Abstract
Cancer is the second leading contributor to global deaths caused by non-communicable diseases. The cancer cells are known to interact with the surrounding non-cancerous cells, including the immune cells and stromal cells, within the tumor microenvironment (TME) to modulate the tumor progression, metastasis and resistance. Currently, chemotherapy and radiotherapy are the standard treatments for cancers. However, these treatments cause a significant number of side effects, as they damage both the cancer cells and the actively dividing normal cells indiscriminately. Hence, a new generation of immunotherapy using natural killer (NK) cells, cytotoxic CD8+ T-lymphocytes or macrophages was developed to achieve tumor-specific targeting and circumvent the adverse effects. However, the progression of cell-based immunotherapy is hindered by the combined action of TME and TD-EVs, which render the cancer cells less immunogenic. Recently, there has been an increase in interest in using immune cell derivatives to treat cancers. One of the highly potential immune cell derivatives is the NK cell-derived EVs (NK-EVs). As an acellular product, NK-EVs are resistant to the influence of TME and TD-EVs, and can be designed for “off-the-shelf” use. In this systematic review, we examine the safety and efficacy of NK-EVs to treat various cancers in vitro and in vivo.
1. Introduction
According to the World Health Organization (WHO), the COVID-19 pandemic has claimed over 6.6 million lives globally [1]. Nonetheless, the incidence of infections and deaths has declined significantly with the availability of vaccines. Contradictorily, solutions for cancer treatment still remain obscure today. In 2021 alone, the WHO recorded 9.3 million global deaths from neoplasms [2]. Chemotherapy and radiotherapy are the standard treatment for cancerous tumors which cannot be removed entirely through surgery, despite the accompanying adverse effects [3,4]. These drugs and radiations damage the actively proliferating cancer cells and dividing normal cell indiscriminately [5,6,7,8], leading to deterioration in patient health by causing severe weight loss, poor skin conditions and hair loss (alopecia) [9,10]. The adverse effects of chemotherapy and radiotherapy in weakening patients’ health and immune system have yet to be resolved despite many years of refinement. In fact, criticisms from patients receiving these treatments discourages many from enrolling as their physical and mental wellbeing are at risk. The percentage of patients refusing to have or continue their chemotherapy is reported to be as high as 3–19% [11]. All these limitations demonstrate the importance of developing novel treatment for cancer which is highly specific and effective in eradicating the cancer cells without affecting the healthy cells.
Stem cells have been studied extensively in the field of regenerative medicine due to their excellent regenerative and anti-inflammatory properties [12]. They have been regarded as a viable solution for many age-related and chronic degenerative illnesses. This development has piqued the interest of utilizing stem cells for other therapeutic applications, including cancer immunotherapy. The development of cell-based immunotherapy can be achieved by establishing cell lines that express tumor selectivity and cytotoxic functions. Several prospective candidates that have been rigorously studied are natural killer (NK) cells, dendritic cells (DC), macrophages and T-lymphocytes [13,14,15,16]. These cells play critical roles in alerting the immune system (e.g., chemotaxis), arming other immune cells (e.g., antibody-dependent and cytokine-dependent activation) and exerting their cytotoxic effect against the cancer cells (e.g., receptor-mediated cell apoptosis). DC and CD4+ T-cells (T-helper cells) are important components of the immune system in that they serve as the moderators of various immune responses [17,18]. They produce molecular signals, e.g., cytokines and chemokines, to guide and activate other immune cells. Conversely, NK cells, macrophages and CD8+ T-cells (cytotoxic T-cells) are effector cells that exert a cytotoxic effect against the cancer cells [19,20,21,22].
The CD8+ T-cell’s mechanism of action is through the antibody-dependent cellular cytotoxicity (ADCC). ADCC is initiated by the binding of T-cell receptors (TCR) to the antigen presented by major histocompatibility complex-1 (MHC-1) [23]. NK cells also can eliminate the target cells via ADCC pathway through the cluster of differentiation-16 (CD16) or FcγRIII on its plasma membrane [24,25]. Upon activation, these cells will release cytolytic enzymes (e.g., caspases, granzymes (Gzm) and perforin (PFN)) to damage the target cells. Meanwhile, macrophages elicit antibody-dependent cellular phagocytosis (ADCP), which involves engulfment and degradation of the internalized cells via phagosome acidification [26,27]. Since mutational activities are frequent in cancer cells, antigen or peptide reconfigurations or direct inhibition of cytotoxic receptor (e.g., CA-125 inhibits Fcy-receptor) can disable ADCC/ADCP entirely [27,28,29,30,31]. In the event ADCC and ADCP are inhibited, the immune cells still can eliminate the cancer cells via the TRAIL and Fas signalling pathways. TRAIL and Fas receptors are “death receptors” which belong to the receptor family of tumor-necrosis factors (TNF) that induce programmed cell death when they are cross-linked by its ligands [32,33,34]. In addition, certain immune cells also secrete lytic granules containing Gzm and PFN that induce lysis or apoptosis of target cells via degranulation [35,36,37].
In spite of its potential, the progression of cell-based immunotherapy has been hampered by the limitations that it may cause life-threatening adverse reactions, laborious production procedure, limited efficacy against solid tumors due to insufficient trafficking and homing, and variable efficacy owing to immune cell inactivation by the tumors [38,39]. Recently, purification of cell biologics was made possible by the new innovations in isolation technique. By using these advanced isolation technologies, researchers have isolated the extracellular vesicles (EVs) produced by the immune cells and use them as cancer immunotherapy. In that regard, immune cells have been repurposed as biological manufacturers of these membrane-bound nanovesicles [40]. NK cells-derived EV (NK-EV) is being explored in many studies as it is rich in cytotoxic proteins, cytokines and miRNAs that selectively kill the tumor cells [41]. In the interest of this development, this systematic review was performed to determine the potential of NK-EVs as cancer immunotherapy based on data collected from the in vitro and in vivo experiments. EVs can be classified based on their biogenesis and size into exosomes (EXO), microvesicles (MVs) and apoptotic bodies [42,43]. In this review, NK-EVs are referring to the EXO and MVs produced by the NK cells.
2. Methods
Search keywords were selected using medical subject headings (MESH) available from PUBMED/MEDLINE. Common terms not available in MESH were also included in the search. Access to SCOPUS, PUBMED and Web of Science (WOS) was provided by the National University of Malaysia (UKM). All searches were filtered to either “research articles” or “journal articles” published in the last 5 years (2017–2022). All search registers were downloaded as bibliographies containing title, keywords and abstract. The bibliographies were labelled appropriately as source, date of access and results (e.g., PUBMED_210622_455 results). The bibliographies were then exported into Mendeley (Elsevier, The Netherlands). The merging of duplicates was performed automatically and manually. The first level of screening was based on relevance of title, abstract and keywords with topic of interest. After that, full-text research articles were downloaded for second level of screening. The final screening was performed following the inclusion and exclusion criteria as briefly described. Inclusion criteria: (i) NK cell, (ii) EV or EXO, (iii) cancer and (iv) controlled experimental studies. Exclusion criteria: (i) other immune cells, (ii) no EV or EXO, (iii) combined therapy and (iv) uncontrolled experimental study. Subsequently, the in vivo studies were reviewed for their risk of bias using SYRCLE’s ROB tool for animal studies. The protocol above has been registered (ID: CRD42022339710) in the international prospective register of systematic reviews (PROSPERO) by the National Institute for Health Research (United Kingdom).
We recorded a combined total of 1002 registers from three databases: PUBMED (330), SCOPUS (438) and WOS (234). A total of 340 registers were removed as a result of duplicate merging, leaving only 662 individual registers. The first process of screening removed 631 registers based on the title, abstract and keywords. This yielded 31 suitable articles for full-text retrieval. One article was not successfully retrieved since no English-text or translation was available. A total of 30 retrieved full-text articles were screened using the inclusion and exclusion criteria stated above. Finally, 18 individual registers containing either in vitro only, in vivo only or both in vitro and in vivo evidence were selected for data extraction and analysis. The article selection was performed by two authors (A.M.L.C and J.M.C.) and any disputes in article inclusion were resolved via discussion to reach mutual consensus. The article selection process is summarized in Figure 1.
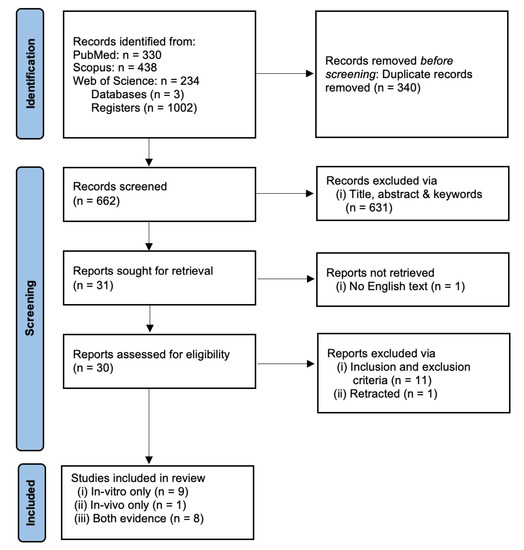
Figure 1.
PRISMA flow chart for systematic review.
3. Results and Discussion
3.1. NK-EVs Express Cytotoxicity against Various Human Cancer Cell Lines
A total of 17 studies performed in vitro experiments to examine the efficacy of NK-EVs against a panel of tumor cell lines, and all of them reported positive outcomes (Table 1). The NK-EVs were found to be effective against a wide range of cancer cell lines, shown in Figure 2 below, such as the brain [44,45,46,47,48,49,50,51], breast [44,45,47,49,50,51,52,53,54,55], blood [44,48,53,55,56,57], cervix [57], colon [50,51], liver [45,49,58], lung [57], ovary [51,54], pancreas [59], prostate [51], skin [51,60], stomach/gastric [50,60], and thyroid [45,49]. The dose of NK-EVs used in these studies ranged from low dose, between 0.3−5 µg, to median dose, between 10−25 µg, and high dose, between 40−100 µg. The NK-EVs were found to kill the cancer cells in a time- and dose-dependent manner [44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60]. Hence, higher cytotoxicity was recorded when the cancer cell lines were exposed to higher dose of NK-EVs for a longer period. Importantly, the NK-EVs showed selective cytotoxicity towards the cancer cell lines without affecting the viability of the healthy or normal cell lines tested. This evidence clearly showed that NK-EVs exhibited selectivity akin to their parental cells, as reported in the previous literature [61].

Table 1.
In vitro evidence demonstrating cytotoxicity of NK-EVs against various cancer cell lines.
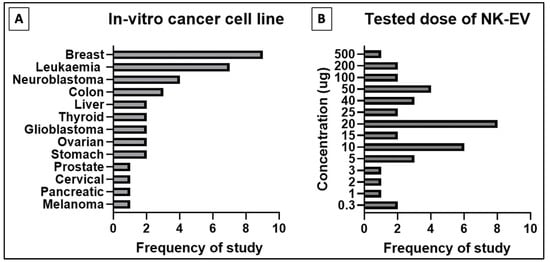
Figure 2.
Distribution of in-vitro studies for (A) type of cancer cell line studied and (B) tested doses of NK-EVs for cancer cell cytotoxic assay.
The impressive findings from the in vitro studies merely demonstrated the fundamental anti-cancer potential of NK-EVs without the interference of other biological systems. Thus, the use of mere in vitro data to show the anti-cancer potential of NK-EVs is totally insufficient, as the model does not replicate the physiological complexities that make up a living organism [62]. For more effective translation, a highly defined and complex model of pharmacodynamics (PD) and pharmacokinetics (PK) is needed, such as the one constructed by Bouhaddou et al. (2020) [63]. In term of cancer, the main debacle that has stumped the progression of many medical innovations is the existence of drug resistance, which is very difficult to replicate in vitro [64]. Tumor cells are known to interact with the surrounding cells and tissues, also known as tumor microenvironment (TME), to ensure their survival and the development of drug resistance. The TME provides important support needed by the tumor to thrive, including the secretion of signalling molecules by the proximally located cells, structural support and biochemical signals granted by the altered extracellular matrices (ECM), protection by the immunosuppressive local immune cells, nutrient supply and waste removal by the sprouting blood vessels, and a dynamic microenvironment (e.g., optimal temperature and pH level) that favor cancer cell growth, invasion and metastasis [65,66,67,68].
3.2. Tumor Microenvironment (TME): A Physicochemical Barrier against Immunotherapy
The TME is described as an extremely hostile ecosystem consisting of both physical and chemical components, as illustrated in Figure 3 [69,70]. Similar to all other cell types, cancerous cells are no exceptions to the innate ability to secrete EVs. In fact, tumor-derived extracellular vesicles (TD-EVs) are expressed in significant quantities, making them a plausible biological marker and progress indicator for cancer patients [71,72,73]. Cancer cells are also more self-sufficient than regular cells since they are capable of producing and responding to their own growth factors [74]. As demonstrated by El-Fattah Ibrahim et al. (2019), these tumor-enabling secretions can operate in an autocrine (own cells), paracrine (neighboring cells) and endocrine (distally located cells) manner [75]. TD-EVs were known to alter the surrounding microenvironment, making it hostile for immune cells and other healthy tissue, but their utility and mechanism of action were never fully comprehended. However, they do share a resemblance to anti-inflammatory secretions by supporting cell proliferation and angiogenesis, while inhibiting cell apoptosis, maturation or differentiation, and suppressing the recruitment of inflammatory-responsive cells (e.g., NK cells, macrophages, B and T lymphocytes) [76]. When healthy cells are replaced or destroyed, the surrounding environment is contaminated by the metabolic waste products, inflammasomes from cell lysis, and cell debris. Active glycolysis in malignant cells consumes the surrounding oxygen and releases acidic products (e.g., pyruvate and hydrogen ions) [77]. After depleting the surrounding oxygen, cancer cells will undergo anaerobic glycolysis, which contributes to further acidification via lactic acid production [78]. These manifest into the well-known acidic and hypoxic properties of TME, forming a chemical barrier that limits immune cell penetration.
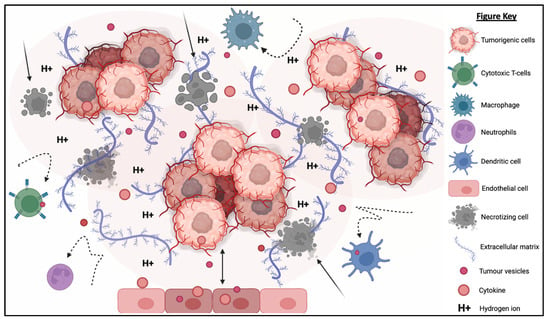
Figure 3.
The tumorigenic cells are centered around faulty extracellular matrices, necrotic bodies, and a hypoxic and acidified microenvironment. This tumor microenvironment discourages immunoreactive cells from responding or otherwise entering the tumor to eradicate the tumor cells. At the same time, the hypoxic and acidic environment will induce cell death and exacerbate the inflammation. Moreover, tumor-derived extracellular vesicles will reprogram the host’s cells (e.g., endothelial cells) to support tumor development via increasing the secretion of growth factor, pro-angiogenic and anti-inflammatory cytokines.
What is also seemingly peculiar about TD-EVs is the manipulation of host cells, akin to a virus. TD-EVs are able to influence or reprogram the recipient cells to cooperate with tumors [79]. For example, TD-EVs can falsely trigger the anti-inflammatory machinery of the healthy cells to reduce the TME inflammation. On the other hand, death cells in TME will continuously release inflammatory particles. Hence, TME paradoxically houses both anti-inflammatory particles and pro-inflammatory particles. How these opposing factors co-exists and modulate the TME inflammation is yet to be fully understood. In addition, studies have shown that the circulating inflammatory bodies passively recruit immunoreactive cells to the tumor region [80,81,82,83]. The immune cells recruited to the TME will be modulated by the TD-EVs to deactivate its tumor cell killing function and/or to maintain its status quo via overstimulation of the inhibition:activation signal ratio [84]. At the same time, the aggregation of naïve/resting or inactivated immune cells interlocked by faulty ECMs obstructs any movement and acts as a shield against activated immune cells or medical interventions (e.g., chemotherapy drugs). Thus, the formation of this pseudo-barrier reinforces the physical impenetrability of the tumor, contributing to drug resistance.
TME and TD-EVs are known to work in harmony to disrupt NK cell function, thus, reducing the effectiveness of NK cell therapy. The acidic pH, as well as the immunosuppressive myeloid derived suppressor cells’ (MDSCs) and M2 macrophages’ presence in the TME, suppress the activation of NK cells, thereby compromising their cytotoxicity against a range of tumor cells [85]. Similarly, the TD-EVs were found to impair the function of NK cells through the transfer of multiple immunosuppressive factors (e.g., miRNAs and TGF-β) [86,87]. The existence of physical and chemical barriers in TEM paired with the inhibitory factors from TD-EVs render the NK cells and other immune cells almost entirely incompetent. Cellular release and uptake of EVs are known to increase in acidic environments [88]. Additionally, the pH-related stress also increases the EVs’ protein content and surface electrokinetic potential (zeta potential) [89]. The EVs with higher surface charges can bind strongly to the cell membrane, therefore increasing its internalization via receptor or non-receptor endocytosis. The higher EV absorption is not restricted to TD-EVs, but applicable to other EVs as well. This implies the possibility of higher absorption of NK-EVs and other immune cell-derived EVs by the cancer cells.
3.3. NK-EVs Show In Vivo Cytotoxicity in Tumor-Bearing Mice
The in vitro to in vivo extrapolation has seen numerous failures due to subpar experimental design and the presence of many limitations that yet to be addressed [62]. Thus, preclinical study needs to be performed to extrapolate animal data to humans despite that it is also unreliable due to species differences. Table 2 describes studies that assess the performance of NK-EV therapy using the in vivo model of tumor-bearing animals and the range of doses per administration [45,46,47,50,53,60,90]. The four studies shown in Figure 4 that performed intra-tumoral (IT) infusion reported significant suppression (p < 0.05) of tumor growth and lowered cancer cell viability in the neoplastic mass [45,53,60,90]. Similar results were reported in the seven studies that performed intravenous (IV) delivery of NK-EVs through the tail vein [45,46,47,49,50,58,90]. In all these studies, NK-EV infusion significantly (p < 0.05) reduced the bioluminescent intensity (BLI) signals and/or physical dimensions of the tumor mass compared to the untreated animals. The study by Zhu et al. (2018) performed a direct comparison between IT and IV administration routes in their animal models [45]. They found that IT administration is more effective (p < 0.05) compared to IV administration in shrinking the tumor mass. Similar results have been reported in previous studies examining the anti-tumor effect of immunotherapies in tumor-bearing mice [91,92]. These findings indicate that the IT route is likely to be more efficacious for NK-EV therapy but the IV route is still viable and may be used in specific situations when the IT route is not accessible.
Table 2.
In vivo evidence demonstrating the antitumor potential of NK-EVs.
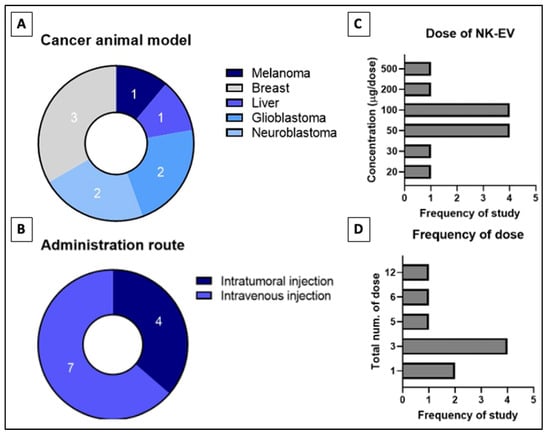
Figure 4.
Distribution of in-vivo studies for (A) type of cancer studied, (B) administration route of NK-EVs, (C) tested dose of NK-EVs per dose and the (D) frequency of dose administered throughout study phase. Numbers indicated in (A,B) refers to the frequency of reviewed studies that fall into the categories.
The SYRCLE’s risk of bias tool adopted from Hooijman 2014 was used to assess the risk of bias of the selected studies (Figure 5) [93]. Generally, most of the elements evaluated showed an unknown risk of bias. While most of the studies demonstrated low risk of bias for some of the elements investigated, a few studies were found to have high risk. Among the nine studies reviewed, five studies (55.6%) explicitly mentioned randomization [45,46,47,50,58] but three studies (33.3%) did not [49,53,60]. A single study (11.1%) was deemed high-risk due to undisclosed sample size and allocation method [90]. The baseline was presented prior to treatment in 6 studies (66.7%) but ambiguous in the remaining three studies (33.3%). None of the studies disclosed information for allocation concealment, random housing, blinding of interventions or caregivers, random outcome assessment, and blinding of outcome assessment. Nearly all studies (n = 8, 88.9%) have unknown risk of bias for selective outcome reporting bias, with one study has high-risk of bias [50]. Five studies (55.5%) reported low risk of attrition bias and three (33.3%) had an unclear risk, with one (11.1%) having high-risk [50]. The unclear risk is mainly attributed to the inconsistency in the sample size of the study’s outcomes with the number of animals allocated per group. Failure to disclosure the number of animals (alive or dead) excluded from the analysis or parameter conducted were also took into account. All studies were at low risk of “other sources of bias” by disclosure of ethics approval, potential conflict of interest, or the collaborating parties and their roles.
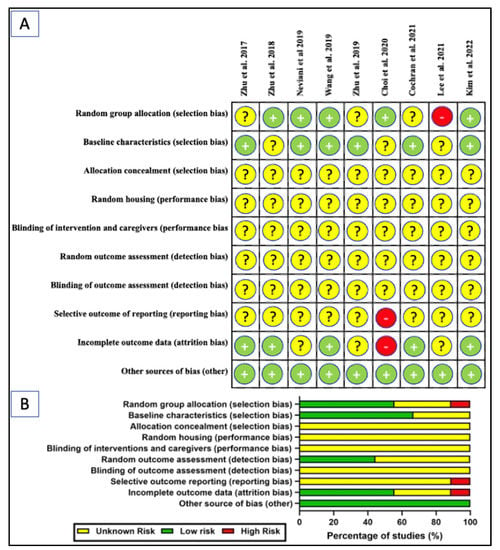
Figure 5.
SYRCLE’s risk of bias tool: (A) scoring of individual studies; (B) overall scores presented as percentage of studies for in-vivo studies [45,46,47,49,50,53,60,90]. Symbols: yellow “?” indicates unclear risk; green “+” indicates low risk; and red “-” indicates high risk of bias.
3.4. NK-EVs Overcome the Chemical and Physical Barriers of TME
NK-EVs inherit the components and functions of NK cells to exert cytotoxicity against tumor cells in vitro and in vivo. NK-EVs not only express tumor-targeting ability but also possess homing and migratory properties via chemotaxis [45,46,47,50,58]. As depicted in Figure 6, NK-EVs’ framework is significantly more compact (30–200 nm) and they are also more biostable than NK cells [94,95]. These properties enable them to have improved infiltration and survival in the hostile TME. It has been demonstrated that NK-EVs express NKG2D, FasL and TRAIL just like the parent cells for receptor-mediated apoptosis. Lytic proteins such as PFN and Gzm also exist ubiquitously in NK-EVs to initiate caspase-dependent apoptosis pathway. All these functions show that the simple configuration of NK-EVs does not compromise their capability to eliminate cancer cells through the multiple cytotoxic pathways utilized by its parent cells. Instead, the NK-EVs might be more effective without the interference of the TME and TD-EVs that halt the production of PFN and Gzm in NK cells. It is believed that the anti-cancer potency of NK-EVs is mainly attributed by these killer proteins. However, the concentration and ratio of NK-EVs in relative to TD-EVs in the TME will likely determine if they will be able to exert anti-tumor effects. It is highly possible that the level of inhibitory signals induced by TD-EVs may overwhelm the activating signals of NK-EVs to prevent any cytotoxic action [84]. This means that the dose and delivery method of NK-EVs in vivo need to be critically evaluated to ensure their high bioavailability in the tumor in order to exert their cytotoxic function.
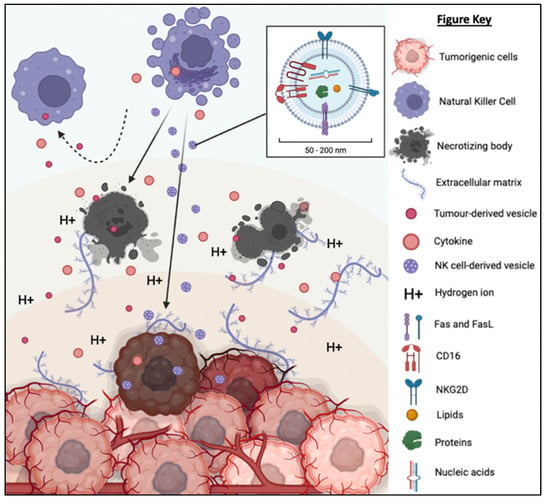
Figure 6.
TD-EVs suppress the recruitment and migration as well as reduce the proliferation, survival and cytolytic function of NK cells. On the other hand, NK-EVs secreted by the NK cells are small and diligent enough to overcome the physical and chemical barriers of TME to reach and exert its cytolytic effect on the tumor cells.
3.5. NK-EVs Are More Clinically Applicable than NK Cells
Results from the in vitro and in vivo experiments demonstrated that NK-EVs are an effective and safe immunotherapy. However, there are several foreseeable issues that need to be addressed in preparing this therapy for future clinical trials and registration. To begin, the functions of NK cells are distinguishable by their cell activation status [24,96,97]. Generally, both naïve and activated NK cells can secrete NK-EVs [98]. However, the activated NK cells are preferable for EV isolation due to their high cell number in in vitro culture. It has been reported that the NK-EVs derived from inactivated NK cells have lower amounts of cytotoxic proteins [46]. In a separate study, the authors found that EVs secreted by the NK92 cell line contain fewer cytotoxic proteins compared to the EVs secreted by in vitro-expanded NK cells [48]. Additionally, Shoae-Hassani et al. (2017) reported that NK cells exposed to neuroblastoma cells produced NK-EVs with higher cytotoxicity towards the tumor cells in vitro and in vivo compared to the NK-EVs derived from naïve NK cells [99]. Priming of NK cells with IL-15 was also reported to increase the cytotoxicity of NK-EVs [49]. These observations show that NK-EVs are highly heterogeneous depending on the cellular origin, culture environment and physiological status. Therefore, it is important to conduct experiments to the optimize the NK cell culture protocol in order to harvest NK-EVs with potent therapeutic potential. Importantly, the optimization should also consider increasing the yield of NK-EVs in order to reduce the cost.
Unlike the stem cell, donor-derived NK cells have variable and limited expansion potential in vitro [100]. Thus, efforts have been made to prepare immortalized NK cell lines, such as the NK92-MI from the American Type Culture Collection (ATCC, USA). From Table 1, eleven of the seventeen reviewed studies purchased this cell line to source their NK-EVs. Wu et al. (2019) and Aarsund et al. (2022) discovered little-to-no difference between the NK-EVs from NK92-MI cell line and those from freshly isolated peripheral blood-derived NK (PB-NK) cells [48,51]. Cryopreservation is widely used for long-term storage of NK cells. However, the cryopreserved NK cells have relatively poor viability albeit different freezing medium have been tested. Many studies have reported cell viability less than 10% after revival [101]. More alarming is that the survived cells ceased proliferating and failed to exert any cytotoxic functions [102]. Failure of cell cryopreservation will definitely hamper the clinical translation of NK-EV therapy as it will limit the potential of producing them on a large scale and consistently. At the moment, new donor-derived NK cells are used to prepare the NK-EVs, which leads to high batch-to-batch variation. Furthermore, it will also increase the production cost and lead time.
Manual cell culture protocol using tissue culture flask is prone to contamination, technical errors and high batch-to-batch variation [103,104]. To overcome these limitations, efforts should be made to shift the NK cell expansion to a bioreactor platform which is highly automated, allows more control over the culture environment (e.g., glucose concentration, oxygen concentration and pH) and requires less manpower [105]. Usage of a highly efficient bioreactor system might also reduce the cost of production [44]. Not least, it is important is to optimize the EV isolation protocol, which should allow quick and reliable isolation of EV subpopulation.
Last but not least, long-term storage and off-the-shelf availability are highly desirable for NK-EV therapy. To achieve this, researchers need to device a storage condition that can preserve EV stability over a long period of time. As an acellular product, NK-EVs can be kept chill (4 °C) for immediate use or frozen (−20 °C or −80 °C) up to 12 months with acceptable loss of proteins and RNAs [106,107,108,109,110]. More importantly, the NK-EVs retain their anti-tumor effect. For easier storage and transportation, NK-EVs could be lyophilized. Even though lyophilization has been widely explored for storage of stem cell-derived EVs, it is still unknown whether lyophilization can be used to preserve the NK-EVs.
3.6. Defining Nomenclature, Isolation Technique, QA/QC Methods and Biomarkers for NK-EVs
In the very beginning, EVs were first thought as a mechanism of disposing cellular waste products [111]. Later, studies found that these minute vesicles are rich in lipids, proteins, nucleic materials (e.g., DNA, RNA, mRNA, miRNA) and other biologically active molecules [112]. On their surface, they express different molecules and proteins adopted from parent cell membrane during the budding or exocytosis process. These physical hallmarks contribute to the diverse functions and heterogeneous properties of EV molecules. Size is commonly used to classify EVs [113]. The largest EVs are the apoptotic bodies (1000–5000 nm), followed by microvesicles (50–1000 nm), with exosomes (30–150 nm) being the smallest [114]. The term “exosome” is often incorrectly used interchangeably with “extracellular vesicle” to describe the small EV preparation [42]. This is because most of the studies isolate the EVs based on their size instead of the specific phenotype that represented their unique biogenesis pathway. Every EV subset has a specific protein phenotype and molecular content. Thus, the Minimal Information for Studies of Extracellular Vesicles (MISEV) 2018 guidelines have recommended the nomenclature “extracellular vesicles” to describe the small membrane-bound particles secreted by the cells. It is crucial to establish a uniform and specific standard for appropriate evaluation of different NK-EV preparations as it would allow researchers to determine the strengths and weaknesses of any modifications or novelties introduced.
Based on Table 3 and Figure 7, the most commonly used EV isolation method is the ultracentrifugation (UC) [44,45,50,52,53,54,55,57,58,60,90]. UC is often used in combination with other EV isolation techniques to improve the purity [45,50,53,58,60]. Alternative isolation techniques used in the reviewed studies include density gradient centrifugation (DGC) [45,49,50,53,60], polyethylene glycol-8000 (PEG8000) precipitation [44,48,53], differential centrifugation (DC) [47,56,58] and size exclusion chromatography (SEC) [46,49,51]. Generally, the existing techniques are unable to achieve both high recovery and high specificity. With the rise in interest in EV-related therapeutics especially mesenchymal stem cell-derived EVs (MSC-EVs) and EV-based immunotherapy, it is vital to develop a novel isolation method which can give high recovery and high purity as well as can be easily scaled up [42,113]. Currently, tangential flow filtration (TFF) is being revisited and improved for more efficient recovery of EVs [58,115,116]. Watson et al. (2018) utilized the combined platforms of TFF and SEC to isolate MSC-EVs and reported that TFF is a reproducible and scalable technique to recover clinical-grade EVs compared to other known techniques [117].
Table 3.
Method of isolation, characterization and functional analysis of NK-EVs.
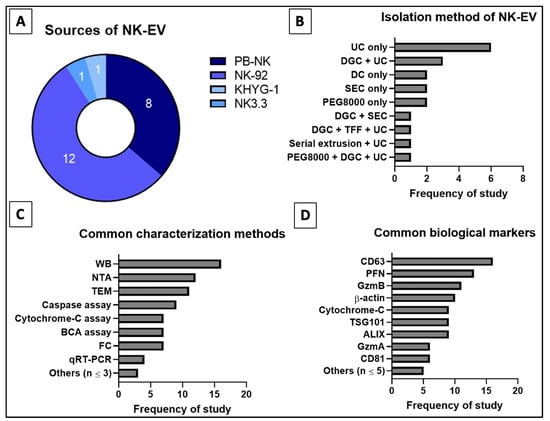
Figure 7.
Distribution of reviewed studies for (A) source, (B) isolation techniques (single or combined), (C) common characterization assays and (D) common biological markers for NK-EVs. Numbers indicated in (A) refer to the frequency of reviewed studies that fall into the categories.
According to MISEV 2018, EVs should be categorized based on size and biochemical composition (e.g., CD9, CD81 and TSG101) [118,119]. Most of the selected articles in this study used small NK-EVs in the range 50–200 nm [44,45,46,47,49,51,52,53,55,56,57,58,59,60,90]. Only Jiang et al. used a subset of EVs that are larger in diameter, i.e., 205.6 ± 29.65 nm [54]. To prevent misreport or misrepresentation of each author’s work, only text-based evidence of the NK-EV sizes were recorded since not all reviewed studies had specifically disclosed the diameter, mean or mode of their NK-EVs. The frequently used assessment tools for EVs in these studies includes nanoparticle tracking analysis (NTA) to examine the size [44,45,46,47,49,51,53,55,56,57,58,90], transmission electron microscopy (TEM) to study the size and morphology [44,45,46,47,49,51,52,53,54,55,58,60], Western blot (WB) [44,45,46,47,48,49,50,51,52,53,56,57,58,60,90] and flow cytometry (FC) [49,54,55,56,57,59,60] to determine the biochemical markers, and bicinchoninic acid (BCA) protein assay [47,50,51,53,54,57,60] to quantify the protein concentration. The biochemical markers consistently tested to characterize the EVs were CD63 [44,45,47,49,50,51,52,53,54,55,56,57,58,59,60,89], CD81 [46,51,56,57,89], ALIX [45,46,47,49,52,53,58,60,90] and TSG101 [46,47,51,52,53,54,56,59,90]. Furthermore, specific markers, i.e., Fas/FasL or Fas/TRAIL [45,48,50,51,54,57,58], granulysin (GNLY) [44,48,52,57], granzymes A (GzmA) and/or B (GzmB) [44,46,48,49,51,53,54,56,57], and PFN [44,45,46,48,49,51,53,54,56,57,59,60] were used to prove that the EVs originated from NK cells. The characterization of NK-EVs using the above techniques is crucial to verify the NK-EVs, as well as to compare and standardize the NK-EV research.
3.7. Future Considerations of NK-EVs as a Tool for Immunotherapy
Table 4 summarizes the established benefits and challenges of NK cells and NK-EVs for immunotherapy. However, the discussion going forward shall emphasize on improving the EV-based immunotherapy as it has yet to be thoroughly explored but already exhibiting significant advantages compared to the former. One of the main benefits of cell-based and EV-based therapies is that they are minimally invasive medical procedures [120]. They are safer alternatives with potentially fewer procedural complications to the high-risk populations such as infants and elderly. In contrast, surgical treatment can lead to many serious complications, including induced hemorrhage, post-operative sepsis and extended rehabilitation [121,122]. Selection of administration route or delivery technique is critical to ensure the safety and efficacy of a therapy [123]. Although IT administration would eliminate any concerns over the homing and migration ability of NK-EVs, the IV administration will be especially useful for regions with poor access or penetrability (e.g., brain) [124]. In actuality, a larger dose is typically needed for IV route to compensate for the accumulated loss of drugs due to unwanted distribution at the other tissues as well as metabolism and excretion primary at the liver and kidney [125,126,127]. Nonetheless, the continual IV administration of NK-EVs after the tumor elimination could be favourable to ensure complete clearing of tumor cells to minimize the risk of relapse [128,129]. To date, it is still unclear on the optimal dosage regime of NK-EVs. More research is needed to determine the ideal frequency, dosage and interval of NK-EV treatment.
Table 4.
Summary of pros and cons of NK cell vs. NK-EV for immunotherapy.
Efforts are also needed to improve the efficacy of NK-EV production in order to reduce the cost. Owing to its niche role, NK cells have high metabolic activity and need specific cytokines to stimulate its proliferation. The commonly used cytokines to expand NK cells are IL-2, IL-15, IL-18, and IL-21. The cytokines can be provided by the peripheral blood mononuclear cells (PBMCs) that serve as the feeder cells [130]. Alternatively, the culture medium can be supplemented with commercially available human recombinant cytokines to maintain the NK cell culture. Although the use of recombinant cytokines is simpler as no purification step is needed to remove the contaminating PBMCs and safer as it is free from PBMC contamination, the cost of recombinant cytokines is very high. The high concentration and cost of recombinant cytokines will significantly escalate the production cost. On top of that, the requirement of using GMP-grade or clinical-grade raw materials and consumables, skilled manpower, stringent quality assurance and quality control (QA/QC), expansive equipment for scalable production, and the use of a cGMP-certified production facility will escalate the treatment cost to unaffordable scales for majority of the patients [131]. This, in turn, will affect the clinical translation and commercial viability of NK-EV therapy.
4. Conclusions
NK-EVs show promising tumor cytotoxicity with no adverse or side effects in vitro and in vivo. NK-EVs are not cytotoxic to the normal or healthy cells and can increase the life span of tumor-bearing animals. Thus, this novel therapy has great clinical potential. In addition, NK-EVs can circumvent some of the limitations of NK cell therapy, such as impediment of NK cell functionality by the TME and TD-EVs. In fact, NK-EVs would see potential benefits due to the increased cellular uptake in the TME. Although IT administration is superior, the IV administration will be meaningful for “difficult-to-reach” tumors and high-risk populations. Additionally, NK-EVs can be readily available off-the-shelf as they are more stable and easier to store compared to NK cells. Nonetheless, NK-EV therapy and its production procedure need further investigation to improve its safety and efficacy as well as to increase the yield and lower the production cost.
Author Contributions
Conceptualization, all authors; methodology, A.M.L.C. and J.M.C.; validation, A.M.L.C. and J.M.C.; formal analysis, A.M.L.C. and J.M.C.; investigation, A.M.L.C. and J.M.C.; data curation, A.M.L.C. and J.M.C.; writing—original draft preparation, A.M.L.C. and J.M.C.; writing—review and editing, all authors; visualization, all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grant provided by Ming Medical Sdn Bhd (FF-2022-330). The granting agencies played no role in activities pertaining to the execution of the study and the submission of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare that there is no conflict of interest.
References
- World Health Organization (WHO). Coronavirus (COVID-19) Dashboard. 2022. Available online: https://covid19.who.int/ (accessed on 13 December 2022).
- World Health Organization (WHO). Non Communicable Diseases. 2022. Available online: https://www.who.int/news-room/fact-sheets/detail/noncommunicable-diseases (accessed on 14 July 2022).
- Liao, G.S.; Apaya, M.K.; Shyur, L.F. Herbal medicine and acupuncture for breast cancer palliative care and adjuvant therapy. Evid. Based Complement. Alternat. Med. 2013, 2013, 437948. [Google Scholar] [CrossRef] [PubMed]
- Altun, İ.; Sonkaya, A. The Most Common Side Effects Experienced by Patients Were Receiving First Cycle of Chemotherapy. Iran. J. Public Health 2018, 47, 1218–1219. [Google Scholar] [PubMed]
- Alcindor, T.; Beauger, N. Oxaliplatin: A review in the era of molecularly targeted therapy. Curr. Oncol. 2011, 18, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Corrie, P.G. Cytotoxic chemotherapy: Clinical aspects. Medicine 2011, 39, 717–722. [Google Scholar] [CrossRef]
- Makovec, T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B. Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: Have we been barking up the wrong tree? Redox Biol. 2020, 29, 101394. [Google Scholar] [CrossRef]
- Smoot, B.; Wampler, M.; Topp, K.S. Breast cancer treatments and complications: Implications for rehabilitation. Rehabilitation Oncology 2009, 27, 16–26. [Google Scholar] [CrossRef]
- Rossi, A.; Fortuna, M.C.; Caro, G.; Pranteda, G.; Garelli, V.; Pompili, U.; Carlesimo, M. Chemotherapy-induced alopecia management: Clinical experience and practical advice. J. Cosmet. Dermatol. 2017, 16, 537–541. [Google Scholar] [CrossRef]
- Frenkel, M. Refusing treatment. Oncologist 2013, 18, 634–636. [Google Scholar] [CrossRef]
- Zakrzewski, W.; Dobrzyński, M.; Szymonowicz, M.; Rybak, Z. Stem cells: Past, present, and future. Stem Cell Res. Ther. 2019, 10, 68. [Google Scholar] [CrossRef]
- Erhart, F.; Buchroithner, J.; Reitermaier, R.; Fischhuber, K.; Klingenbrunner, S.; Sloma, I.; Hibsh, D.; Kozol, R.; Efroni, S.; Ricken, G.; et al. Immunological analysis of phase II glioblastoma dendritic cell vaccine (Audencel) trial: Immune system characteristics influence outcome and Audencel up-regulates Th1-related immunovariables. Acta Neuropathol. Commun. 2018, 6, 135. [Google Scholar] [CrossRef]
- Margolin, K.; Morishima, C.; Velcheti, V.; Miller, J.S.; Lee, S.M.; Silk, A.W.; Holtan, S.G.; Lacroix, A.M.; Fling, S.P.; Kaiser, J.C.; et al. Phase I Trial of ALT-803, A Novel Recombinant IL15 Complex, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2018, 24, 5552–5561. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.; Oliveira, R.; Cavadas, B.; Pinto, F.; Cardoso, A.P.; Castro, F.; Sousa, B.; Pinto, M.L.; Silva, A.J.; Adão, D.; et al. Hypoxia and Macrophages Act in Concert Towards a Beneficial Outcome in Colon Cancer. Cancers 2020, 12, 818. [Google Scholar] [CrossRef] [PubMed]
- Gumrukcu, S.; Nguyen, T.X.; White, R.L.; Howell, G.T.; Musikanth, P. Allogeneic Natural Killer and Cytomegalovirus (CMV)-pp65 Pulsed Dendritic Cells Induced Complete Response Through 15 Months in a Patient with Recurrent Glioblastoma: A Case Study. Am. J. Case Rep. 2021, 22, e931030, Retraction in Am. J. Case Rep. 2022, 23, e937680. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I. Dendritic cells: Master regulators of the immune response. Cancer Immunol. Res. 2013, 1, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4+T cells: Differentiation and functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Lenz, L.L.; Harris, R.A. A Breakthrough: Macrophage-Directed Cancer Immunotherapy. Cancer Res. 2016, 76, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Oh, S.; Lee, J.H.; Kwack, K.; Choi, S.W. Natural Killer Cell Therapy: A New Treatment Paradigm for Solid Tumors. Cancers 2019, 11, 1534. [Google Scholar] [CrossRef]
- Komatsu, F.; Tamiya, H. Relationship between antibody-dependent cell-mediated cytotoxicity due to anti-HTLV-1 and negative signal of major histocompatibility complex class I antigens on adult T-cell leukemia cell lines. Oncol. Res. 1998, 10, 59–67. [Google Scholar]
- Høydahl, L.S.; Frick, R.; Sandlie, I.; Løset, G.Å. Targeting the MHC Ligandome by Use of TCR-Like Antibodies. Antibodies 2019, 8, 32. [Google Scholar] [CrossRef]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed]
- Lo Nigro, C.; Macagno, M.; Sangiolo, D.; Bertolaccini, L.; Aglietta, M.; Merlano, M.C. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: Biological evidence and clinical perspectives. Ann. Transl. Med. 2019, 7, 105. [Google Scholar] [CrossRef] [PubMed]
- Kurdi, A.T.; Glavey, S.V.; Bezman, N.A.; Jhatakia, A.; Guerriero, J.L.; Manier, S.; Moschetta, M.; Mishima, Y.; Roccaro, A.; Detappe, A.; et al. Antibody-Dependent Cellular Phagocytosis by Macrophages is a Novel Mechanism of Action of Elotuzumab. Mol. Cancer Ther. 2018, 17, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Somiya, M.; Kuroda, S. Enhancing antibody-dependent cellular phagocytosis by Re-education of tumor-associated macrophages with resiquimod-encapsulated liposomes. Biomaterials 2021, 268, 120601. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zheng, H.; Duan, Z.; Liu, H.; Hu, D.; Bode, A.; Dong, Z.; Cao, Y. Promotion of cell proliferation and inhibition of ADCC by cancerous immunoglobulin expressed in cancer cell lines. Cell Mol Immunol. 2012, 9, 54–61. [Google Scholar] [CrossRef]
- Kline, J.B.; Kennedy, R.P.; Albone, E.; Chao, Q.; Fernando, S.; McDonough, J.M.; Rybinski, K.; Wang, W.; Somers, E.B.; Schweizer, C.; et al. Tumor antigen CA125 suppresses antibody-dependent cellular cytotoxicity (ADCC) via direct antibody binding and suppressed Fc-γ receptor engagement. Oncotarget 2017, 8, 52045–52060. [Google Scholar] [CrossRef]
- Darwich, A.; Silvestri, A.; Benmebarek, M.R.; Mouriès, J.; Cadilha, B.; Melacarne, A.; Morelli, L.; Supino, D.; Taleb, A.; Obeck, H.; et al. Paralysis of the cytotoxic granule machinery is a new cancer immune evasion mechanism mediated by chitinase 3-like-1. J. Immunother. Cancer 2021, 9, e003224. [Google Scholar] [CrossRef]
- Tsuchiya, H.; Shiota, G. Immune evasion by cancer stem cells. Regen. Ther. 2021, 17, 20–33. [Google Scholar] [CrossRef]
- Cullen, S.P.; Martin, S.J. Fas and TRAIL “death receptors” as initiators of inflammation: Implications for cancer. Semin. Cell Dev. Biol. 2015, 39, 26–34. [Google Scholar] [CrossRef]
- Rossin, A.; Miloro, G.; Hueber, A.O. TRAIL and FasL Functions in Cancer and Autoimmune Diseases: Towards an Increasing Complexity. Cancers 2019, 11, 639. [Google Scholar] [CrossRef] [PubMed]
- Snajdauf, M.; Havlova, K.; Vachtenheim, J., Jr.; Ozaniak, A.; Lischke, R.; Bartunkova, J.; Smrz, D.; Strizova, Z. The TRAIL in the Treatment of Human Cancer: An Update on Clinical Trials. Front. Mol. Biosci. 2021, 8, 628332. [Google Scholar] [CrossRef]
- Clark, R.; Griffiths, G.M. Lytic granules, secretory lysosomes and disease. Curr. Opin. Immunol. 2003, 15, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Krzewski, K.; Coligan, J.E. Human NK cell lytic granules and regulation of their exocytosis. Front. Immunol. 2012, 3, 335. [Google Scholar] [CrossRef] [PubMed]
- Kabanova, A.; Zurli, V.; Baldari, C.T. Signals Controlling Lytic Granule Polarization at the Cytotoxic Immune Synapse. Front. Immunol. 2018, 9, 307. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. Cancer Immunotherapy, Part 3: Challenges and Future Trends. Pharm. Ther. 2017, 42, 514–521. [Google Scholar]
- Tan, S.; Li, D.; Zhu, X. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef] [PubMed]
- Pugholm, L.H.; Bæk, R.; Søndergaard, E.K.; Revenfeld, A.L.; Jørgensen, M.M.; Varming, K. Phenotyping of Leukocytes and Leukocyte-Derived Extracellular Vesicles. J. Immunol. Res. 2016, 2016, 6391264. [Google Scholar] [CrossRef]
- Wu, F.; Xie, M.; Hun, M.; She, Z.; Li, C.; Luo, S.; Chen, X.; Wan, W.; Wen, C.; Tian, J. Natural Killer Cell-Derived Extracellular Vesicles: Novel Players in Cancer Immunotherapy. Front. Immunol. 2021, 12, 658698. [Google Scholar] [CrossRef]
- Ng, C.Y.; Kee, L.T.; Al-Masawa, M.E.; Lee, Q.H.; Subramaniam, T.; Kok, D.; Ng, M.H.; Law, J.X. Scalable Production of Extracellular Vesicles and Its Therapeutic Values: A Review. Int. J. Mol. Sci. 2022, 23, 7986. [Google Scholar] [CrossRef]
- Kee, L.T.; Ng, C.Y.; Al-Masawa, M.E.; Foo, J.B.; How, C.W.; Ng, M.H.; Law, J.X. Extracellular Vesicles in Facial Aesthetics: A Review. Int. J. Mol. Sci. 2022, 23, 6742. [Google Scholar] [CrossRef] [PubMed]
- Jong, A.Y.; Wu, C.H.; Li, J.; Sun, J.; Fabbri, M.; Wayne, A.S.; Seeger, R.C. Large-scale isolation and cytotoxicity of extracellular vesicles derived from activated human natural killer cells. J. Extracell Vesicles 2017, 6, 1294368. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Gangadaran, P.; Kalimuthu, S.; Oh, J.M.; Baek, S.H.; Jeong, S.Y.; Lee, S.W.; Lee, J.; Ahn, B.C. Novel alternatives to extracellular vesicle-based immunotherapy—Exosome mimetics derived from natural killer cells. Artif. Cells Nanomed. Biotechnol. 2018, 46 (Suppl. 3), S166–S179. [Google Scholar] [CrossRef] [PubMed]
- Neviani, P.; Wise, P.M.; Murtadha, M.; Liu, C.W.; Wu, C.H.; Jong, A.Y.; Seeger, R.C.; Fabbri, M. Natural Killer-Derived Exosomal miR-186 Inhibits Neuroblastoma Growth and Immune Escape Mechanisms. Cancer Res. 2019, 79, 1151–1164. [Google Scholar] [CrossRef]
- Wang, G.; Hu, W.; Chen, H.; Shou, X.; Ye, T.; Xu, Y. Cocktail Strategy Based on NK Cell-Derived Exosomes and Their Biomimetic Nanoparticles for Dual Tumor Therapy. Cancers 2019, 11, 1560. [Google Scholar] [CrossRef]
- Wu, C.H.; Li, J.; Li, L.; Sun, J.; Fabbri, M.; Wayne, A.S.; Seeger, R.C.; Jong, A.Y. Extracellular vesicles derived from natural killer cells use multiple cytotoxic proteins and killing mechanisms to target cancer cells. J. Extracell Vesicles 2019, 8, 1588538. [Google Scholar] [CrossRef]
- Zhu, L.; Kalimuthu, S.; Oh, J.M.; Gangadaran, P.; Baek, S.H.; Jeong, S.Y.; Lee, S.W.; Lee, J.; Ahn, B.C. Enhancement of antitumor potency of extracellular vesicles derived from natural killer cells by IL-15 priming. Biomaterials 2019, 190–191, 38–50. [Google Scholar] [CrossRef]
- Choi, J.W.; Lim, S.; Kang, J.H.; Hwang, S.H.; Hwang, K.C.; Kim, S.W.; Lee, S. Proteome Analysis of Human Natural Killer Cell Derived Extracellular Vesicles for Identification of Anticancer Effectors. Molecules 2020, 25, 5216. [Google Scholar] [CrossRef]
- Aarsund, M.; Segers, F.M.; Wu, Y.; Inngjerdingen, M. Comparison of characteristics and tumor targeting properties of extracellular vesicles derived from primary NK cells or NK-cell lines stimulated with IL-15 or IL-12/15/18. Cancer Immunol. Immunother. 2022, 71, 2227–2238. [Google Scholar] [CrossRef]
- Han, D.; Wang, K.; Zhang, T.; Gao, G.C.; Xu, H. Natural killer cell-derived exosome-entrapped paclitaxel can enhance its anti-tumor effect. Eur. Rev. Med. Pharmacol Sci. 2020, 24, 5703–5713. [Google Scholar] [CrossRef]
- Cochran, A.M.; Kornbluth, J. Extracellular Vesicles From the Human Natural Killer Cell Line NK3.3 Have Broad and Potent Anti-Tumor Activity. Front. Cell Dev. Biol. 2021, 9, 698639. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Jiang, H.; Wang, K.; Liu, C.; Man, X.; Fu, Q. Hypoxia enhances the production and antitumor effect of exosomes derived from natural killer cells. Ann. Transl. Med. 2021, 9, 473. [Google Scholar] [CrossRef] [PubMed]
- Kaban, K.; Hinterleitner, C.; Zhou, Y.; Salva, E.; Kantarci, A.G.; Salih, H.R.; Märklin, M. Therapeutic Silencing of BCL-2 Using NK Cell-Derived Exosomes as a Novel Therapeutic Approach in Breast Cancer. Cancers 2021, 13, 2397. [Google Scholar] [CrossRef]
- Di Pace, A.L.; Tumino, N.; Besi, F.; Alicata, C.; Conti, L.A.; Munari, E.; Maggi, E.; Vacca, P.; Moretta, L. Characterization of Human NK Cell-Derived Exosomes: Role of DNAM1 Receptor In Exosome-Mediated Cytotoxicity Against Tumor. Cancers 2020, 12, 661. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, Y.; Li, P.; Jenkins, L.M.; Anastasakis, D.; Lyons, G.C.; Hafner, M.; Leonard, W.J. Cytokine-enhanced cytolytic activity of exosomes from NK Cells. Cancer Gene Ther. 2022, 29, 734–749. [Google Scholar] [CrossRef]
- Kim, H.Y.; Min, H.K.; Song, H.W.; Yoo, A.; Lee, S.; Kim, K.P.; Park, J.O.; Choi, Y.H.; Choi, E. Delivery of human natural killer cell-derived exosomes for liver cancer therapy: An in vivo study in subcutaneous and orthotopic animal models. Drug Deliv. 2022, 29, 2897–2911. [Google Scholar] [CrossRef]
- Sun, H.; Shi, K.; Qi, K.; Kong, H.; Zhang, J.; Dai, S.; Ye, W.; Deng, T.; He, Q.; Zhou, M. Natural Killer Cell-Derived Exosomal miR-3607-3p Inhibits Pancreatic Cancer Progression by Targeting IL-26. Front. Immunol. 2019, 10, 2819. [Google Scholar] [CrossRef]
- Zhu, L.; Kalimuthu, S.; Gangadaran, P.; Oh, J.M.; Lee, H.W.; Baek, S.H.; Jeong, S.Y.; Lee, S.W.; Lee, J.; Ahn, B.C. Exosomes Derived From Natural Killer Cells Exert Therapeutic Effect in Melanoma. Theranostics 2017, 7, 2732–2745. [Google Scholar] [CrossRef]
- Farcas, M.; Inngjerdingen, M. Natural killer cell-derived extracellular vesicles in cancer therapy. Scand. J. Immunol. 2020, 92, e12938. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Goldstein, D.B. In vitro assays fail to predict in vivo effects of regulatory polymorphisms. Hum. Mol. Genet. 2007, 16, 1931–1939. [Google Scholar] [CrossRef]
- Bouhaddou, M.; Yu, L.J.; Lunardi, S.; Stamatelos, S.K.; Mack, F.; Gallo, J.M.; Birtwistle, M.R.; Walz, A.C. Predicting In Vivo Efficacy from In Vitro Data: Quantitative Systems Pharmacology Modeling for an Epigenetic Modifier Drug in Cancer. Clin. Transl. Sci. 2020, 13, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.S.; Holland, A.J. The impact of mitotic errors on cell proliferation and tumorigenesis. Genes Dev. 2018, 32, 620–638. [Google Scholar] [CrossRef] [PubMed]
- Rani, B.; Cao, Y.; Malfettone, A.; Tomuleasa, C.; Fabregat, I.; Giannelli, G. Role of the tissue microenvironment as a therapeutic target in hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 4128–4140. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.J.; Lau, P.K.H.; Unsworth, A.S.; Loi, S.; Darcy, P.K.; Kershaw, M.H.; Slaney, C.Y. Tissue-Dependent Tumor Microenvironments and Their Impact on Immunotherapy Responses. Front. Immunol. 2018, 9, 70. [Google Scholar] [CrossRef]
- Zhuang, X.; Zhang, H.; Hu, G. Cancer and Microenvironment Plasticity: Double-Edged Swords in Metastasis. Trend. Pharmacol. Sci. 2019, 40, 419–429. [Google Scholar] [CrossRef]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.A.M.A.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Xue, H.; Lu, B.; Lai, M. The cancer secretome: A reservoir of biomarkers. J. Transl. Med. 2008, 6, 52. [Google Scholar] [CrossRef]
- Lin, J.; Ma, L.; Zhang, D.; Gao, J.; Jin, Y.; Han, Z.; Lin, D. Tumour biomarkers—Tracing the molecular function and clinical implication. Cell Prolif. 2019, 52, e12589. [Google Scholar] [CrossRef]
- Kartikasari, A.E.R.; Huertas, C.S.; Mitchell, A.; Plebanski, M. Tumor-Induced Inflammatory Cytokines and the Emerging Diagnostic Devices for Cancer Detection and Prognosis. Front. Oncol. 2021, 11, 692142. [Google Scholar] [CrossRef] [PubMed]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar] [PubMed]
- El-Fattah Ibrahim, S.A.; Abudu, A.; Johnson, E.; Aftab, N.; Conrad, S.; Fluck, M. Correction: The role of AP-1 in self-sufficient proliferation and migration of cancer cells and its potential impact on an autocrine/paracrine loop. Oncotarget 2019, 10, 799, Erratum for: Oncotarget 2018, 9, 34259–34278. [Google Scholar] [CrossRef] [PubMed]
- Qiao, F.; Pan, P.; Yan, J.; Sun, J.; Zong, Y.; Wu, Z.; Lu, X.; Chen, N.; Mi, R.; Ma, Y.; et al. Role of tumor-derived extracellular vesicles in cancer progression and their clinical applications (Review). Int. J. Oncol. 2019, 54, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13, 89. [Google Scholar] [CrossRef]
- Ribeiro Franco, P.I.; Rodrigues, A.P.; de Menezes, L.B.; Pacheco Miguel, M. Tumor microenvironment components: Allies of cancer progression. Pathol. Res. Pract. 2020, 216, 152729. [Google Scholar] [CrossRef]
- Li, P.; Lu, M.; Shi, J.; Hua, L.; Gong, Z.; Li, Q.; Shultz, L.D.; Ren, G. Dual roles of neutrophils in metastatic colonization are governed by the host NK cell status. Nat. Commun. 2020, 11, 4387. [Google Scholar] [CrossRef]
- Nowak, M.; Klink, M. The Role of Tumor-Associated Macrophages in the Progression and Chemoresistance of Ovarian Cancer. Cells 2020, 9, 1299. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]
- Cendrowicz, E.; Sas, Z.; Bremer, E.; Rygiel, T.P. The Role of Macrophages in Cancer Development and Therapy. Cancers 2021, 13, 1946. [Google Scholar] [CrossRef] [PubMed]
- Hodge, G.; Barnawi, J.; Jurisevic, C.; Moffat, D.; Holmes, M.; Reynolds, P.N.; Jersmann, H.; Hodge, S. Lung cancer is associated with decreased expression of perforin, granzyme B and interferon (IFN)-γ by infiltrating lung tissue T cells, natural killer (NK) T-like and NK cells. Clin. Exp. Immunol. 2014, 178, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Huang, T.; Chen, X.; Li, W.; Yang, X.; Zhang, W.; Li, M.; Gao, R. Uncovering the interplay between pH receptors and immune cells: Potential drug targets (Review). Oncol. Rep. 2021, 46, 228. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Schlößer, H.A.; Wang, Z.; Qin, J.; Li, J.; Popp, F.; Popp, M.C.; Alakus, H.; Chon, S.H.; Hansen, H.P.; et al. Tumor-Derived Extracellular Vesicles Inhibit Natural Killer Cell Function in Pancreatic Cancer. Cancers 2019, 11, 874. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cai, S.; Li, M.; Salma, K.I.; Zhou, X.; Han, F.; Chen, J.; Huyan, T. Tumor-Derived Extracellular Vesicles: Their Role in Immune Cells and Immunotherapy. Int. J. Nanomed. 2021, 16, 5395–5409. [Google Scholar] [CrossRef]
- Nakase, I.; Ueno, N.; Matsuzawa, M.; Noguchi, K.; Hirano, M.; Omura, M.; Takenaka, T.; Sugiyama, A.; Bailey Kobayashi, N.; Hashimoto, T.; et al. Environmental pH stress influences cellular secretion and uptake of extracellular vesicles. FEBS Open Biol. 2021, 11, 753–767. [Google Scholar] [CrossRef]
- Honary, S.; Zahir, F. Effect of Zeta Potential on the Properties of Nano-Drug Delivery Systems—A Review (Part 1). Trop. J. Pharm. Res. 2013, 12, 255–264. [Google Scholar] [CrossRef]
- Lee, J.; Lee, S.A.; Gu, N.Y.; Jeong, S.Y.; Byeon, J.S.; Jeong, D.U.; Ouh, I.O.; Lee, Y.H.; Hyun, B.H. Canine Natural Killer Cell-Derived Exosomes Exhibit Antitumor Activity in a Mouse Model of Canine Mammary Tumor. Biomed. Res. Int. 2021, 2021, 6690704. [Google Scholar] [CrossRef]
- Kim, Y.I.; Ahn, B.C.; Ronald, J.A.; Katzenberg, R.; Singh, A.; Paulmurugan, R.; Ray, S.; Gambhir, S.S.; Hofmann, L.V. Intratumoral versus intravenous gene therapy using a transcriptionally targeted viral vector in an orthotopic hepatocellular carcinoma rat model. J. Vasc. Interv. Radiol. 2012, 23, 704–711. [Google Scholar] [CrossRef]
- Baniel, C.C.; Sumiec, E.G.; Hank, J.A.; Bates, A.M.; Erbe, A.K.; Pieper, A.A.; Hoefges, A.G.; Patel, R.B.; Rakhmilevich, A.L.; Morris, Z.S.; et al. Intratumoral injection reduces toxicity and antibody-mediated neutralization of immunocytokine in a mouse melanoma model. J. Immunother. Cancer 2020, 8, e001262. [Google Scholar] [CrossRef]
- Hooijmans, C.R.; Rovers, M.M.; de Vries, R.B.; Leenaars, M.; Ritskes-Hoitinga, M.; Langendam, M.W. SYRCLE’s risk of bias tool for animal studies. BMC Med. Res. Methodol. 2014, 14, 43. [Google Scholar] [CrossRef] [PubMed]
- van Dommelen, S.M.; Vader, P.; Lakhal, S.; Kooijmans, S.A.; van Solinge, W.W.; Wood, M.J.; Schiffelers, R.M. Microvesicles and exosomes: Opportunities for cell-derived membrane vesicles in drug delivery. J. Control Release 2012, 161, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.Y.; Papoutsakis, E.T. Extracellular vesicles: Exosomes, microparticles, their parts, and their targets to enable their biomanufacturing and clinical applications. Curr. Opin. Biotechnol. 2019, 60, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Bryceson, Y.T.; March, M.E.; Ljunggren, H.G.; Long, E.O. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol Rev. 2006, 214, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Hood, S.P.; Foulds, G.A.; Imrie, H.; Reeder, S.; McArdle, S.E.B.; Khan, M.; Pockley, A.G. Phenotype and Function of Activated Natural Killer Cells From Patients With Prostate Cancer: Patient-Dependent Responses to Priming and IL-2 Activation. Front. Immunol. 2019, 9, 3169. [Google Scholar] [CrossRef] [PubMed]
- Lugini, L.; Cecchetti, S.; Huber, V.; Luciani, F.; Macchia, G.; Spadaro, F.; Paris, L.; Abalsamo, L.; Colone, M.; Molinari, A.; et al. Immune surveillance properties of human NK cell-derived exosomes. J. Immunol. 2012, 189, 2833–2842. [Google Scholar] [CrossRef]
- Shoae-Hassani, A.; Hamidieh, A.A.; Behfar, M.; Mohseni, R.; Mortazavi-Tabatabaei, S.A.; Asgharzadeh, S. NK Cell-derived Exosomes From NK Cells Previously Exposed to Neuroblastoma Cells Augment the Antitumor Activity of Cytokine-activated NK Cells. J. Immunother. 2017, 40, 265–276. [Google Scholar] [CrossRef]
- Fehniger, T.A.; Bluman, E.M.; Porter, M.M.; Mrózek, E.; Cooper, M.A.; VanDeusen, J.B.; Frankel, S.R.; Stock, W.; Caligiuri, M.A. Potential mechanisms of human natural killer cell expansion in vivo during low-dose IL-2 therapy. J. Clin. Invest. 2000, 106, 117–124. [Google Scholar] [CrossRef]
- Ishikawa, T.; Okayama, T.; Sakamoto, N.; Ideno, M.; Oka, K.; Enoki, T.; Mineno, J.; Yoshida, N.; Katada, K.; Kamada, K.; et al. Phase I clinical trial of adoptive transfer of expanded natural killer cells in combination with IgG1 antibody in patients with gastric or colorectal cancer. Int. J. Cancer 2018, 142, 2599–2609. [Google Scholar] [CrossRef]
- Sanchez-Martinez, D.; Allende-Vega, N.; Orecchioni, S.; Talarico, G.; Cornillon, A.; Vo, D.N.; Rene, C.; Lu, Z.Y.; Krzywinska, E.; Anel, A.; et al. Expansion of allogeneic NK cells with efficient antibody-dependent cell cytotoxicity against multiple tumors. Theranostics 2018, 8, 3856–3869. [Google Scholar] [CrossRef]
- Manuel, J. Low cost tissue culture technology for the regeneration of some economically important plants for developing countries. Int. J. Agric. Environ. Biotechnol. 2013, 6, 703–711. [Google Scholar]
- Daniszewski, M.; Crombie, D.E.; Henderson, R.; Liang, H.H.; Wong, R.C.B.; Hewitt, A.W.; Pébay, A. Automated Cell Culture Systems and Their Applications to Human Pluripotent Stem Cell Studies. SLAS Technol. 2018, 23, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.N.F.B.; Yazid, M.D.; Yunus, M.H.M.; Chowdhury, S.R.; Lokanathan, Y.; Idrus, R.B.H.; Ng, A.M.H.; Law, J.X. Large-Scale Expansion of Human Mesenchymal Stem Cells. Stem Cells Int. 2020, 2020, 9529465. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Li, Y.J.; Hu, X.B.; Huang, S.; Xiang, D.X. Preservation of small extracellular vesicles for functional analysis and therapeutic applications: A comparative evaluation of storage conditions. Drug Deliv. 2021, 28, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Gelibter, S.; Marostica, G.; Mandelli, A.; Siciliani, S.; Podini, P.; Finardi, A.; Furlan, R. The impact of storage on extracellular vesicles: A systematic study. J. Extracell Vesicles 2022, 11, e12162. [Google Scholar] [CrossRef]
- Sivanantham, A.; Jin, Y. Impact of Storage Conditions on EV Integrity/Surface Markers and Cargos. Life 2022, 12, 697. [Google Scholar] [CrossRef]
- Palay, S.L.; Palade, G.E. The fine structure of neurons. J. Biophys. Biochem. Cytol. 1955, 1, 69–88. [Google Scholar] [CrossRef]
- Rashed, M.H.; Bayraktar, E.; Helal, G.K.; Abd-Ellah, M.F.; Amero, P.; Chavez-Reyes, A.; Rodriguez-Aguayo, C. Exosomes: From Garbage Bins to Promising Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 538. [Google Scholar] [CrossRef]
- Vidal, M. Exosomes: Revisiting their role as “garbage bags”. Traffic 2019, 20, 815–828. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef]
- Koh, B.; Tan, K.L.; Chan, H.H.; Daniel Looi, Q.H.; Lim, M.N.; How, C.W.; Law, J.X.; Foo, J.B. A Simple Benchtop Filtration Method to Isolate Small Extracellular Vesicles from Human Mesenchymal Stem Cells. J. Vis. Exp. 2022, 184, e64106. [Google Scholar] [CrossRef] [PubMed]
- Al-Masawa, M.E.; Alshawsh, M.A.; Ng, C.Y.; Ng, A.M.H.; Foo, J.B.; Vijakumaran, U.; Subramaniam, R.; Ghani, N.A.A.; Witwer, K.W.; Law, J.X. Efficacy and safety of small extracellular vesicle interventions in wound healing and skin regeneration: A systematic review and meta-analysis of animal studies. Theranostics 2022, 12, 6455–6508. [Google Scholar] [CrossRef] [PubMed]
- van Reis, R.; Leonard, L.C.; Hsu, C.C.; Builder, S.E. Industrial scale harvest of proteins from mammalian cell culture by tangential flow filtration. Biotechnol. Bioeng. 1991, 38, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Busatto, S.; Vilanilam, G.; Ticer, T.; Lin, W.L.; Dickson, D.W.; Shapiro, S.; Bergese, P.; Wolfram, J. Tangential Flow Filtration for Highly Efficient Concentration of Extracellular Vesicles from Large Volumes of Fluid. Cells 2018, 7, 273. [Google Scholar] [CrossRef] [PubMed]
- Watson, D.C.; Yung, B.C.; Bergamaschi, C.; Chowdhury, B.; Bear, J.; Stellas, D.; Morales-Kastresana, A.; Jones, J.C.; Felber, B.K.; Chen, X.; et al. Scalable, cGMP-compatible purification of extracellular vesicles carrying bioactive human heterodimeric IL-15/lactadherin complexes. J. Extracell Vesicles 2018, 7, 1442088. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin–Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Kazemi, N.Y.; Gendrot, B.; Berishvili, E.; Markovic, S.N.; Cohen, M. The Role and Clinical Interest of Extracellular Vesicles in Pregnancy and Ovarian Cancer. Biomedicines 2021, 9, 1257. [Google Scholar] [CrossRef]
- Witwer, K.W.; Théry, C. Extracellular vesicles or exosomes? On primacy, precision, and popularity influencing a choice of nomenclature. J. Extracell Vesicles 2019, 8, 1648167. [Google Scholar] [CrossRef]
- Ashammakhi, N.; Ahadian, S.; Darabi, M.A.; El Tahchi, M.; Lee, J.; Suthiwanich, K.; Sheikhi, A.; Dokmeci, M.R.; Oklu, R.; Khademhosseini, A. Minimally Invasive and Regenerative Therapeutics. Adv. Mater. 2019, 31, e1804041. [Google Scholar] [CrossRef]
- Pinto, A.; Faiz, O.; Davis, R.; Almoudaris, A.; Vincent, C. Surgical complications and their impact on patients’ psychosocial well-being: A systematic review and meta-analysis. BMJ Open 2016, 6, e007224. [Google Scholar] [CrossRef]
- Cheng, H.; Clymer, J.W.; Po-Han Chen, B.; Sadeghirad, B.; Ferko, N.C.; Cameron, C.G.; Hinoul, P. Prolonged operative duration is associated with complications: A systematic review and meta-analysis. J. Surg. Res. 2018, 229, 134–144. [Google Scholar] [CrossRef]
- Sultana, A.; Zare, M.; Thomas, V.; Kumar, T.S.; Ramakrishna, S. Nano-based drug delivery systems: Conventional drug delivery routes, recent developments and future prospects. Med. Drug Discov. 2022, 15, 100134. [Google Scholar] [CrossRef]
- Jin, J.F.; Zhu, L.L.; Chen, M.; Xu, H.M.; Wang, H.F.; Feng, X.Q.; Zhu, X.P.; Zhou, Q. The optimal choice of medication administration route regarding intravenous, intramuscular, and subcutaneous injection. Patient Prefer. Adherence 2015, 9, 923–942. [Google Scholar] [CrossRef] [PubMed]
- Lam, W.J.; Bhowmick, T.; Gross, A.; Vanschooneveld, T.C.; Weinstein, M.P. Using higher doses to compensate for tubing residuals in extended-infusion piperacillin-tazobactam. Ann. Pharmacother. 2013, 47, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Bolla, B.; Buxani, Y.; Wong, R.; Jones, L.; Dube, M. Understanding IV antimicrobial drug losses: The importance of flushing infusion administration sets. JAC Antimicrob. Resist. 2020, 2, dlaa061. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.L.; Zhang, M.; Tang, Y.L.; Liang, X.H. Cancer cell dormancy: Mechanisms and implications of cancer recurrence and metastasis. OncoTarget. Ther. 2017, 10, 5219–5228. [Google Scholar] [CrossRef]
- Gomis, R.R.; Gawrzak, S. Tumor cell dormancy. Mol. Oncol. 2017, 11, 62–78. [Google Scholar] [CrossRef]
- Becker, P.S.; Suck, G.; Nowakowska, P.; Ullrich, E.; Seifried, E.; Bader, P.; Tonn, T.; Seidl, C. Selection and expansion of natural killer cells for NK cell-based immunotherapy. Cancer Immunol. Immunother. 2016, 65, 477–484. [Google Scholar] [CrossRef]
- Bt Hj Idrus, R.; Abas, A.; Ab Rahim, F.; Saim, A.B. Clinical Translation of Cell Therapy, Tissue Engineering, and Regenerative Medicine Product in Malaysia and Its Regulatory Policy. Tissue Eng. Part A 2015, 21, 2812–2816. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).