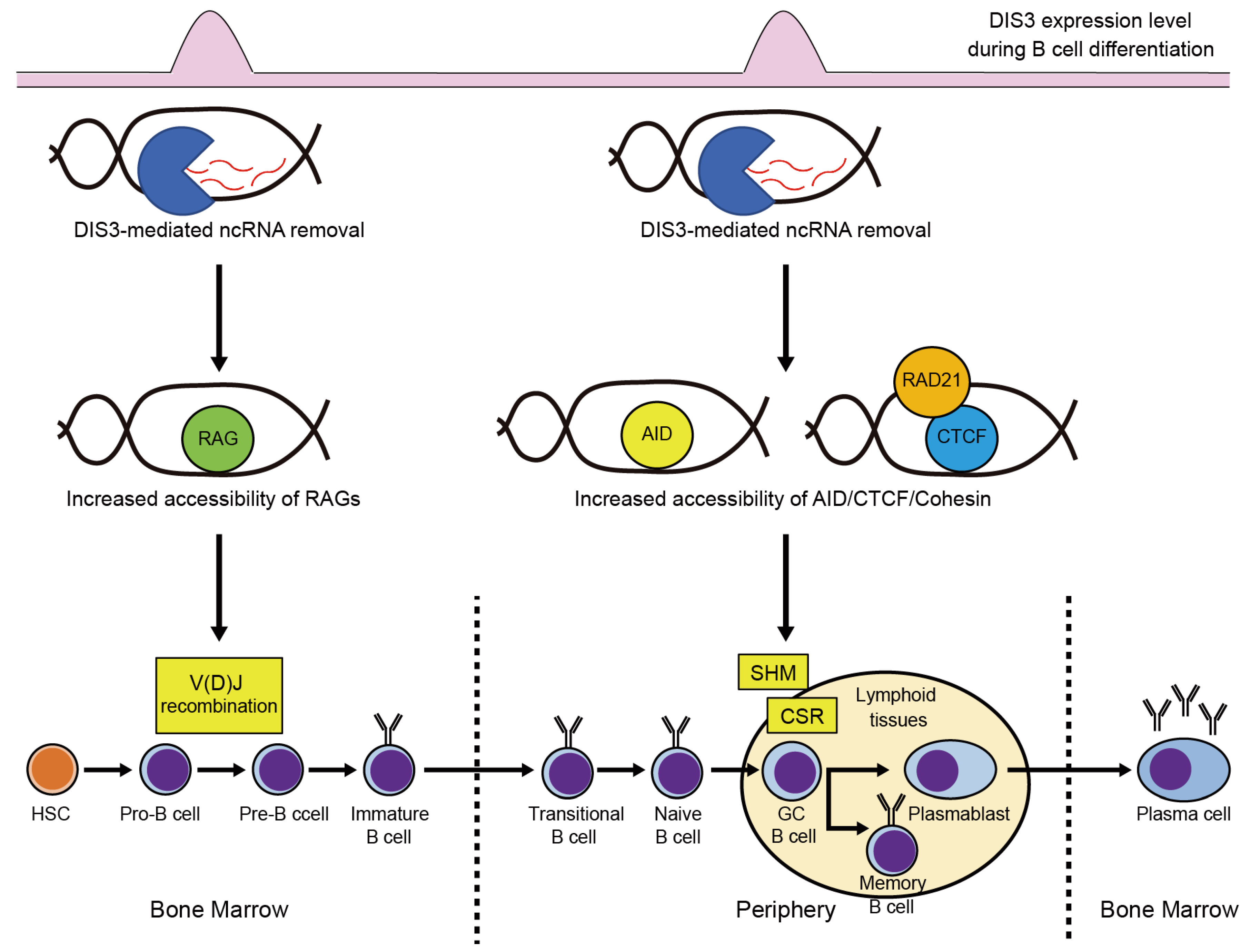
3.1. DIS3 Functions in B Cell Development
3.2. DIS3 Functions in Erythropoiesis
4. Characteristics and Clinical Impact of DIS3 Mutations in MM
4.1. Characteristics of DIS3 Mutations in MM
4.2. Correlations of DIS3 Mutations with Cytogenetic Abnormalities and Other Mutations in MM
4.3. Association of DIS3 Mutations with Prognosis in MM
4.4. Transcriptional Profiles of MM Patients with DIS3 Mutations
5. Pathological Functions of DIS3 Hypomorphism in MM
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Prim. 2017, 3, 17046. [Google Scholar] [CrossRef]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef]
- Pawlyn, C.; Morgan, G.J. Evolutionary biology of high-risk multiple myeloma. Nat. Rev. Cancer 2017, 17, 543–556. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef]
- Mroczek, S.; Chlebowska, J.; Kulinski, T.M.; Gewartowska, O.; Gruchota, J.; Cysewski, D.; Liudkovska, V.; Borsuk, E.; Nowis, D.; Dziembowski, A. The non-canonical poly(A) polymerase FAM46C acts as an onco-suppressor in multiple myeloma. Nat. Commun. 2017, 8, 619. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Shi, C.X.; Bruins, L.A.; Jedlowski, P.; Wang, X.; Kortum, K.M.; Luo, M.; Ahmann, J.M.; Braggio, E.; Stewart, A.K. Loss of FAM46C Promotes Cell Survival in Myeloma. Cancer Res. 2017, 77, 4317–4327. [Google Scholar] [CrossRef]
- Bilska, A.; Kusio-Kobialka, M.; Krawczyk, P.S.; Gewartowska, O.; Tarkowski, B.; Kobylecki, K.; Nowis, D.; Golab, J.; Gruchota, J.; Borsuk, E.; et al. Immunoglobulin expression and the humoral immune response is regulated by the non-canonical poly(A) polymerase TENT5C. Nat. Commun. 2020, 11, 2032. [Google Scholar] [CrossRef]
- Fucci, C.; Resnati, M.; Riva, E.; Perini, T.; Ruggieri, E.; Orfanelli, U.; Paradiso, F.; Cremasco, F.; Raimondi, A.; Pasqualetto, E.; et al. The Interaction of the Tumor Suppressor FAM46C with p62 and FNDC3 Proteins Integrates Protein and Secretory Homeostasis. Cell Rep. 2020, 32, 108162. [Google Scholar] [CrossRef]
- Kanasugi, J.; Hanamura, I.; Ota, A.; Karnan, S.; Lam, V.Q.; Mizuno, S.; Wahiduzzaman, M.; Rahman, M.L.; Hyodo, T.; Konishi, H.; et al. Biallelic loss of FAM46C triggers tumor growth with concomitant activation of Akt signaling in multiple myeloma cells. Cancer Sci. 2020, 111, 1663–1675. [Google Scholar] [CrossRef]
- Manfrini, N.; Mancino, M.; Miluzio, A.; Oliveto, S.; Balestra, M.; Calamita, P.; Alfieri, R.; Rossi, R.L.; Sassoe-Pognetto, M.; Salio, C.; et al. FAM46C and FNDC3A Are Multiple Myeloma Tumor Suppressors That Act in Concert to Impair Clearing of Protein Aggregates and Autophagy. Cancer Res. 2020, 80, 4693–4706. [Google Scholar] [CrossRef]
- Perini, T.; Materozzi, M.; Milan, E. The Immunity-malignancy equilibrium in multiple myeloma: Lessons from oncogenic events in plasma cells. FEBS J. 2022, 289, 4383–4397. [Google Scholar] [CrossRef]
- Houseley, J.; LaCava, J.; Tollervey, D. RNA-quality control by the exosome. Nat. Rev. Mol. Cell Biol. 2006, 7, 529–539. [Google Scholar] [CrossRef]
- Lykke-Andersen, S.; Tomecki, R.; Jensen, T.H.; Dziembowski, A. The eukaryotic RNA exosome: Same scaffold but variable catalytic subunits. RNA Biol. 2011, 8, 61–66. [Google Scholar] [CrossRef]
- Kilchert, C.; Wittmann, S.; Vasiljeva, L. The regulation and functions of the nuclear RNA exosome complex. Nat. Rev. Mol. Cell Biol. 2016, 17, 227–239. [Google Scholar] [CrossRef]
- Schmid, M.; Jensen, T.H. Controlling nuclear RNA levels. Nat. Rev. Genet. 2018, 19, 518–529. [Google Scholar] [CrossRef]
- Makino, D.L.; Baumgartner, M.; Conti, E. Crystal structure of an RNA-bound 11-subunit eukaryotic exosome complex. Nature 2013, 495, 70–75. [Google Scholar] [CrossRef]
- Bonneau, F.; Basquin, J.; Ebert, J.; Lorentzen, E.; Conti, E. The yeast exosome functions as a macromolecular cage to channel RNA substrates for degradation. Cell 2009, 139, 547–559. [Google Scholar] [CrossRef]
- Wasmuth, E.V.; Lima, C.D. Exo- and endoribonucleolytic activities of yeast cytoplasmic and nuclear RNA exosomes are dependent on the noncatalytic core and central channel. Mol. Cell 2012, 48, 133–144. [Google Scholar] [CrossRef]
- Tomecki, R.; Kristiansen, M.S.; Lykke-Andersen, S.; Chlebowski, A.; Larsen, K.M.; Szczesny, R.J.; Drazkowska, K.; Pastula, A.; Andersen, J.S.; Stepien, P.P.; et al. The human core exosome interacts with differentially localized processive RNases: hDIS3 and hDIS3L. EMBO J. 2010, 29, 2342–2357. [Google Scholar] [CrossRef]
- Staals, R.H.; Bronkhorst, A.W.; Schilders, G.; Slomovic, S.; Schuster, G.; Heck, A.J.; Raijmakers, R.; Pruijn, G.J. Dis3-like 1: A novel exoribonuclease associated with the human exosome. EMBO J 2010, 29, 2358–2367. [Google Scholar] [CrossRef]
- Drazkowska, K.; Tomecki, R.; Stodus, K.; Kowalska, K.; Czarnocki-Cieciura, M.; Dziembowski, A. The RNA exosome complex central channel controls both exonuclease and endonuclease Dis3 activities in vivo and in vitro. Nucleic Acids Res. 2013, 41, 3845–3858. [Google Scholar] [CrossRef] [PubMed]
- Wasmuth, E.V.; Januszyk, K.; Lima, C.D. Structure of an Rrp6-RNA exosome complex bound to poly(A) RNA. Nature 2014, 511, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Makino, D.L.; Schuch, B.; Stegmann, E.; Baumgartner, M.; Basquin, C.; Conti, E. RNA degradation paths in a 12-subunit nuclear exosome complex. Nature 2015, 524, 54–58. [Google Scholar] [CrossRef]
- Schneider, C.; Kudla, G.; Wlotzka, W.; Tuck, A.; Tollervey, D. Transcriptome-wide analysis of exosome targets. Mol. Cell 2012, 48, 422–433. [Google Scholar] [CrossRef]
- Mitchell, P. Exosome substrate targeting: The long and short of it. Biochem. Soc. Trans. 2014, 42, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef]
- Ng, D.; Toure, O.; Wei, M.H.; Arthur, D.C.; Abbasi, F.; Fontaine, L.; Marti, G.E.; Fraumeni, J.F., Jr.; Goldin, L.R.; Caporaso, N.; et al. Identification of a novel chromosome region, 13q21.33-q22.2, for susceptibility genes in familial chronic lymphocytic leukemia. Blood 2007, 109, 916–925. [Google Scholar] [CrossRef]
- Morton, D.J.; Kuiper, E.G.; Jones, S.K.; Leung, S.W.; Corbett, A.H.; Fasken, M.B. The RNA exosome and RNA exosome-linked disease. RNA 2018, 24, 127–142. [Google Scholar] [CrossRef]
- Di Donato, N.; Neuhann, T.; Kahlert, A.K.; Klink, B.; Hackmann, K.; Neuhann, I.; Novotna, B.; Schallner, J.; Krause, C.; Glass, I.A.; et al. Mutations in EXOSC2 are associated with a novel syndrome characterised by retinitis pigmentosa, progressive hearing loss, premature ageing, short stature, mild intellectual disability and distinctive gestalt. J. Med. Genet. 2016, 53, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.R.; Oliver, A.W.; Chevassut, T.J.; Newbury, S.F. The 3’ to 5’ Exoribonuclease DIS3: From Structure and Mechanisms to Biological Functions and Role in Human Disease. Biomolecules 2015, 5, 1515–1539. [Google Scholar] [CrossRef]
- Dziembowski, A.; Lorentzen, E.; Conti, E.; Seraphin, B. A single subunit, Dis3, is essentially responsible for yeast exosome core activity. Nat. Struct. Mol. Biol. 2007, 14, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Lebreton, A.; Tomecki, R.; Dziembowski, A.; Seraphin, B. Endonucleolytic RNA cleavage by a eukaryotic exosome. Nature 2008, 456, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, D.; Tsanova, B.; Barbas, A.; Reis, F.P.; Dastidar, E.G.; Sanchez-Rotunno, M.; Arraiano, C.M.; van Hoof, A. The exosome contains domains with specific endoribonuclease, exoribonuclease and cytoplasmic mRNA decay activities. Nat. Struct. Mol. Biol. 2009, 16, 56–62. [Google Scholar] [CrossRef]
- Schneider, C.; Leung, E.; Brown, J.; Tollervey, D. The N-terminal PIN domain of the exosome subunit Rrp44 harbors endonuclease activity and tethers Rrp44 to the yeast core exosome. Nucleic Acids Res. 2009, 37, 1127–1140. [Google Scholar] [CrossRef] [PubMed]
- Szczepinska, T.; Kalisiak, K.; Tomecki, R.; Labno, A.; Borowski, L.S.; Kulinski, T.M.; Adamska, D.; Kosinska, J.; Dziembowski, A. DIS3 shapes the RNA polymerase II transcriptome in humans by degrading a variety of unwanted transcripts. Genome Res. 2015, 25, 1622–1633. [Google Scholar] [CrossRef]
- Frazao, C.; McVey, C.E.; Amblar, M.; Barbas, A.; Vonrhein, C.; Arraiano, C.M.; Carrondo, M.A. Unravelling the dynamics of RNA degradation by ribonuclease II and its RNA-bound complex. Nature 2006, 443, 110–114. [Google Scholar] [CrossRef]
- Schaeffer, D.; Reis, F.P.; Johnson, S.J.; Arraiano, C.M.; van Hoof, A. The CR3 motif of Rrp44p is important for interaction with the core exosome and exosome function. Nucleic Acids Res. 2012, 40, 9298–9307. [Google Scholar] [CrossRef]
- Chang, H.M.; Triboulet, R.; Thornton, J.E.; Gregory, R.I. A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28-let-7 pathway. Nature 2013, 497, 244–248. [Google Scholar] [CrossRef]
- Davidson, L.; Francis, L.; Cordiner, R.A.; Eaton, J.D.; Estell, C.; Macias, S.; Caceres, J.F.; West, S. Rapid Depletion of DIS3, EXOSC10, or XRN2 Reveals the Immediate Impact of Exoribonucleolysis on Nuclear RNA Metabolism and Transcriptional Control. Cell Rep. 2019, 26, 2779–2791. [Google Scholar] [CrossRef]
- Wyers, F.; Rougemaille, M.; Badis, G.; Rousselle, J.C.; Dufour, M.E.; Boulay, J.; Regnault, B.; Devaux, F.; Namane, A.; Seraphin, B.; et al. Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell 2005, 121, 725–737. [Google Scholar] [CrossRef]
- Preker, P.; Nielsen, J.; Kammler, S.; Lykke-Andersen, S.; Christensen, M.S.; Mapendano, C.K.; Schierup, M.H.; Jensen, T.H. RNA exosome depletion reveals transcription upstream of active human promoters. Science 2008, 322, 1851–1854. [Google Scholar] [CrossRef]
- Fox, A.H.; Lamond, A.I. Paraspeckles. Cold Spring Harb. Perspect. Biol. 2010, 2, a000687. [Google Scholar] [CrossRef]
- Laffleur, B.; Lim, J.; Zhang, W.; Chen, Y.; Pefanis, E.; Bizarro, J.; Batista, C.R.; Wu, L.; Economides, A.N.; Wang, J.; et al. Noncoding RNA processing by DIS3 regulates chromosomal architecture and somatic hypermutation in B cells. Nat. Genet. 2021, 53, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Laffleur, B.; Batista, C.R.; Zhang, W.; Lim, J.; Yang, B.; Rossille, D.; Wu, L.; Estrella, J.; Rothschild, G.; Pefanis, E.; et al. RNA exosome drives early B cell development via noncoding RNA processing mechanisms. Sci. Immunol. 2022, 7, eabn2738. [Google Scholar] [CrossRef] [PubMed]
- Klein, U.; Dalla-Favera, R. Germinal centres: Role in B-cell physiology and malignancy. Nat. Rev. Immunol. 2008, 8, 22–33. [Google Scholar] [CrossRef]
- Milbury, K.L.; Paul, B.; Lari, A.; Fowler, C.; Montpetit, B.; Stirling, P.C. Exonuclease domain mutants of yeast DIS3 display genome instability. Nucleus 2019, 10, 21–32. [Google Scholar] [CrossRef]
- McIver, S.C.; Kang, Y.A.; DeVilbiss, A.W.; O’Driscoll, C.A.; Ouellette, J.N.; Pope, N.J.; Camprecios, G.; Chang, C.J.; Yang, D.; Bouhassira, E.E.; et al. The exosome complex establishes a barricade to erythroid maturation. Blood 2014, 124, 2285–2297. [Google Scholar] [CrossRef] [PubMed]
- Mehta, C.; Fraga de Andrade, I.; Matson, D.R.; Dewey, C.N.; Bresnick, E.H. RNA-regulatory exosome complex confers cellular survival to promote erythropoiesis. Nucleic Acids Res. 2021, 49, 9007–9025. [Google Scholar] [CrossRef]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef]
- Weissbach, S.; Langer, C.; Puppe, B.; Nedeva, T.; Bach, E.; Kull, M.; Bargou, R.; Einsele, H.; Rosenwald, A.; Knop, S.; et al. The molecular spectrum and clinical impact of DIS3 mutations in multiple myeloma. Br. J. Haematol. 2015, 169, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, M.; Barbieri, M.; Todoerti, K.; Agnelli, L.; Fabris, S.; Tonon, G.; Segalla, S.; Cifola, I.; Pinatel, E.; Tassone, P.; et al. A compendium of DIS3 mutations and associated transcriptional signatures in plasma cell dyscrasias. Oncotarget 2015, 6, 26129–26141. [Google Scholar] [CrossRef]
- Todoerti, K.; Ronchetti, D.; Favasuli, V.; Maura, F.; Morabito, F.; Bolli, N.; Taiana, E.; Neri, A. DIS3 mutations in multiple myeloma impact the transcriptional signature and clinical outcome. Haematologica 2022, 107, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Pertesi, M.; Vallee, M.; Wei, X.; Revuelta, M.V.; Galia, P.; Demangel, D.; Oliver, J.; Foll, M.; Chen, S.; Perrial, E.; et al. Exome sequencing identifies germline variants in DIS3 in familial multiple myeloma. Leukemia 2019, 33, 2324–2330. [Google Scholar] [CrossRef] [PubMed]
- Lorentzen, E.; Basquin, J.; Tomecki, R.; Dziembowski, A.; Conti, E. Structure of the active subunit of the yeast exosome core, Rrp44: Diverse modes of substrate recruitment in the RNase II nuclease family. Mol. Cell 2008, 29, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Tomecki, R.; Drazkowska, K.; Kucinski, I.; Stodus, K.; Szczesny, R.J.; Gruchota, J.; Owczarek, E.P.; Kalisiak, K.; Dziembowski, A. Multiple myeloma-associated hDIS3 mutations cause perturbations in cellular RNA metabolism and suggest hDIS3 PIN domain as a potential drug target. Nucleic Acids Res. 2014, 42, 1270–1290. [Google Scholar] [CrossRef]
- Boyle, E.M.; Ashby, C.; Tytarenko, R.G.; Deshpande, S.; Wang, H.; Wang, Y.; Rosenthal, A.; Sawyer, J.; Tian, E.; Flynt, E.; et al. BRAF and DIS3 Mutations Associate with Adverse Outcome in a Long-term Follow-up of Patients with Multiple Myeloma. Clin. Cancer Res. 2020, 26, 2422–2432. [Google Scholar] [CrossRef] [PubMed]
- Gritti, I.; Basso, V.; Rinchai, D.; Corigliano, F.; Pivetti, S.; Gaviraghi, M.; Rosano, D.; Mazza, D.; Barozzi, S.; Roncador, M.; et al. Loss of ribonuclease DIS3 hampers genome integrity in myeloma by disrupting DNA:RNA hybrid metabolism. EMBO J. 2022, 41, e108040. [Google Scholar] [CrossRef]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef]
- Roush, S.; Slack, F.J. The let-7 family of microRNAs. Trends Cell Biol. 2008, 18, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Segalla, S.; Pivetti, S.; Todoerti, K.; Chudzik, M.A.; Giuliani, E.C.; Lazzaro, F.; Volta, V.; Lazarevic, D.; Musco, G.; Muzi-Falconi, M.; et al. The ribonuclease DIS3 promotes let-7 miRNA maturation by degrading the pluripotency factor LIN28B mRNA. Nucleic Acids Res. 2015, 43, 5182–5193. [Google Scholar] [CrossRef] [PubMed]
- Snee, M.J.; Wilson, W.C.; Zhu, Y.; Chen, S.Y.; Wilson, B.A.; Kseib, C.; O’Neal, J.; Mahajan, N.; Tomasson, M.H.; Arur, S.; et al. Collaborative Control of Cell Cycle Progression by the RNA Exonuclease Dis3 and Ras Is Conserved Across Species. Genetics 2016, 203, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Yates, L.R.; Campbell, P.J. Evolution of the cancer genome. Nat. Rev. Genet. 2012, 13, 795–806. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Target RNAs | Exosome Function |
|---|---|---|
| Nucleus | Pervasive transcripts (PROMPTs and eRNAs) | Degradation |
| Nucleus | Defective RNAs (rRNAs, tRNAs, mRNAs, snRNAs, and snoRNAs) | Degradation (surveillance) |
| Nucleus | Precursor RNAs (rRNAs, snRNAs, and snoRNAs) | Processing (maturation) |
| Cytoplasm | Normal mRNAs | Degradation (turnover) |
| Cytoplasm | Defective mRNAs | Degradation (surveillance) |
| Cytoplasm | mRNAs harboring ARE | Degradation (ARE-mediated decay) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohguchi, Y.; Ohguchi, H. DIS3: The Enigmatic Gene in Multiple Myeloma. Int. J. Mol. Sci. 2023, 24, 4079. https://doi.org/10.3390/ijms24044079
Ohguchi Y, Ohguchi H. DIS3: The Enigmatic Gene in Multiple Myeloma. International Journal of Molecular Sciences. 2023; 24(4):4079. https://doi.org/10.3390/ijms24044079
Chicago/Turabian StyleOhguchi, Yasuyo, and Hiroto Ohguchi. 2023. "DIS3: The Enigmatic Gene in Multiple Myeloma" International Journal of Molecular Sciences 24, no. 4: 4079. https://doi.org/10.3390/ijms24044079
APA StyleOhguchi, Y., & Ohguchi, H. (2023). DIS3: The Enigmatic Gene in Multiple Myeloma. International Journal of Molecular Sciences, 24(4), 4079. https://doi.org/10.3390/ijms24044079