
Modern Advances in CARs Therapy and Creating a New Approach to Future Treatment

Abstract
:1. Introduction
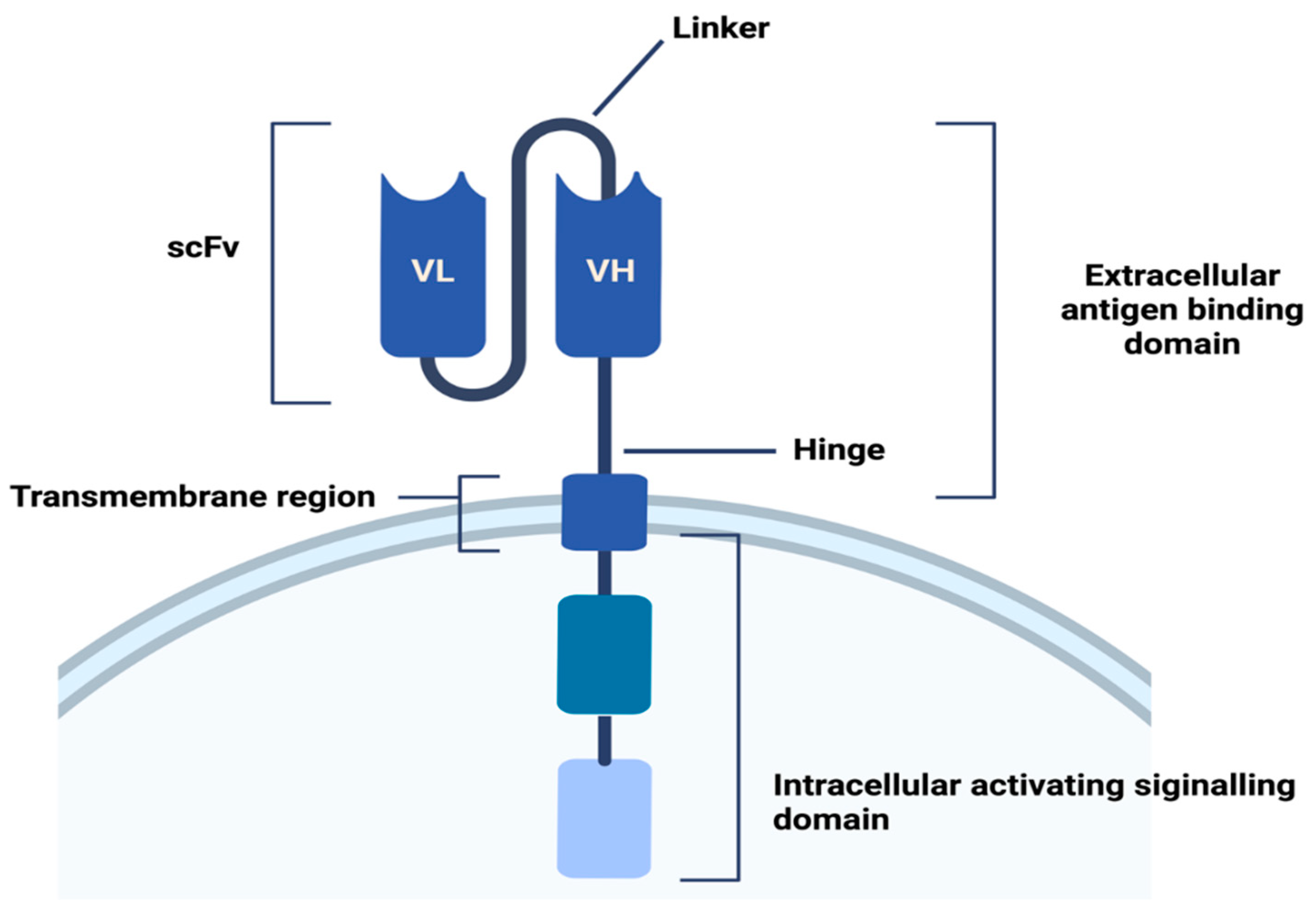
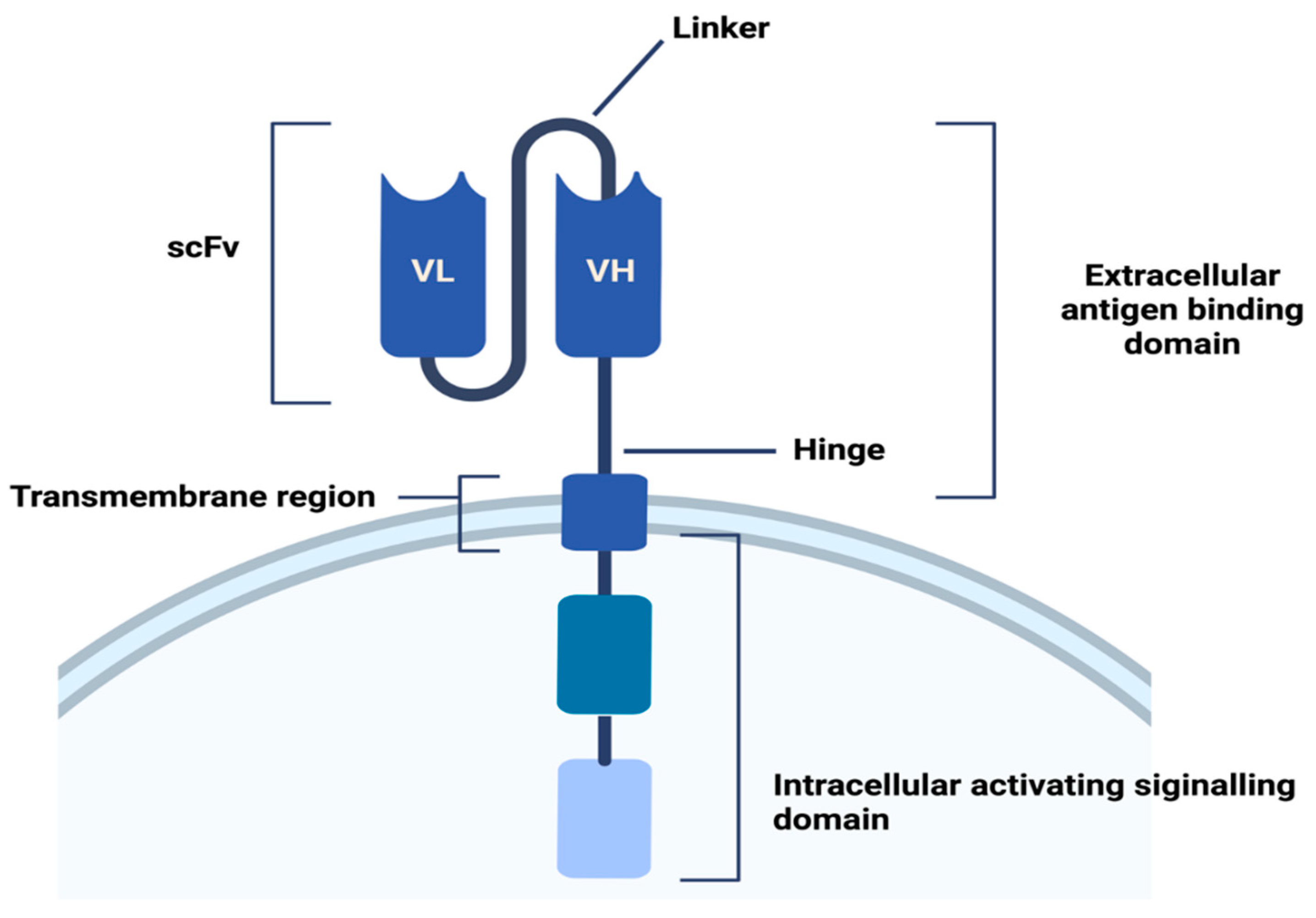
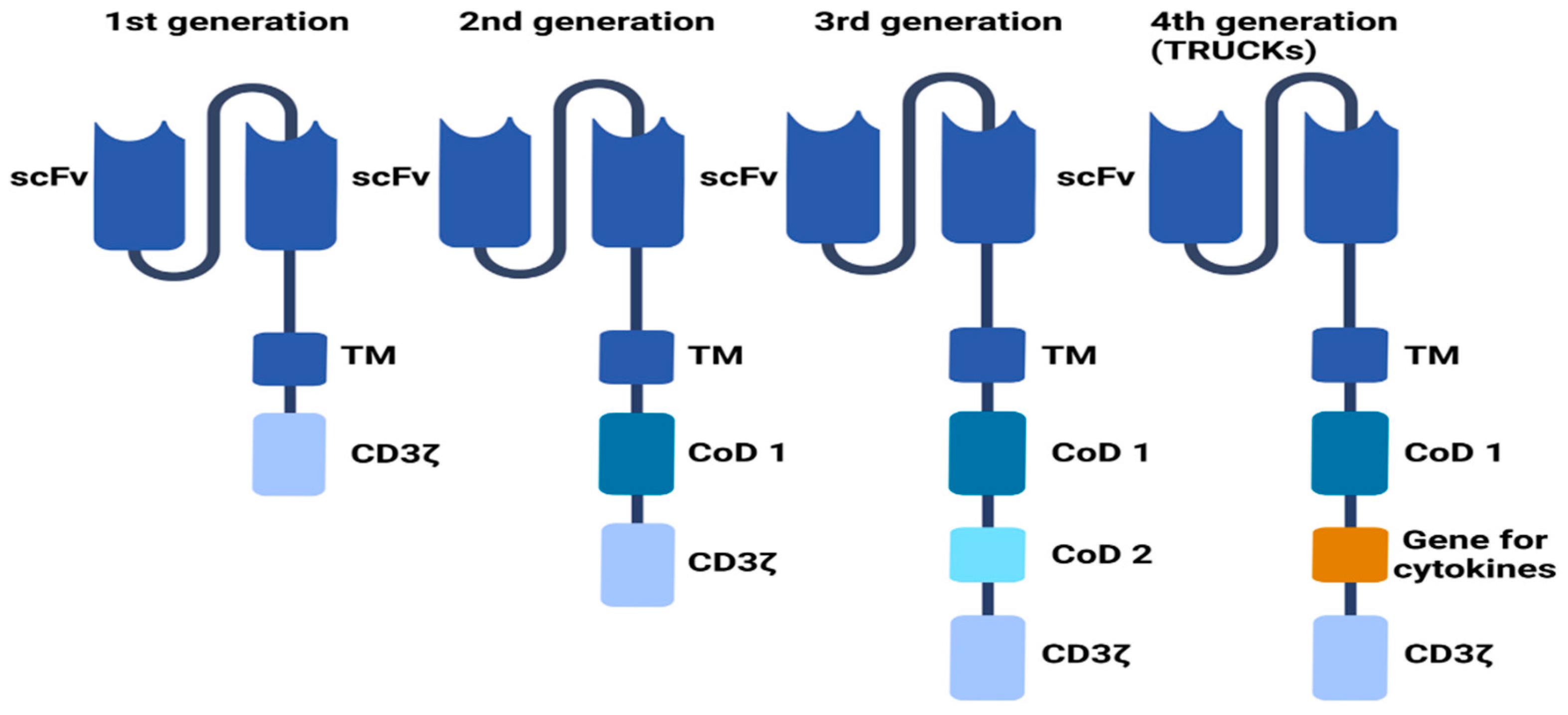
2. CARs
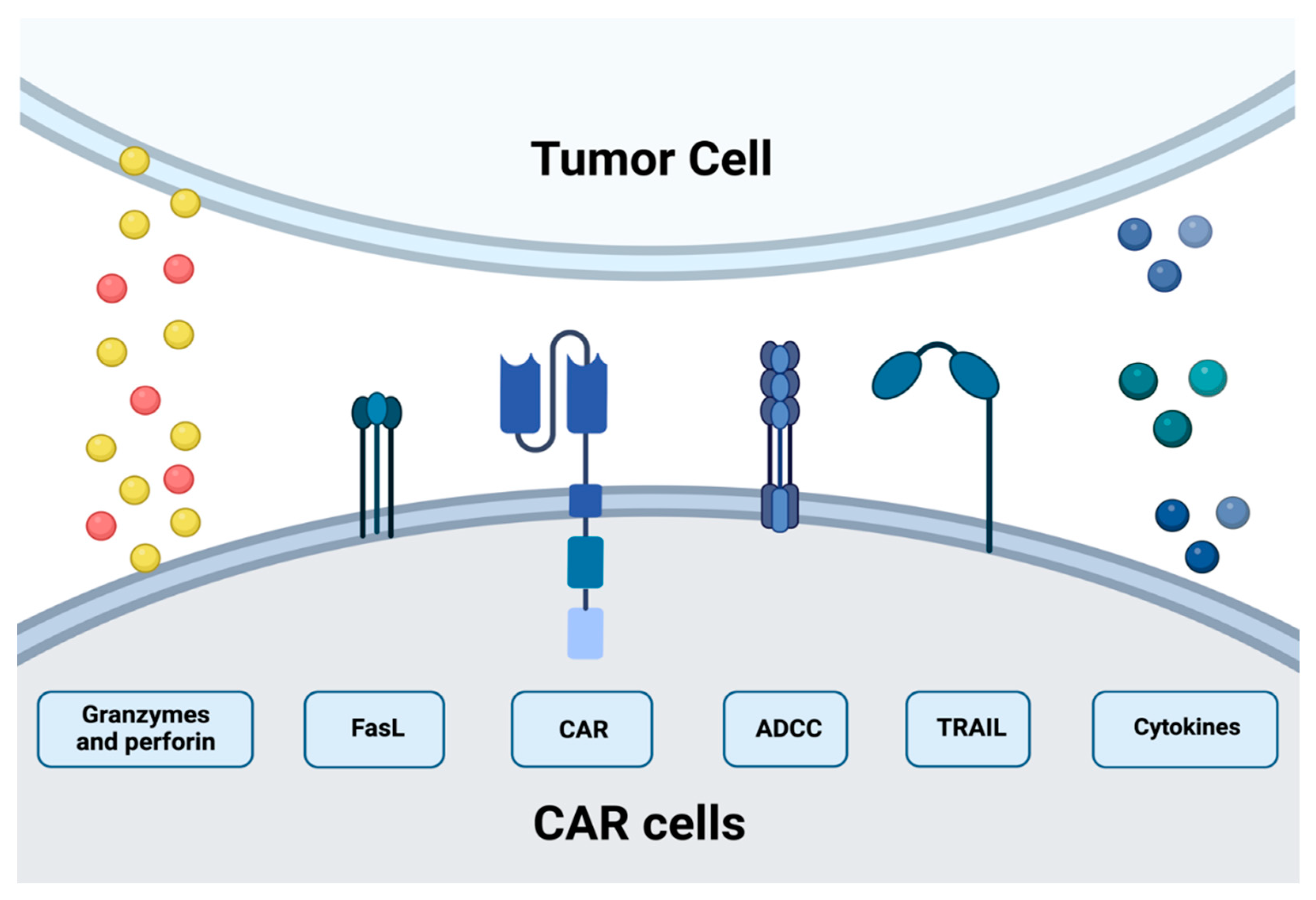
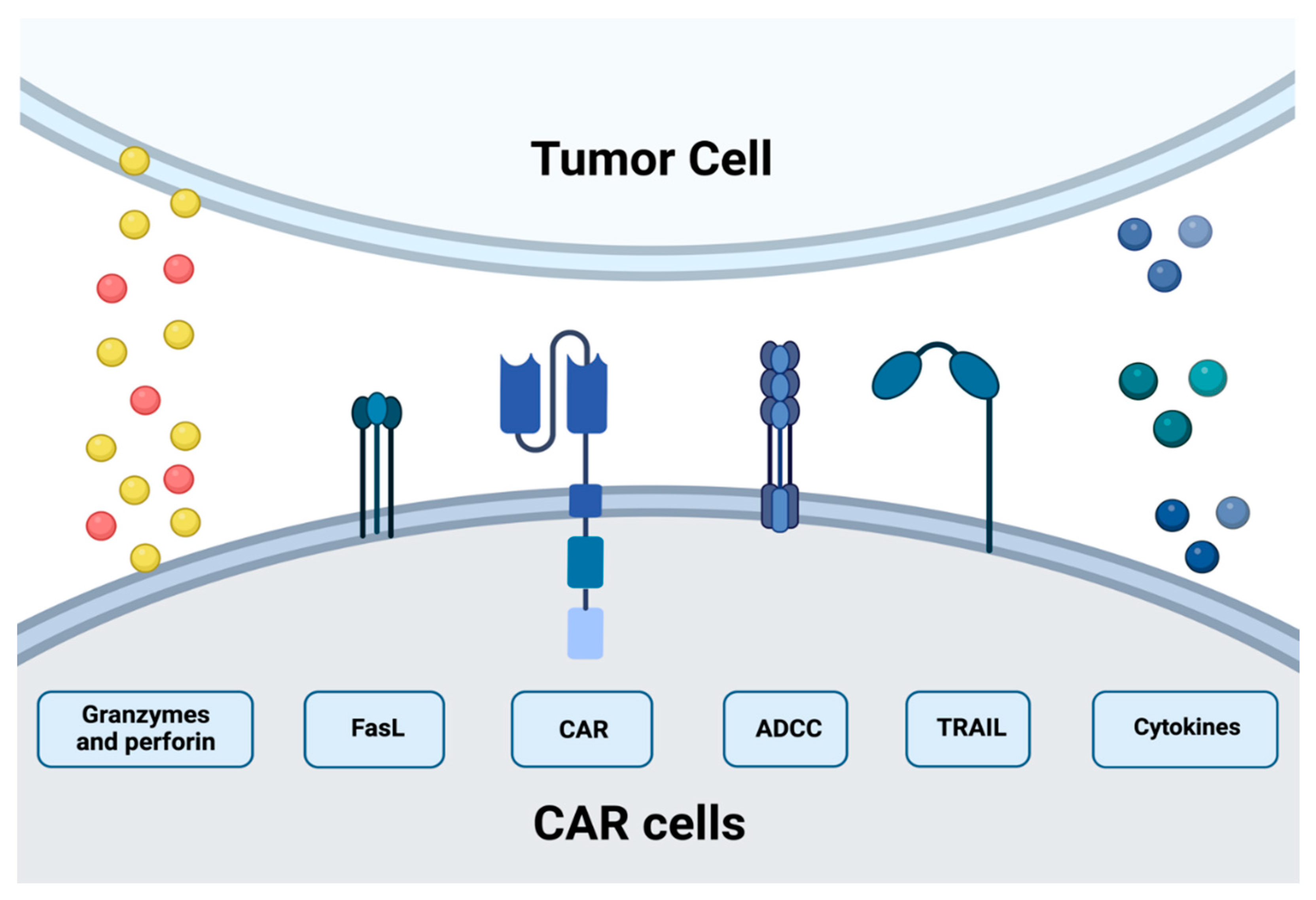
3. CAR-T Cytotoxic Mechanism
4. CAR-T Therapy
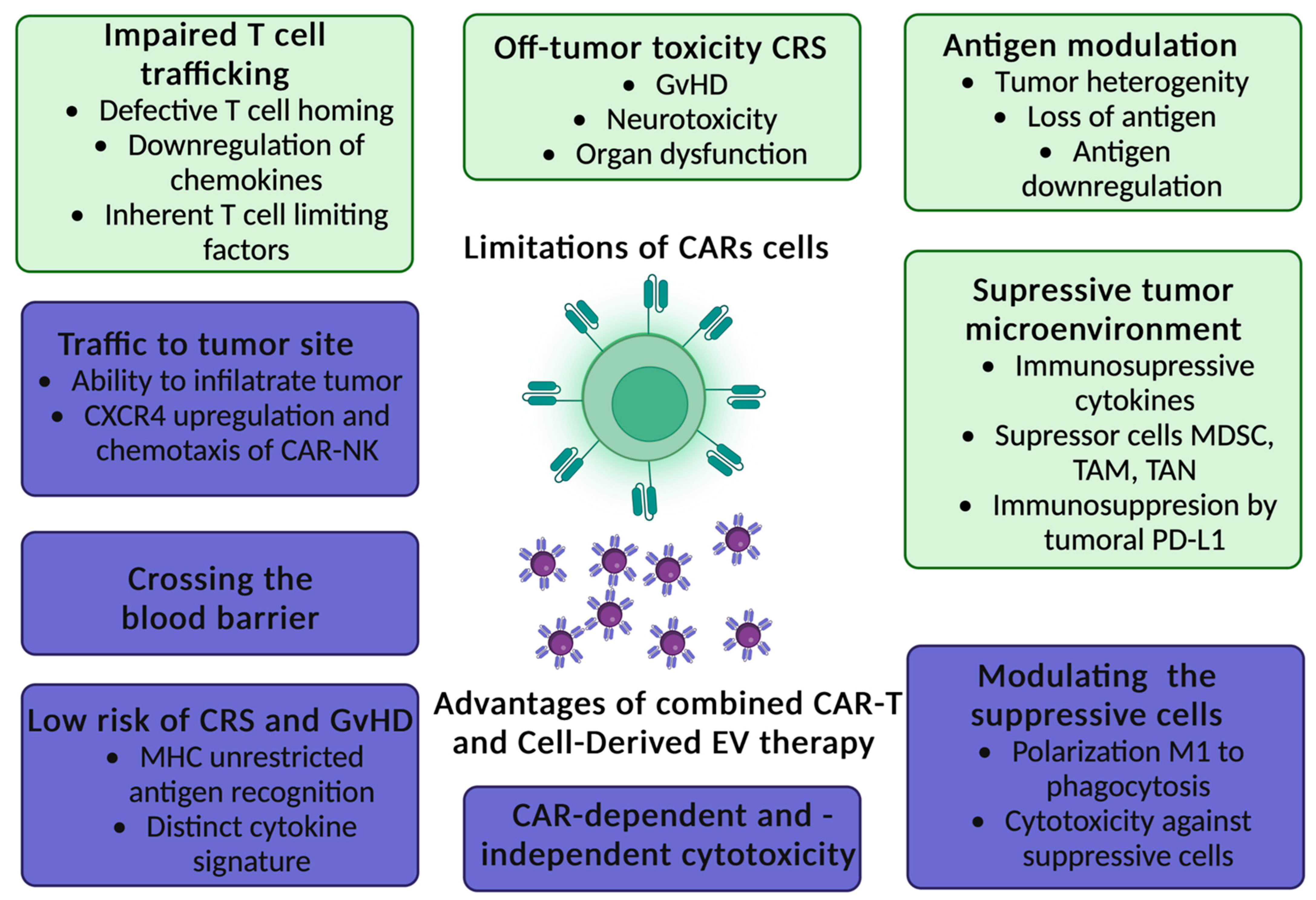
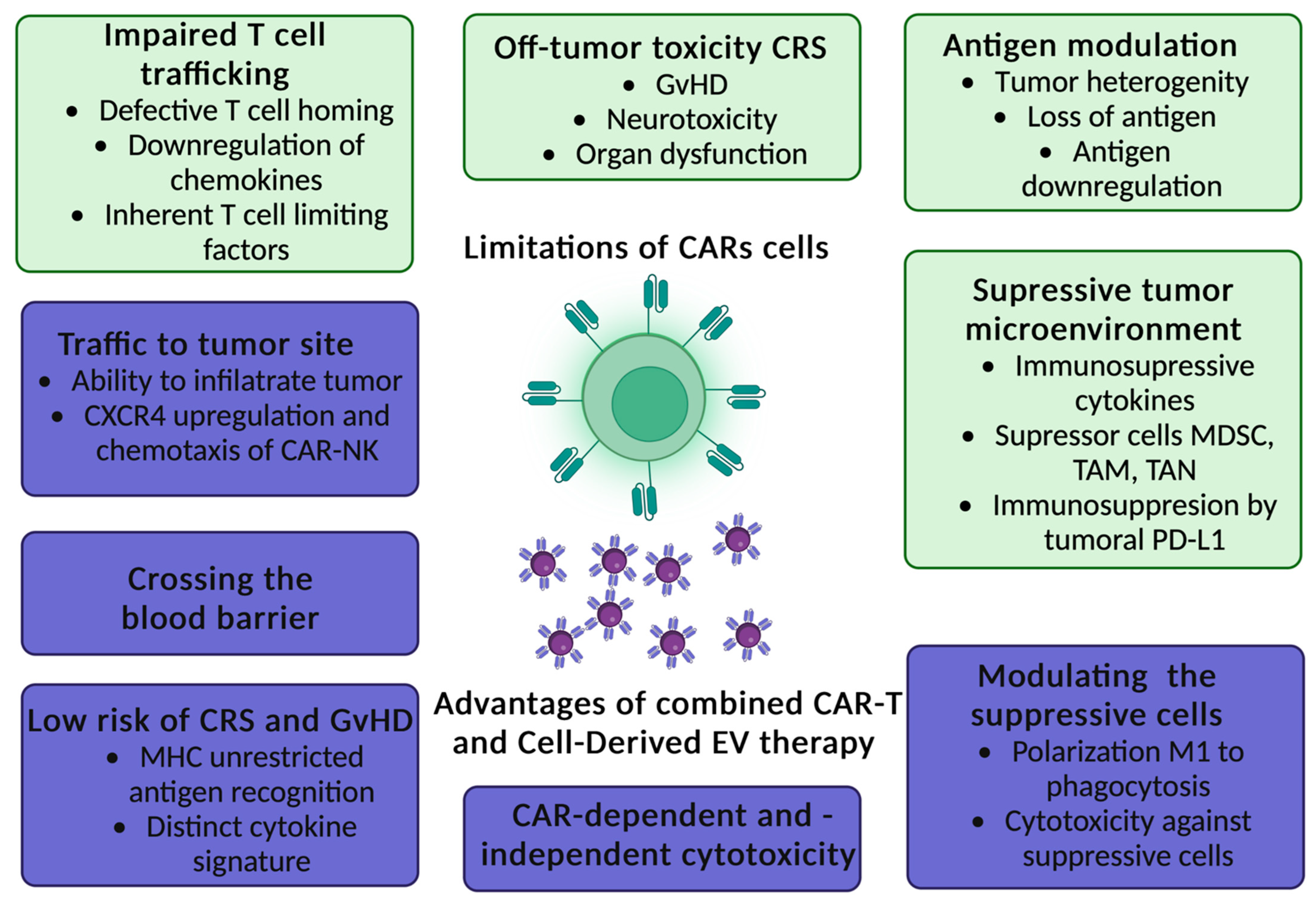
5. Limitations of CAR-T Cells Therapy
6. CAR-NK Cytotoxic Mechanism
7. CAR-NK Therapy
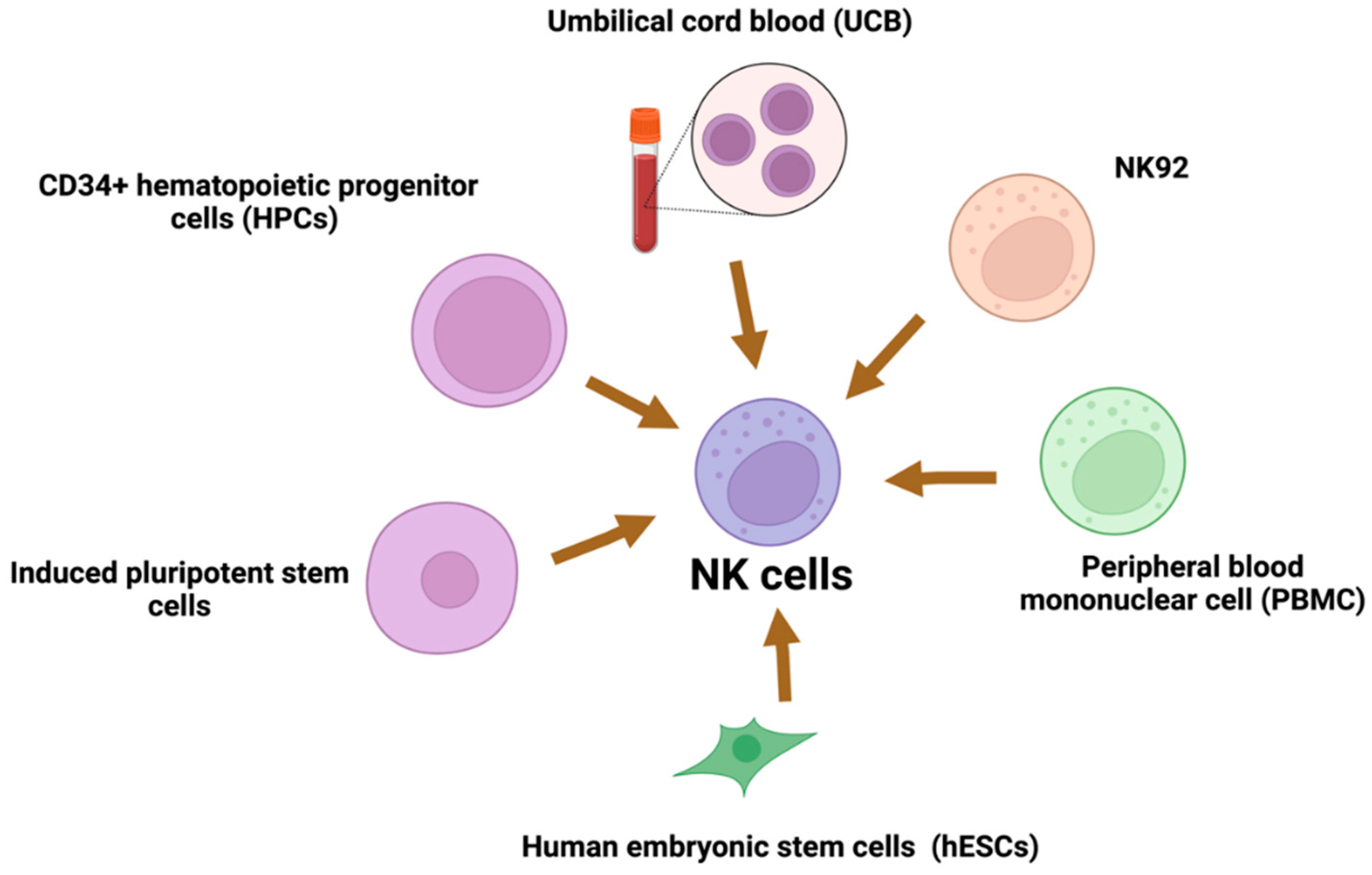
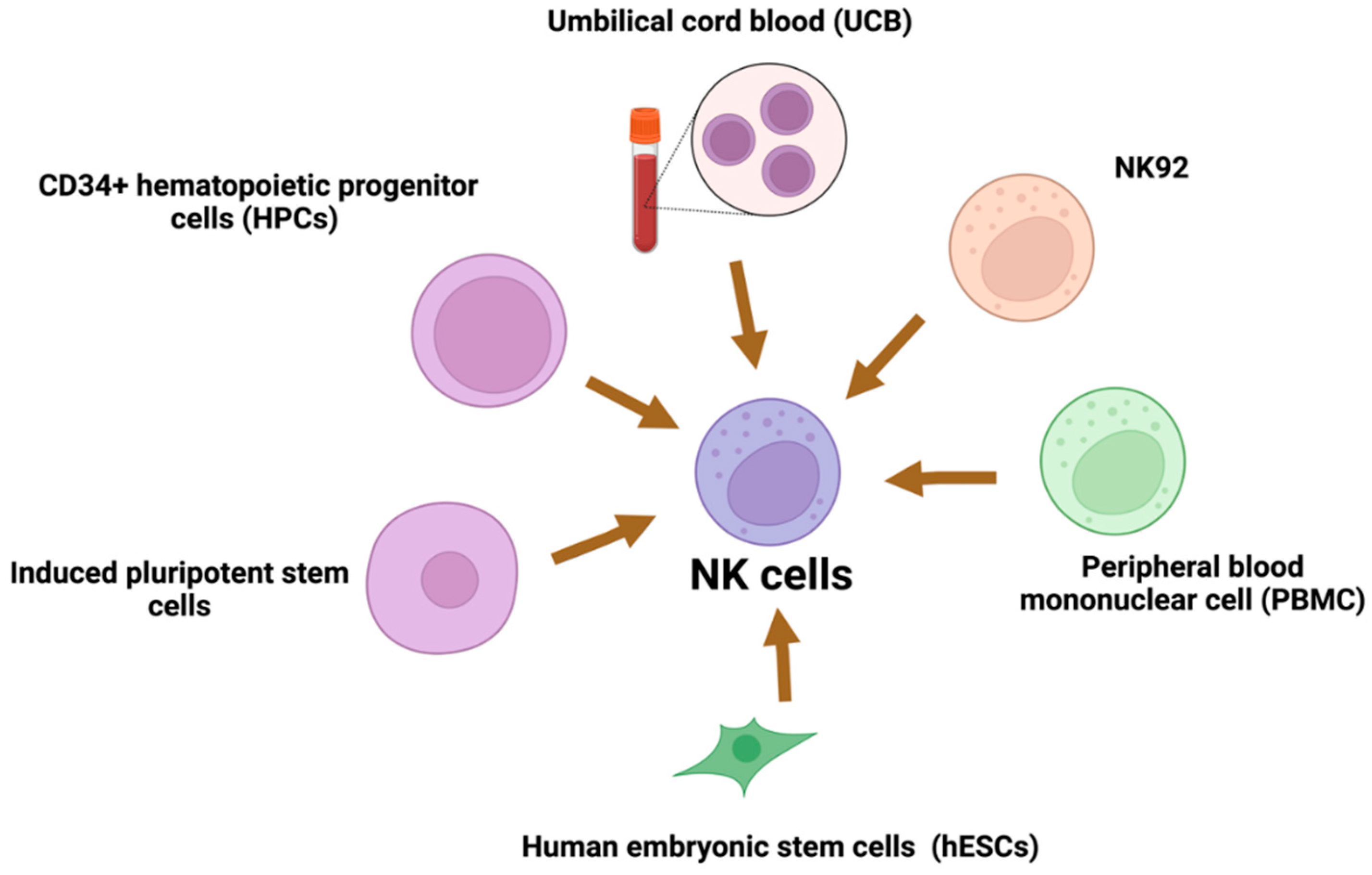
8. Sources of NK Cells
9. Limitations CAR-NK Cells Therapy
10. Nanobodies Based CARs
11. CAR Exosomes in Cancer Therapy as a Novel Anti-Cancer Strategy
12. Comparison to Other Immunotherapies
13. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, D.; Li, X.; Zhou, W.-L.; Huang, Y.; Liang, X.; Jiang, L.; Yang, X.; Sun, J.; Li, Z.; Han, W.-D.; et al. Genetically engineered T cells for cancer immunotherapy. Signal Transduct. Target. Ther. 2019, 4, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, V.; Shamalov, K.; Meir, S.; Hoogi, S.; Sarkar, R.; Pinker, S.; Markel, G.; Porgador, A.; Cohen, C.J. Targeting Multiple Tumors Using T-Cells Engineered to Express a Natural Cytotoxicity Receptor 2-Based Chimeric Receptor. Front. Immunol. 2017, 8, 1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daher, M.; Rezvani, K. Outlook for New CAR-Based Therapies with a Focus on CAR NK Cells: What Lies Beyond CAR-Engineered T Cells in the Race against Cancer. Cancer Discov. 2021, 11, 45–58. [Google Scholar] [CrossRef]
- Benmebarek, M.-R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20, 1283. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Sun, H.-Y.; Xiao, W.-H.; Zhang, C.; Tian, Z.-G. Natural killer cell dysfunction in hepatocellular carcinoma and NK cell-based immunotherapy. Acta Pharmacol. Sin. 2015, 36, 1191–1199. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Mishra, H.K.; Walcheck, B. Role of ADAM17 as a regulatory checkpoint of CD16A in NK cells and as a potential target for cancer immunotherapy. J. Leukoc. Biol. 2019, 105, 1297–1303. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.C.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef]
- Skorka, K.; Ostapinska, K.; Malesa, A.; Giannopoulos, K. The Application of CAR-T Cells in Haematological Malignancies. Arch. Immunol. Ther. Exp. 2020, 68, 34. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Minamino, S.; Kuwabara, K. CAR-expressing NK cells for cancer therapy: A new hope. Biosci. Trends 2020, 14, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Caruso, H.G.; Hurton, L.V.; Najjar, A.; Rushworth, D.; Ang, S.; Olivares, S.; Mi, T.; Switzer, K.; Singh, H.; Huls, H.; et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res. 2015, 75, 3505–35018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.C.; Cogdill, A.P.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.A.; Büning, H.; Sauer, M.; Schambach, A. Use of Cell and Genome Modification Technologies to Generate Improved "Off-the-Shelf" CAR T and CAR NK Cells. Front. Immunol. 2020, 11, 1965. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef] [Green Version]
- Bridgeman, J.S.; Hawkins, R.E.; Bagley, S.; Blaylock, M.; Holland, M.; Gilham, D.E. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J. Immunol. 2010, 184, 6938–6949. [Google Scholar] [CrossRef] [Green Version]
- Alabanza, L.; Pegues, M.; Geldres, C.; Shi, V.; Wiltzius, J.J.; Sievers, S.A.; Yang, S.; Kochenderfer, J.N. Function of Novel Anti-CD19 Chimeric Antigen Receptors with Human Variable Regions Is Affected by Hinge and Transmembrane Domains. Mol. Ther. 2017, 25, 2452–2465. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Bridgeman, J.S.; Hawkins, R.E.; Hombach, A.A.; Abken, H.; Gilham, D.E. Building better chimeric antigen receptors for adoptive T cell therapy. Curr. Gene Ther. 2010, 10, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Kowolik, C.M.; Topp, M.S.; Gonzalez, S.; Pfeiffer, T.; Olivares, S.; Gonzalez, N.; Smith, D.D.; Forman, S.J.; Jensen, M.C.; Cooper, L.J. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006, 66, 10995–11004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T cells. Biomark. Res. 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, D.-G.; Ye, Q.; Poussin, M.; Harms, G.M.; Figini, M.; Powell, D.J., Jr. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood 2012, 119, 696–706. [Google Scholar] [CrossRef]
- Elahi, R.; Khosh, E.; Tahmasebi, S.; Esmaeilzadeh, A. Cell Hacking: Challenges and Clinical Approaches to Create Smarter Generations of Chimeric Antigen Receptor T Cells. Front. Immunol. 2018, 9, 1717. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Ping, J.; Huang, Z.; Zhang, X.; Zhou, J.; Wang, G.; Liu, S.; Ma, J. CAR-T Cell Therapy in Cancer: Tribulations and Road Ahead. J. Immunol. Res. 2020, 2020, 1924379. [Google Scholar] [CrossRef] [Green Version]
- Petersen, T.C.; Krenciute, G. Next Generation CAR T Cells for the Immunotherapy of High-Grade Glioma. Front. Oncol. 2019, 9, 69. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Riddell, S.R. Engineering CAR-T cells: Design concepts. Trends Immunol. 2015, 36, 494–502. [Google Scholar] [CrossRef] [Green Version]
- Harris, T.D.; Kranz, D.M. Adoptive T Cell Therapies: A Comparison of T Cell Receptors and Chimeric Antigen Receptors. Trends Pharmacol. Sci. 2016, 37, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Meiraz, A.; Garber, O.G.; Harari, S.; Hassin, D.; Berke, G. Switch from perforin-expressing to perforin-deficient CD8(+) T cells accounts for two distinct types of effector cytotoxic T lymphocytes in vivo. Immunology 2009, 128, 69–82. [Google Scholar] [CrossRef]
- Davenport, A.J.; Cross, R.S.; Watson, K.A.; Liao, Y.; Shi, W.; Prince, H.M.; Beavis, P.A.; Trapani, J.A.; Kershaw, M.H.; Ritchie, D.S.; et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl. Acad. Sci. USA 2018, 115, E2068–E2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stinchcombe, J.C.; Majorovits, E.; Bossi, G.; Fuller, S.; Griffiths, G.M. Centrosome polarization delivers secretory granules to the immunological synapse. Nature 2006, 443, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Kägi, D.; Ledermann, B.; Bürki, K.; Seiler, P.; Odermatt, B.; Olsen, K.J.; Podack, E.R.; Zinkernagel, R.M.; Hengartner, H. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 1994, 369, 31–37. [Google Scholar] [CrossRef] [PubMed]
- de Saint Basile, G.; Menasche, G.; Fischer, A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat. Rev. Immunol. 2010, 10, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Koehler, H.; Kofler, D.; Hombach, A.; Abken, H. CD28 costimulation overcomes transforming growth factor-beta-mediated repression of proliferation of redirected human CD4+ and CD8+ T cells in an antitumor cell attack. Cancer Res. 2007, 67, 2265–2273. [Google Scholar] [CrossRef] [Green Version]
- Davenport, A.J.; Jenkins, M.R.; Cross, R.S.; Yong, C.S.; Prince, H.M.; Ritchie, D.S.; Trapani, J.A.; Kershaw, M.H.; Darcy, P.K.; Neeson, P.J. CAR-T Cells Inflict Sequential Killing of Multiple Tumor Target Cells. Cancer Immunol. Res. 2015, 3, 483–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumaresan, P.R.; Manuri, P.R.; Albert, N.D.; Maiti, S.; Singh, H.; Mi, T.; Roszik, J.; Rabinovich, B.; Olivares, S.; Krishnamurthy, J.; et al. Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc. Natl. Acad. Sci. USA 2014, 111, 10660–10665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef]
- Voskoboinik, I.; Whisstock, J.C.; Trapani, J.A. Trapani, Perforin and granzymes: Function, dysfunction and human pathology. Nat. Rev. Immunol. 2015, 15, 388–400. [Google Scholar] [CrossRef]
- Hong, L.K.; Chen, Y.; Smith, C.C.; Montgomery, S.A.; Vincent, B.G.; Dotti, G.; Savoldo, B. CD30-Redirected Chimeric Antigen Receptor T Cells Target CD30(+) and CD30(-) Embryonal Carcinoma via Antigen-Dependent and Fas/FasL Interactions. Cancer Immunol. Res. 2018, 6, 1274–1287. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.-H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Textor, A.; Listopad, J.J.; Wührmann, L.L.; Perez, C.; Kruschinski, A.; Chmielewski, M.; Abken, H.; Blankenstein, T.; Charo, J. Efficacy of CAR T-cell therapy in large tumors relies upon stromal targeting by IFNgamma. Cancer Res. 2014, 74, 6796–6805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braendstrup, P.; Levine, B.L.; Ruella, M. The long road to the first FDA-approved gene therapy: Chimeric antigen receptor T cells targeting CD19. Cytotherapy 2020, 22, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Yakoub-Agha, I.; Chabannon, C.; Bader, P.; Basak, G.W.; Bonig, H.; Ciceri, F.; Corbacioglu, S.; Duarte, R.F.; Einsele, H.; Hudecek, M.; et al. Management of adults and children undergoing chimeric antigen receptor T-cell therapy: Best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Haematologica 2020, 105, 297–316. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.; Wang, Y.; Munoz, J.; Jain, P. Brexucabtagene autoleucel: A breakthrough in the treatment of mantle cell lymphoma. Drugs Today 2022, 58, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.V. Approval of brexucabtagene autoleucel for adults with relapsed and refractory acute lymphocytic leukemia. Blood 2022, 140, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Lamb, M.; Cairo, M.S.; Lee, D.A. The Future of Natural Killer Cell Immunotherapy for B Cell Non-Hodgkin Lymphoma (B Cell NHL). Curr. Treat. Options Oncol. 2022, 23, 381–403. [Google Scholar] [CrossRef]
- Jasinski, M.; Basak, G.W.; Jedrzejczak, W.W. Perspectives for the Use of CAR-T Cells for the Treatment of Multiple Myeloma. Front. Immunol. 2021, 12, 632937. [Google Scholar] [CrossRef]
- Mikkilineni, L.; Kochenderfer, J.N. CAR T cell therapies for patients with multiple myeloma. Nat. Rev. Clin. Oncol. 2021, 18, 71–84. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, Y.; Francisco, N.M.; Zhang, Y.; Wu, M. The application of CAR-T cell therapy in hematological malignancies: Advantages and challenges. Acta Pharm. Sin. B 2018, 8, 539–551. [Google Scholar] [CrossRef]
- Hay, K.A. Cytokine release syndrome and neurotoxicity after CD19 chimeric antigen receptor-modified (CAR-) T cell therapy. Br. J. Haematol. 2018, 183, 364–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poorebrahim, M.; Melief, J.; de Coaña, Y.P.; Wickström, S.L.; Cid-Arregui, A.; Kiessling, R. Counteracting CAR T cell dysfunction. Oncogene 2021, 40, 421–435. [Google Scholar] [CrossRef]
- Franco, F.; Jaccard, A.; Romero, P.; Yu, Y.-R.; Ho, P.-C. Metabolic and epigenetic regulation of T-cell exhaustion. Nat. Metab. 2020, 2, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Pietrobon, V.; Todd, L.A.; Goswami, A.; Stefanson, O.; Yang, Z.; Marincola, F. Improving CAR T-Cell Persistence. Int. J. Mol. Sci. 2021, 22, 10828. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Cheng, J.; Mu, W.; Zhou, J.; Zhu, L. Advances in Universal CAR-T Cell Therapy. Front. Immunol. 2021, 12, 744823. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, J.K.; Gottschalk, S.; Talleur, A.C. Allogeneic CAR Cell Therapy-More Than a Pipe Dream. Front. Immunol. 2020, 11, 618427. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef]
- Zamai, L.; Del Zotto, G.; Buccella, F.; Gabrielli, S.; Canonico, B.; Artico, M.; Ortolani, C.; Papa, S. Understanding the Synergy of NKp46 and Co-Activating Signals in Various NK Cell Subpopulations: Paving the Way for More Successful NK-Cell-Based Immunotherapy. Cells 2020, 9, 753. [Google Scholar] [CrossRef]
- Bryceson, Y.; March, M.; Ljunggren, H.-G.; Long, E.O. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol. Rev. 2006, 214, 73–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingemann, H. Are natural killer cells superior CAR drivers? Oncoimmunology 2014, 3, e28147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, D.B.; Jacobson, C.A. CAR T-Cell Associated Neurotoxicity: Mechanisms, Clinicopathologic Correlates, and Future Directions. J. Natl. Cancer Inst. 2019, 111, 646–654. [Google Scholar] [CrossRef]
- Vitale, M.; Cantoni, C.; Della Chiesa, M.; Ferlazzo, G.; Carlomagno, S.; Pende, D.; Falco, M.; Pessino, A.; Muccio, L.; De Maria, A.; et al. An Historical Overview: The Discovery of How NK Cells Can Kill Enemies, Recruit Defense Troops, and More. Front. Immunol. 2019, 10, 1415. [Google Scholar] [CrossRef] [Green Version]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef]
- Oberoi, P.; Jabulowsky, R.A.; Bähr-Mahmud, H.; Wels, W.S. EGFR-targeted granzyme B expressed in NK cells enhances natural cytotoxicity and mediates specific killing of tumor cells. PLoS ONE 2013, 8, e61267. [Google Scholar] [CrossRef]
- Oberoi, P.; Wels, W.S. Arming NK cells with enhanced antitumor activity: CARs and beyond. Oncoimmunology 2013, 2, e25220. [Google Scholar] [CrossRef] [Green Version]
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.J.; Dougan, M.; Ingram, J.R.; Pishesha, N.; Fang, T.; Momin, N.; Ploegh, H.L. Improved Antitumor Efficacy of Chimeric Antigen Receptor T Cells that Secrete Single-Domain Antibody Fragments. Cancer Immunol. Res. 2020, 8, 518–529. [Google Scholar] [CrossRef] [PubMed]
- John, L.B.; Kershaw, M.; Darcy, P.K. Darcy, Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunology 2013, 2, e26286. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J., Jr.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, 1109. [Google Scholar] [CrossRef]
- Hosseinkhani, N.; Derakhshani, A.; Kooshkaki, O.; Abdoli Shadbad, M.; Hajiasgharzadeh, K.; Baghbanzadeh, A.; Safarpour, H.; Mokhtarzadeh, A.; Brunetti, O.; Yue, S.C.; et al. Immune Checkpoints and CAR-T Cells: The Pioneers in Future Cancer Therapies? Int. J. Mol. Sci. 2020, 21, 8305. [Google Scholar] [CrossRef] [PubMed]
- Oelsner, S.; Friede, M.E.; Zhang, C.; Wagner, J.; Badura, S.; Bader, P.; Ullrich, E.; Ottmann, O.G.; Klingemann, H.; Tonn, T.; et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy 2017, 19, 235–249. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.P.; Perry, E.H.; Price, T.H.; Bolan, C.D.; Karanes, C.; Boyd, T.M.; Chitphakdithai, P.; King, R.J. Recovery and safety profiles of marrow and PBSC donors: Experience of the National Marrow Donor Program. Biol. Blood Marrow Transplant. 2008, 14 (Suppl. S9), 29–36. [Google Scholar] [CrossRef] [Green Version]
- Wilber, A.; Linehan, J.L.; Tian, X.; Woll, P.S.; Morris, J.K.; Belur, L.R.; McIvor, R.S.; Kaufman, D.S. Efficient and stable transgene expression in human embryonic stem cells using transposon-mediated gene transfer. Stem Cells 2007, 25, 2919–2927. [Google Scholar] [CrossRef]
- Rezvani, K.; Rouce, R.; Liu, E.; Shpall, E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol. Ther. 2017, 25, 1769–1781. [Google Scholar] [CrossRef]
- Tonn, T.; Becker, S.; Esser, R.; Schwabe, D.; Seifried, E. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J. Hematother Stem Cell Res. 2001, 10, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Maki, G.; Klingemann, H.-G.; Martinson, J.A.; Tam, Y.K. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J. Hematother Stem Cell Res. 2001, 10, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; DeFor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geller, M.A.; Cooley, S.; Judson, P.L.; Ghebre, R.; Carson, L.F.; Argenta, P.A.; Jonson, A.L.; Panoskaltsis-Mortari, A.; Curtsinger, J.; McKenna, D.; et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy 2011, 13, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafei, H.; Daher, M.; Rezvani, K. Chimeric antigen receptor (CAR) natural killer (NK)-cell therapy: Leveraging the power of innate immunity. Br. J. Haematol. 2021, 193, 216–230. [Google Scholar] [CrossRef] [PubMed]
- Domogala, A.; Madrigal, J.A.; Saudemont, A. Cryopreservation has no effect on function of natural killer cells differentiated in vitro from umbilical cord blood CD34(+) cells. Cytotherapy 2016, 18, 754–759. [Google Scholar] [CrossRef]
- van Ostaijen-ten Dam, M.M.; Prins, H.-J.; Boerman, G.H.; Vervat, C.; Pende, D.; Putter, H.; Lankester, A.; van Tol, M.J.D.; Zwaginga, J.J.; Schilham, M.W. Preparation of Cytokine-activated NK Cells for Use in Adoptive Cell Therapy in Cancer Patients: Protocol Optimization and Therapeutic Potential. J. Immunother. 2016, 39, 90–100. [Google Scholar] [CrossRef]
- Klingemann, H. Challenges of cancer therapy with natural killer cells. Cytotherapy 2015, 17, 245–249. [Google Scholar] [CrossRef]
- Murray, S.; Lundqvist, A. Targeting the tumor microenvironment to improve natural killer cell-based immunotherapies: On being in the right place at the right time, with resilience. Hum. Vaccin. Immunother 2016, 12, 607–611. [Google Scholar] [CrossRef] [Green Version]
- Bi, J.; Tian, Z. NK Cell Dysfunction and Checkpoint Immunotherapy. Front. Immunol. 2019, 10, 1999. [Google Scholar] [CrossRef]
- Konjević, G.M.; Vuletić, A.M.; Mirjačić Martinović, K.M.; Larsen, A.K.; Jurišić, V.B. The role of cytokines in the regulation of NK cells in the tumor environment. Cytokine 2019, 117, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chu, J.; Yu, J.; Wei, W. Cellular and molecular mechanisms in graft-versus-host disease. J. Leukoc. Biol. 2016, 99, 279–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonetta, F.; Alvarez, M.; Negrin, R.S. Natural Killer Cells in Graft-versus-Host-Disease after Allogeneic Hematopoietic Cell Transplantation. Front. Immunol. 2017, 8, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.R.; Lee, Y.S.; Yang, S.H.; Ahn, K.H.; Lee, J.-H.; Lee, J.-H.; Kim, D.Y.; Kang, Y.A.; Jeon, M.; Seol, M.; et al. Generation of donor natural killer cells from CD34(+) progenitor cells and subsequent infusion after HLA-mismatched allogeneic hematopoietic cell transplantation: A feasibility study. Bone Marrow Transpl. 2010, 45, 1038–1046. [Google Scholar] [CrossRef]
- Rubnitz, J.E.; Inaba, H.; Ribeiro, R.C.; Pounds, S.; Rooney, B.; Bell, T.; Pui, C.-H.; Leung, W. NKAML: A pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 955–959. [Google Scholar] [CrossRef] [Green Version]
- Ingegnere, T.; Mariotti, F.R.; Pelosi, A.; Quintarelli, C.; DE Angelis, B.; Tumino, N.; Besi, F.; Cantoni, C.; Locatelli, F.; Vacca, P.; et al. Human CAR NK Cells: A New Non-viral Method Allowing High Efficient Transfection and Strong Tumor Cell Killing. Front. Immunol. 2019, 10, 957. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Freud, A.G.; Caligiuri, M.A. Location and cellular stages of natural killer cell development. Trends Immunol. 2013, 34, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Zhao, X.; Li, Z.; Hu, Y.; Wang, H. From CAR-T Cells to CAR-NK Cells: A Developing Immunotherapy Method for Hematological Malignancies. Front. Oncol. 2021, 11, 720501. [Google Scholar] [CrossRef]
- Imai, C.; Iwamoto, S.; Campana, D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 2005, 106, 376–383. [Google Scholar] [CrossRef] [Green Version]
- Genssler, S.; Burger, M.C.; Zhang, C.; Oelsner, S.; Mildenberger, I.; Wagner, M. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology 2016, 5, e1119354. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Liu, Q.; Zhong, M.; Wang, Z.; Chen, Z.; Zhang, Y.; Xing, H.; Tian, Z.; Tang, K.; Liao, X.; et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J. Hematol. Oncol. 2019, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Romanski, A.; Uherek, C.; Bug, G.; Seifried, E.; Klingemann, H.; Wels, W.S.; Ottmann, O.G.; Tonn, T. CD19-CAR engineered NK-92 cells are sufficient to overcome NK cell resistance in B-cell malignancies. J. Cell Mol. Med. 2016, 20, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, M.; Yang, S.; Wang, J.; Feng, X.; Han, Z. Natural killer cells: Of-the-shelf cytotherapy for cancer immunosurveillance. Am. J. Cancer Res. 2021, 11, 1770–1791. [Google Scholar] [PubMed]
- Geller, A.M.; Miller, J.S. Use of allogeneic NK cells for cancer immunotherapy. Immunotherapy 2011, 3, 1445–1459. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimiyan, H.; Tamimi, A.; Shokoohian, B.; Minaei, N.; Memarnejadian, A.; Hossein-Khannazer, N.; Hassan, M.; Vosough, M. Novel insights in CAR-NK cells beyond CAR-T cell technology; promising advantages. Int. Immunopharmacol. 2022, 106, 108587. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.E.; Shah, K. Nanobodies: Next Generation of Cancer Diagnostics and Therapeutics. Front. Oncol. 2020, 10, 1182. [Google Scholar] [CrossRef]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T design: Elements and their synergistic function. eBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef]
- Strohl, R.W.; Naso, M. Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells. Antibodies 2019, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Barakat, S.; Berksöz, M.; Zahedimaram, P.; Piepoli, S.; Erman, B. Nanobodies as molecular imaging probes. Free Radic. Biol. Med. 2022, 182, 260–275. [Google Scholar] [CrossRef]
- Greenberg, A.S.; Avila, D.; Hughes, M.; Hughes, A.; McKinney, E.C.; Flajnik, M.F. A new antigen receptor gene family that undergoes rearrangement and extensive somatic diversification in sharks. Nature 1995, 374, 168–173. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, C.; Gao, Q.; Li, L.-L.; Han, L.; Zhang, B.; Ding, Y.; Song, Z.; Zhang, R.; Zhang, J.; Wu, X.-H. The Application of Nanobody in CAR-T Therapy. Biomolecules 2021, 11, 238. [Google Scholar] [CrossRef] [PubMed]
- Völkel, T.; Korn, T.; Bach, M.; Müller, R.; Kontermann, R.E. Optimized linker sequences for the expression of monomeric and dimeric bispecific single-chain diabodies. Protein Eng. 2001, 14, 815–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Gorovits, B.; Koren, E. Immunogenicity of Chimeric Antigen Receptor T-Cell Therapeutics. BioDrugs 2019, 33, 275–284. [Google Scholar] [CrossRef]
- Denardo, G.L.; Bradt, B.M.; Denardo, M. Human antiglobulin response to foreign antibodies: Therapeutic benefit? Cancer Immunol. Immunother. 2003, 52, 309–316. [Google Scholar] [CrossRef]
- Klee, G.G. Human anti-mouse antibodies. Arch. Pathol. Lab. Med. 2000, 124, 921–923. [Google Scholar] [CrossRef]
- Ackaert, C.; Smiejkowska, N.; Xavier, C.; Sterckx, Y.G.J.; Denies, S.; Stijlemans, B.; Elkrim, Y.; Devoogdt, N.; Caveliers, V.; Lahoutte, T.; et al. Immunogenicity Risk Profile of Nanobodies. Front. Immunol. 2021, 12, 632687. [Google Scholar] [CrossRef]
- Hegde, M.; Gottschalk, S.; Grada, Z.; Brawley, V.S.; Byrd, T.T.; Ahmed, N. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [Green Version]
- Nisbet, R.M.; Van der Jeugd, A.; Leinenga, G.; Evans, H.T.; Janowicz, P.W.; Götz, J. Combined effects of scanning ultrasound and a tau-specific single chain antibody in a tau transgenic mouse model. Brain 2017, 140, 1220–1230. [Google Scholar] [CrossRef]
- Weatherill, E.E.; Cain, K.L.; Heywood, S.P.; Compson, J.E.; Heads, J.T.; Adams, R.; Humphreys, D.P. Towards a universal disulphide stabilised single chain Fv format: Importance of interchain disulphide bond location and vL-vH orientation. Protein Eng. Des. Sel. 2012, 25, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, N.A.; Romanowska, M.; Haritonova, H.; Foerster, J. Optimized production and concentration of lentiviral vectors containing large inserts. J. Gene Med. 2007, 9, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Bos, T.J.; De Bruyne, E.; Van Lint, S.; Heirman, C.; Vanderkerken, K. Large double copy vectors are functional but show a size-dependent decline in transduction efficiency. J. Biotechnol. 2010, 150, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Keller, B.; Makalou, N.; Sutton, R.E. Systematic determination of the packaging limit of lentiviral vectors. Hum. Gene Ther. 2001, 12, 1893–1905. [Google Scholar] [CrossRef]
- Vu, K.B.; Ghahroudi, M.A.; Wyns, L.; Muyldermans, S. Comparison of llama VH sequences from conventional and heavy chain antibodies. Mol. Immunol. 1997, 34, 1121–1131. [Google Scholar] [CrossRef]
- Bannas, P.; Hambach, J.; Koch-Nolte, F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front. Immunol. 2017, 8, 1603. [Google Scholar] [CrossRef]
- Gulati, S.; Jin, H.; Masuho, I.; Orban, T.; Cai, Y.; Pardon, E.; Martemyanov, K.A.; Kiser, P.D.; Stewart, P.L.; Ford, C.P.; et al. Targeting G protein-coupled receptor signaling at the G protein level with a selective nanobody inhibitor. Nat. Commun. 2018, 9, 1996. [Google Scholar] [CrossRef] [Green Version]
- Safarzadeh Kozani, P.; Naseri, A.; Mirarefin, S.M.J.; Salem, F.; Nikbakht, M.; Evazi Bakhshi, S.; Safarzadeh Kozani, P. Nanobody-based CAR-T cells for cancer immunotherapy. Biomark. Res. 2022, 10, 24. [Google Scholar] [CrossRef]
- Hambach, J.; Riecken, K.; Cichutek, S.; Schütze, K.; Albrecht, B.; Petry, K.; Röckendorf, J.L.; Baum, N.; Kröger, N.; Hansen, T.; et al. Targeting CD38-Expressing Multiple Myeloma and Burkitt Lymphoma Cells In Vitro with Nanobody-Based Chimeric Antigen Receptors (Nb-CARs). Cells 2020, 9, 321. [Google Scholar] [CrossRef] [Green Version]
- You, F.; Wang, Y.; Jiang, L.; Zhu, X.; Chen, D.; Yuan, L.; An, G.; Meng, H.; Yang, L. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am. J. Cancer Res. 2019, 9, 64–78. [Google Scholar]
- Yuan, K.-M.; Zhang, P.-H.; Qi, S.-S.; Zhu, Q.-Z.; Li, P. Emerging Role for Exosomes in the Progress of Stem Cell Research. Am. J. Med. Sci. 2018, 356, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Lei, C.; Liu, S.; Cui, Y.; Wang, C.; Qian, K.; Li, T.; Shen, Y.; Fan, X.; Lin, F.; et al. CAR exosomes derived from effector CAR-T cells have potent antitumour effects and low toxicity. Nat. Commun. 2019, 10, 4355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olejarz, W.; Dominiak, A.; Żołnierzak, A.; Kubiak-Tomaszewska, G.; Lorenc, T. Tumor-Derived Exosomes in Immunosuppression and Immunotherapy. J. Immunol. Res. 2020, 2020, 6272498. [Google Scholar] [CrossRef] [PubMed]
- Olejarz, W.; Kubiak-Tomaszewska, G.; Chrzanowska, A.; Lorenc, T. Exosomes in Angiogenesis and Anti-angiogenic Therapy in Cancers. Int. J. Mol. Sci. 2020, 21, 5840. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Jiang, L. Exosomes in cancer therapy: A novel experimental strategy. Am. J. Cancer Res. 2018, 8, 2165–2175. [Google Scholar]
- Wang, C.; Fu, W.; Lei, C.; Hu, S. Generation and functional characterization of CAR exosomes. Methods Cell Biol. 2022, 167, 123–131. [Google Scholar]
- Peters, P.J.; Geuze, H.J.; van der Donk, H.A.; Borst, J. A new model for lethal hit delivery by cytotoxic T lymphocytes. Immunol. Today 1990, 11, 28–32. [Google Scholar] [CrossRef]
- Tang, X.-J.; Sun, X.-Y.; Huang, K.-M.; Zhang, L.; Yang, Z.-S.; Zou, D.-D.; Wang, B.; Warnock, G.L.; Dai, L.-J.; Luo, J. Therapeutic potential of CAR-T cell-derived exosomes: A cell-free modality for targeted cancer therapy. Oncotarget 2015, 6, 44179–44190. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Choi, S.-J.; Cho, H.; Yea, K.; Baek, K.Y. Immune cell-derived small extracellular vesicles in cancer treatment. BMB Rep. 2022, 55, 48–56. [Google Scholar] [CrossRef]
- Calvo, V.; Izquierdo, M. T Lymphocyte and CAR-T Cell-Derived Extracellular Vesicles and Their Applications in Cancer Therapy. Cells 2022, 11, 790. [Google Scholar] [CrossRef] [PubMed]
- Ventimiglia, L.N.; Fernández-Martín, L.; Martínez-Alonso, E.; Antón, O.M.; Guerra, M.; Martínez-Menárguez, J.A.; Andrés, G.; Alonso, M.A. Cutting Edge: Regulation of Exosome Secretion by the Integral MAL Protein in T Cells. J. Immunol. 2015, 195, 810–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.; Cao, X.; Cai, H.; Feng, P.; Chen, X.; Zhu, Y.; Yang, Y.; An, W.; Yang, Y.; Jie, J. The exosomes derived from CAR-T cell efficiently target mesothelin and reduce triple-negative breast cancer growth. Cell Immunol. 2021, 360, 104262. [Google Scholar] [CrossRef] [PubMed]
- Mittelbrunn, M.; Gutiérrez-Vázquez, C.; Villarroya-Beltri, C.; González, S.; Sánchez-Cabo, F.; González, M.Á.; Bernad, A.; Sánchez-Madrid, F. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2011, 2, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlgren, J.; Karlson, T.D.L.; Glader, P.; Telemo, E.; Valadi, H. Activated human T cells secrete exosomes that participate in IL-2 mediated immune response signaling. PLoS ONE 2012, 7, e49723. [Google Scholar] [CrossRef] [Green Version]
- Ventimiglia, N.L.; Alonso, M.A. Biogenesis and Function of T Cell-Derived Exosomes. Front. Cell Dev. Biol. 2016, 4, 84. [Google Scholar] [CrossRef] [Green Version]
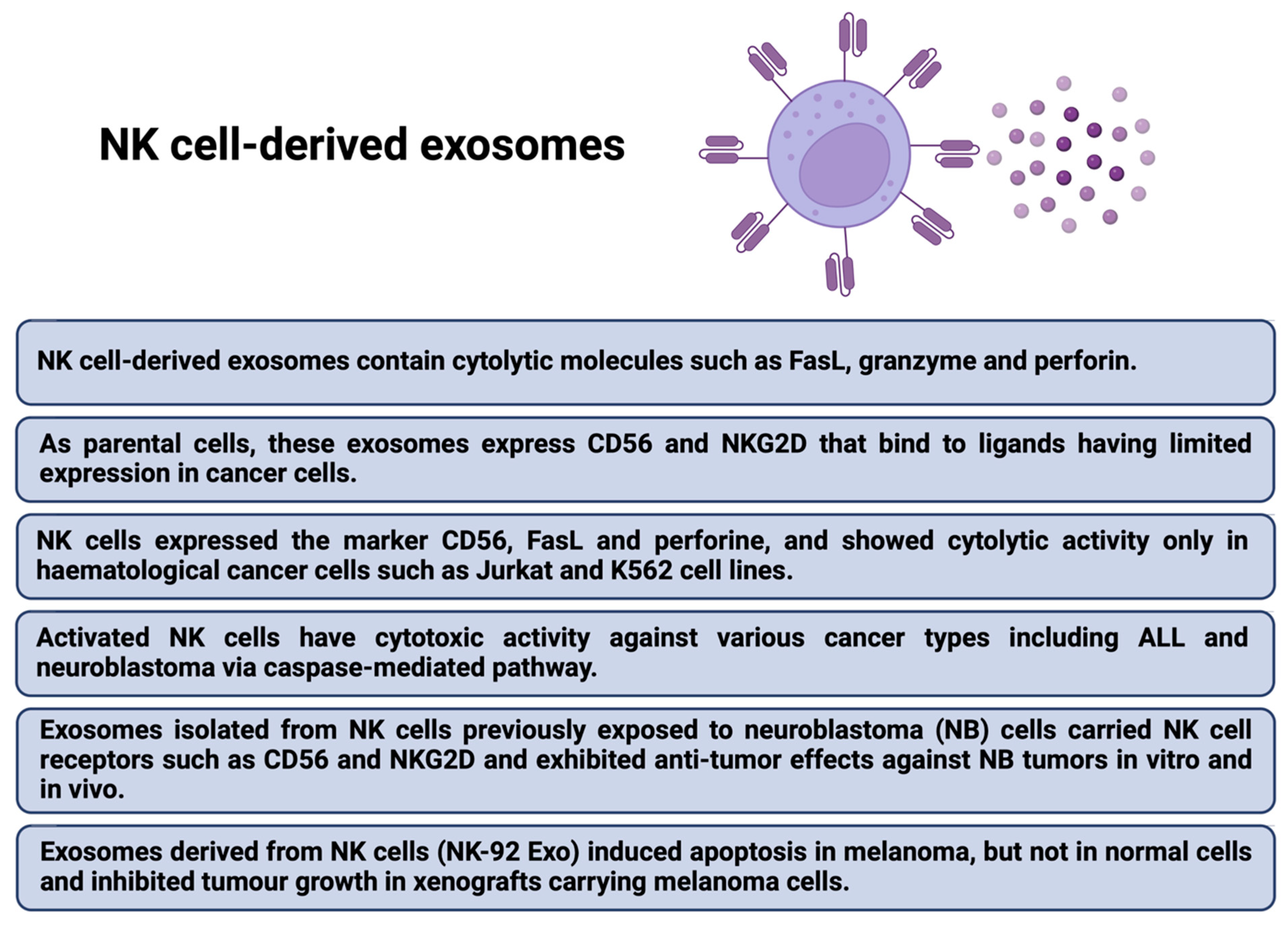
- Shoae-Hassani, A.; Hamidieh, A.A.; Behfar, M.; Mohseni, R.; Mortazavi-Tabatabaei, S.A.; Asgharzadeh, S. NK Cell-derived Exosomes From NK Cells Previously Exposed to Neuroblastoma Cells Augment the Antitumor Activity of Cytokine-activated NK Cells. J. Immunother. 2017, 40, 265–276. [Google Scholar] [CrossRef]
- Zhu, L.; Kalimuthu, S.; Gangadaran, P.; Oh, J.M.; Lee, H.W.; Baek, S.H.; Jeong, S.Y.; Lee, S.-W.; Lee, J.; Ahn, B.-C. Exosomes Derived From Natural Killer Cells Exert Therapeutic Effect in Melanoma. Theranostics 2017, 7, 2732–2745. [Google Scholar] [CrossRef]
- Jong, A.Y.; Wu, C.-H.; Li, J.; Sun, J.; Fabbri, M.; Wayne, A.S.; Seeger, R.C. Large-scale isolation and cytotoxicity of extracellular vesicles derived from activated human natural killer cells. J. Extracell. Vesicles 2017, 6, 1294368. [Google Scholar] [CrossRef] [Green Version]
- Lugini, L.; Cecchetti, S.; Huber, V.; Luciani, F.; Macchia, G.; Spadaro, F.; Paris, L.; Abalsamo, L.; Colone, M.; Molinari, A.; et al. Immune surveillance properties of human NK cell-derived exosomes. J. Immunol. 2012, 189, 2833–2842. [Google Scholar] [CrossRef] [Green Version]
- Fais, S. NK cell-released exosomes: Natural nanobullets against tumors. Oncoimmunology 2013, 2, e22337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurenzana, I.; Lamorte, D.; Trino, S.; de Luca, L.; Ambrosino, C.; Zoppoli, P.; Ruggieri, V.; del Vecchio, L.; Musto, P.; Caivano, A.; et al. Extracellular Vesicles: A New Prospective in Crosstalk between Microenvironment and Stem Cells in Hematological Malignancies. Stem Cells Int. 2018, 2018, 9863194. [Google Scholar] [CrossRef] [PubMed]
- Aharon, A.; Horn, G.; Bar-Lev, T.H.; Yohay, M.E.Z.; Waks, M.T.; Levin, M.; Unger, N.D.; Avivi, I.; Levin, A.G. Extracellular Vesicles Derived from Chimeric Antigen Receptor-T Cells: A Potential Therapy for Cancer. Hum. Gene Ther. 2021, 32, 1224–1241. [Google Scholar] [CrossRef]
- Kennedy, L.B.; Salama, A.K.S. A review of cancer immunotherapy toxicity. CA Cancer J. Clin. 2020, 70, 86–104. [Google Scholar] [CrossRef] [Green Version]
- Yoon, D.H.; Osborn, M.J.; Tolar, J.; Kim, C.J. Incorporation of Immune Checkpoint Blockade into Chimeric Antigen Receptor T Cells (CAR-Ts): Combination or Built-In CAR-T. Int. J. Mol. Sci. 2018, 19, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; Van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Ellerman, D. Bispecific T-cell engagers: Towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods 2019, 154, 102–117. [Google Scholar] [CrossRef]
- Przepiorka, D.; Ko, C.-W.; Deisseroth, A.; Yancey, C.L.; Candau-Chacon, R.; Chiu, H.-J.; Gehrke, B.J.; Gomez-Broughton, C.; Kane, R.C.; Kirshner, S.; et al. FDA Approval: Blinatumomab. Clin. Cancer Res. 2015, 21, 4035–4039. [Google Scholar] [CrossRef] [Green Version]
- Curran, E.; Stock, W. Taking a "BiTE out of ALL": Blinatumomab approval for MRD-positive ALL. Blood 2019, 133, 1715–1719. [Google Scholar] [CrossRef]
- Gökbuget, N.; Dombret, H.; Bonifacio, M.; Reichle, A.; Graux, C.; Faul, C.; Diedrich, H.; Topp, M.S.; Brüggemann, M.; Horst, H.-A.; et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood 2018, 131, 1522–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bargou, R.; Leo, E.; Zugmaier, G.; Klinger, M.; Goebeler, M.; Knop, S.; Noppeney, R.; Viardot, A.; Hess, G.; Schuler, M.; et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008, 321, 974–977. [Google Scholar] [CrossRef]
- Goebeler, E.M.; Bargou, R.C. T cell-engaging therapies-BiTEs and beyond. Nat. Rev. Clin. Oncol. 2020, 17, 418–434. [Google Scholar] [CrossRef]
- Choudhry, A.; O’Brien, S. Inotuzumab ozogamicin for the treatment of patients with acute lymphocytic leukemia. Drugs Today 2017, 53, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Porter, D. Cytokine Release Syndrome with Chimeric Antigen Receptor T Cell Therapy. Biol. Blood Marrow Transpl. 2019, 25, e123–e127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotch, C.; Barrett, D.; Teachey, D.T. Tocilizumab for the treatment of chimeric antigen receptor T cell-induced cytokine release syndrome. Expert Rev. Clin. Immunol. 2019, 15, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Frey, V.N.; Porter, D.L. Cytokine release syndrome with novel therapeutics for acute lymphoblastic leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Jain, K.K. Personalized Immuno-Oncology. Med. Princ. Pract. 2021, 30, 1–16. [Google Scholar] [CrossRef]
- Labanieh, L.; Majzner, R.G.; Mackall, C.L. Programming CAR-T cells to kill cancer. Nat. Biomed. Eng. 2018, 2, 377–391. [Google Scholar] [CrossRef]
- Johnson, C.P.; Abramson, J.S. Engineered T Cells: CAR T Cell Therapy and Beyond. Curr. Oncol. Rep. 2022, 24, 23–31. [Google Scholar] [CrossRef]
- Sterner, C.R.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Janelle, V.; Delisle, J.S. T-Cell Dysfunction as a Limitation of Adoptive Immunotherapy: Current Concepts and Mitigation Strategies. Cancers 2021, 13, 598. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.J.; Vervoort, S.J.; Ramsbottom, K.M.; Kelly, M.J.; Michie, J.; Pijpers, L.; Johnstone, R.W.; Kearney, C.J.; Oliaro, J. Natural Killer Cells Suppress T Cell-Associated Tumor Immune Evasion. Cell Rep. 2019, 28, 2784–2794.e5. [Google Scholar] [CrossRef]
- Kasakovski, D.; Xu, L.; Li, Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J. Hematol. Oncol. 2018, 11, 91. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Generic Name | Brand Name | Target Antigen | Targeted Disease | Patient Population |
|---|---|---|---|---|
| Tisagenlecleucel | Kymriah | CD19 | B-cell acute lymphoblastic leukemia (ALL) | Children and young adults with refractory or relapsed B-cell ALL |
| B-cell non-Hodgkin lymphoma (NHL) | Adults with relapsed or refractory B-cell NHL | |||
| Axicabtagene ciloleucel | Yescarta | CD19 | B-cell non-Hodgkin lymphoma (NHL) | Adults with relapsed or refractory B-cell NHL |
| Follicular lymphoma | Adults with relapsed or refractory follicular lymphoma | |||
| Brexucabtagene autoleucel | Tecartus | CD19 | B-cell acute lymphoblastic leukemia (ALL) | Adults with refractory or relapsed B-cell ALL |
| Mantle cell lymphoma (MCL) | Adults with refractory or relapsed MCL | |||
| Lisocabtagene maraleucel | Breyanzi | CD19 | B-cell non-Hodgkin lymphoma (NHL) | Adults with relapsed or refractory B-cell NHL |
| Ciltabtagene autoleucel | Carvykti | BCMA | Multiple myeloma | Adults with relapsed or refractory multiple myeloma |
| Idecabtagene vicleucel | Abecma | BCMA | Multiple myeloma | Adults with relapsed or refractory multiple myeloma |
| CAR-T Cells | ||
|---|---|---|
| Sources | Different mixes of helper CD4+ and cytotoxic CD8+ cells T cells. | |
| Autologous T cells | Allogeneic T cells | |
| Quality of source | Limitations of quality and quantity T cell number. Various donors of T cells. | Multiple T cell sources from many healthy donors (PB or UCB). Standardized source of cell. |
| Manufacture | Risk of manufacture in a group for heavily pretreated patients. The limited potency of the CAR-T cellular product is because the patient’s T lymphocytes treated with chemotherapy are more differentiated with lower proliferation capacity and rapid exhaustion. | The starting material is high quality from a healthy donor. |
| Risk of contamination | Risk of contamination with cancer cells in patient blood. | Minimal risks of cancer cell contamination, source form healthy donor blood. |
| Persistence | Increased in vivo persistence compared with allogeneic CAR-T cells due to lack of immune rejection from the host. | Decreased in vivo persistence due to higher immunogenicity. |
| Risk of GVHD | Low | High |
| Scalability | Low-personalized product for one patient | High—one product for many patients |
| Acute side effects | It may cause GVHD, CRS and neurotoxicity. | Barely cause GVHD, may even protect against GVHD. Lack of CRS and neurotoxicity. |
| Mechanism | CAR-NK Cells | CAR-T Cells |
|---|---|---|
| Chimeric antigen receptor | CARs cells can aim for specific tumor antigen | |
| Antigen presentation | NK cells can specifically recognize the cells that lack the expression of self-MHC class I molecules [65]. Enhancing the antigen presentation to T cells by killing the immature DC while promoting the IFN g and TNF-a mediated maturation of DC [66]. | They can recognize antigens regardless of MHC presentation. However, they are limited to the recognition of structures expressed at the surface [28,29]. |
| Transduction efficiency | lower | higher |
| In vivo persistence | worse | better |
| Fas/FasL | The Fas-FasL is a major apoptosis pathway via caspase-dependent activation. The antigen-negative cancer cells can be targeted via FAS and Fas L axis, independent of presenting death receptors by the cancer cell. It is estimated that the functions of this pathway may be pivotal in the heterogeneous environment of the tumor [38,39]. | |
| Cytolytic granules | CARs cells lyse the antigen-positive cancer cells mainly by the cytolytic granules. The perforins are inducing pore formation in the cancer membrane, forming the access for granzymes. In the cytoplasm, they could induce apoptotic cell death in a caspase-dependent or independent way. Therefore, cytolytic degranulation is assumed to be the most important mechanism of cell killing by CAR-T cells [6]. Cytokine production induces cell death via secondary mechanisms, such as enhancing CARs, Fas, or TRAIL pathways. They trigger several anti-tumor immune responses, including the enhancement of the cytotoxic response, recruitment, and activation of innate immune cells [67]. | |
| There were attempts to use ectopically expressed chimeric granzyme B. This approach could enhance NK-cell degranulation and efficient producing cytolytic granules [68,69]. | Cytolytic granules secretion by CAR-T cells mediates tumor lysis via upregulating IFN-gamma on stromal cells [41]. That leads to immune cell modulations, such as the polarization of macrophages to the antitumoral M1 phenotype [42]. | |
| Checkpoint inhibitors | Prevention of the interaction of inhibitory receptors with their respective ligands leads to inhibition of NK cell suppression [70]. Additionally, checkpoint molecules can enable tumor escape from NK cell vigilance [71]. | CAR-T cells can secrete immune checkpoint inhibitors to overcome immunosuppression of tumors (e.g., anti-PD-1/PD-L1/CTLA-4) for enhanced strength, effectiveness, and persistence of CAR-T therapy [72,73,74,75,76]. |
| CAR-T Cells | CAR-NK Cells | ||
|---|---|---|---|
| Advantages | Disadvantages | Advantages | Disadvantages |
|
|
|
|
| Event | CAR-T Cells | CAR-T Cell-Derived Exosomes |
|---|---|---|
| Cross the blood barrier | − | ++ |
| Cytokine releasing syndrome | ++ | − |
| Neurotoxicity and GvHD | ++ | − |
| Reprograming and act against suppressive cells | − | ++ |
| Efficiency against solid tumors | + | ++ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadowski, K.; Olejarz, W.; Basak, G. Modern Advances in CARs Therapy and Creating a New Approach to Future Treatment. Int. J. Mol. Sci. 2022, 23, 15006. https://doi.org/10.3390/ijms232315006
Sadowski K, Olejarz W, Basak G. Modern Advances in CARs Therapy and Creating a New Approach to Future Treatment. International Journal of Molecular Sciences. 2022; 23(23):15006. https://doi.org/10.3390/ijms232315006
Chicago/Turabian StyleSadowski, Karol, Wioletta Olejarz, and Grzegorz Basak. 2022. "Modern Advances in CARs Therapy and Creating a New Approach to Future Treatment" International Journal of Molecular Sciences 23, no. 23: 15006. https://doi.org/10.3390/ijms232315006
APA StyleSadowski, K., Olejarz, W., & Basak, G. (2022). Modern Advances in CARs Therapy and Creating a New Approach to Future Treatment. International Journal of Molecular Sciences, 23(23), 15006. https://doi.org/10.3390/ijms232315006