
Cytosolic Release of Mitochondrial DNA and Associated cGAS Signaling Mediates Radiation-Induced Hematopoietic Injury of Mice


 and
and
Abstract
1. Introduction
2. Results
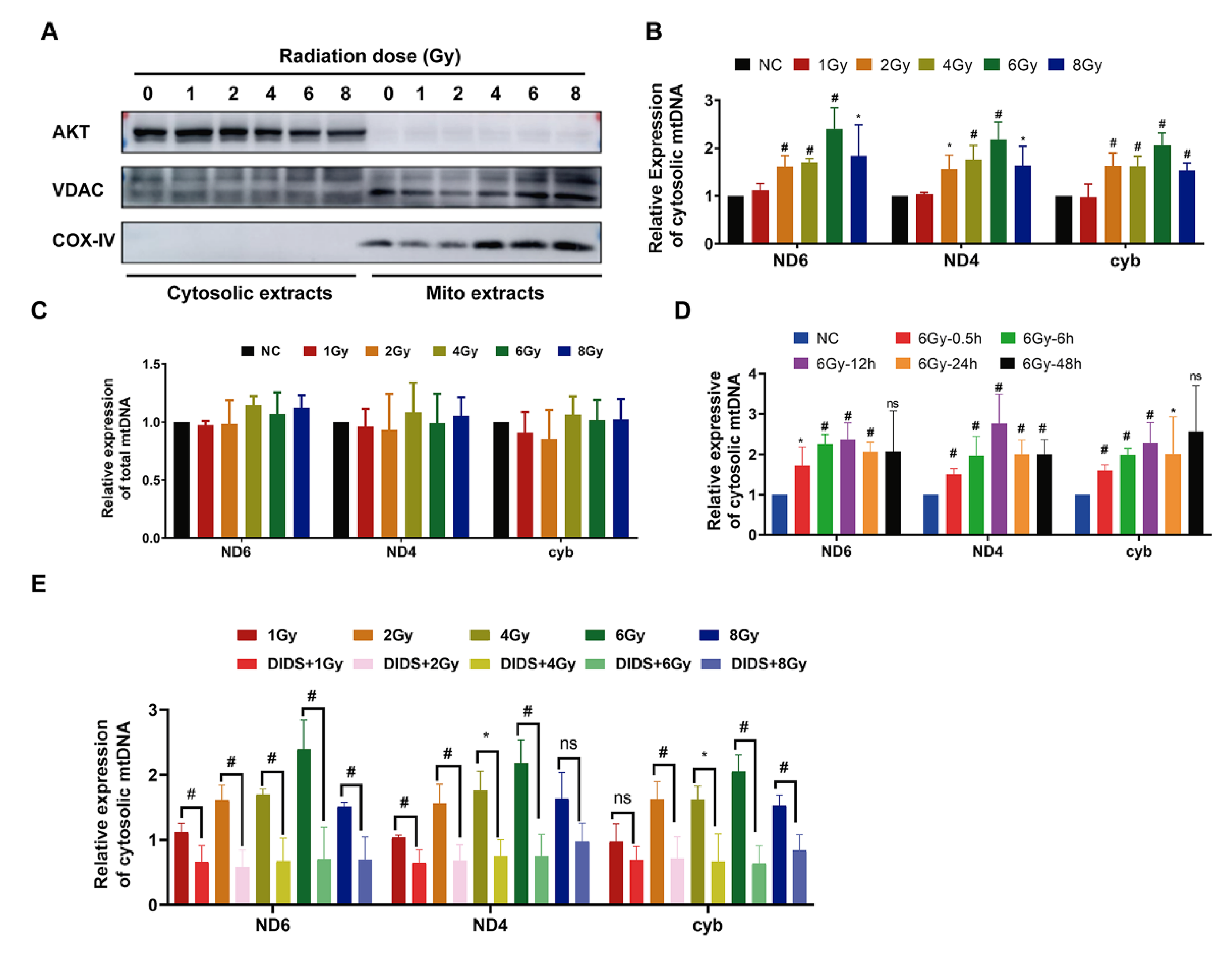
2.1. Increased Release of Mitochondrial DNA into Cytosol by γ-ray Irradiation
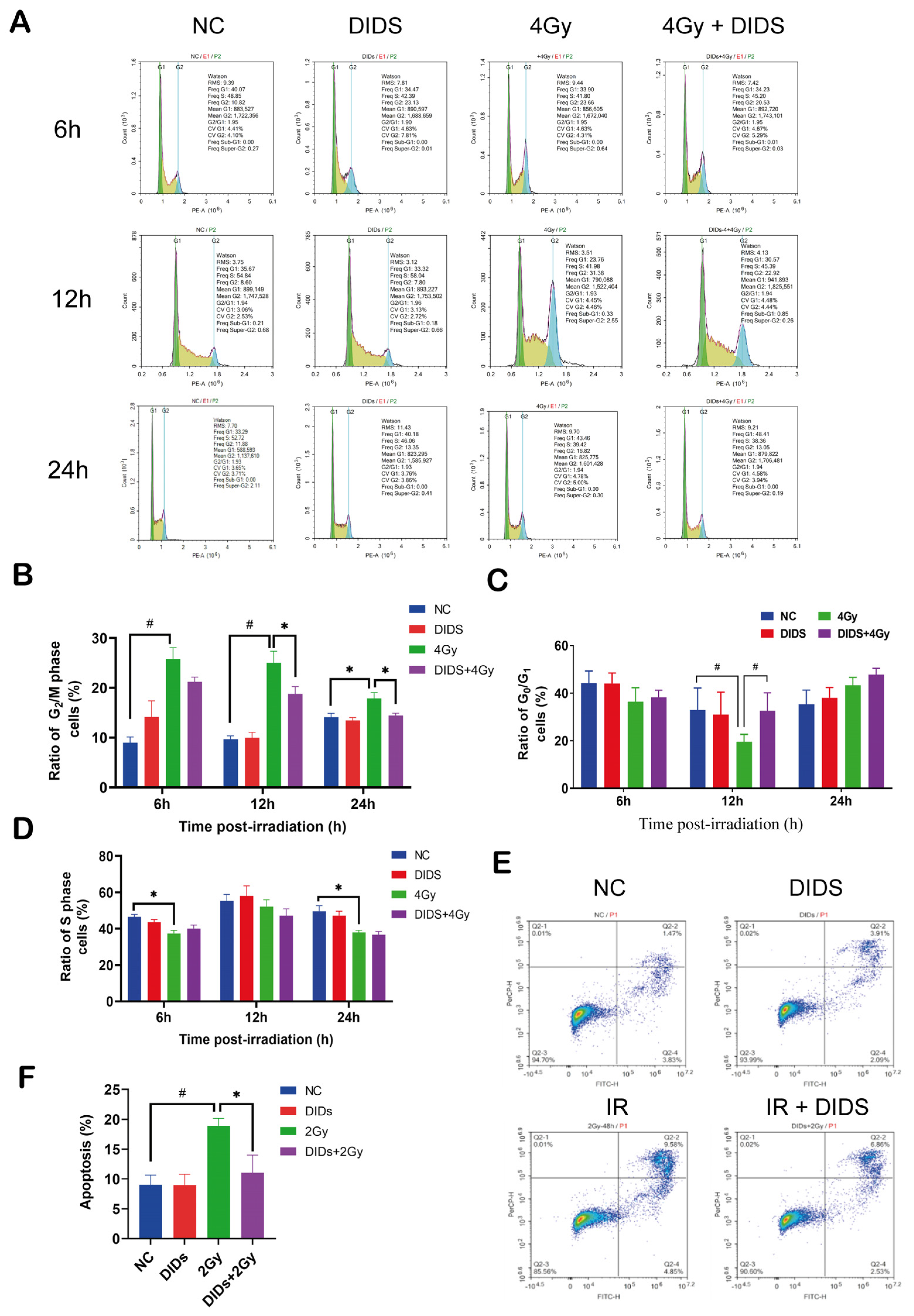
2.2. The Released Cytosolic mtDNA Involves in Regulation of FDC-P1 Cells’ Responses to γ-ray Irradiation
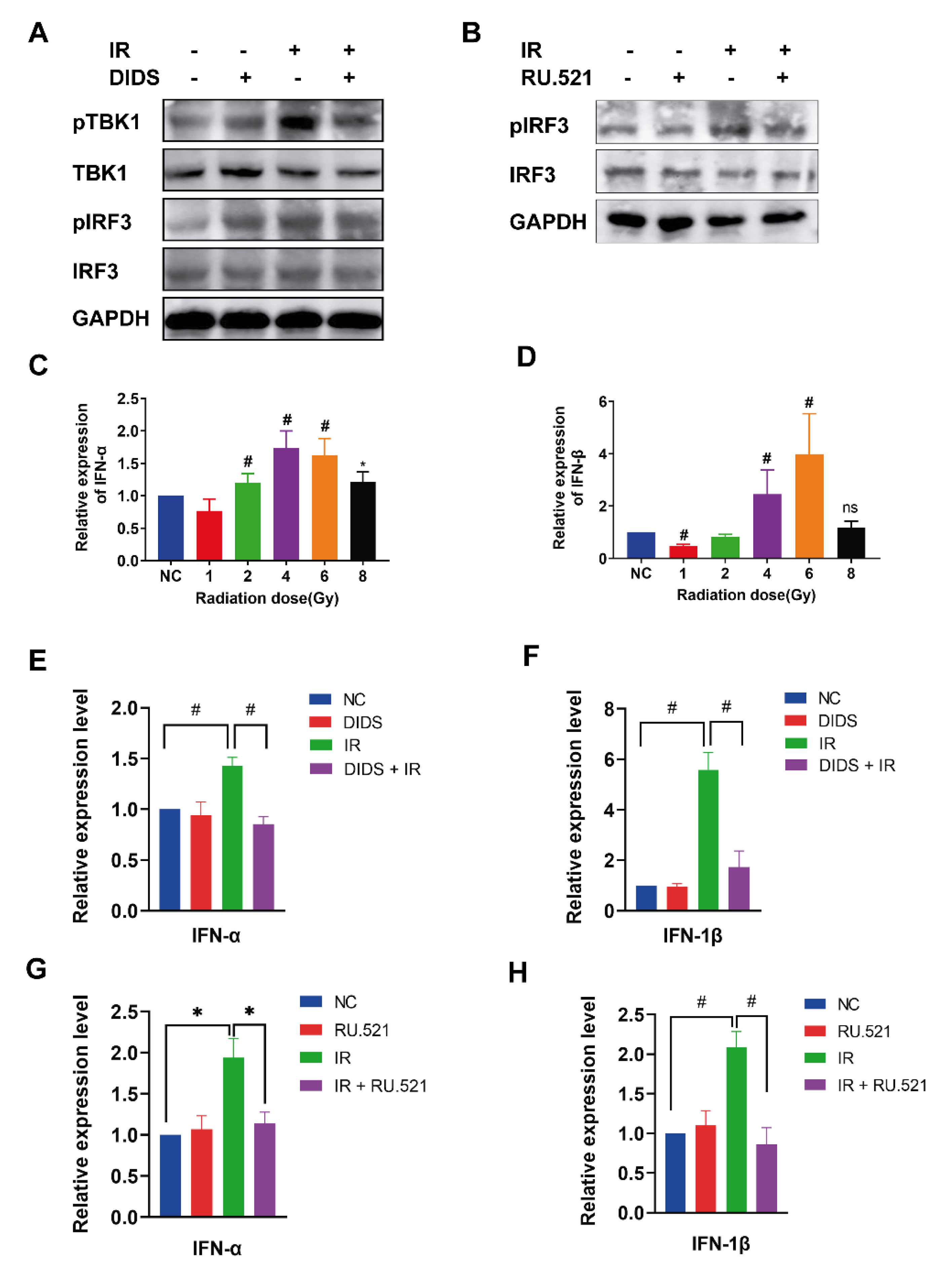
2.3. Activation of the cGAS Signaling Pathway in Irradiated Cells by Cytosolic Mitochondrial DNA
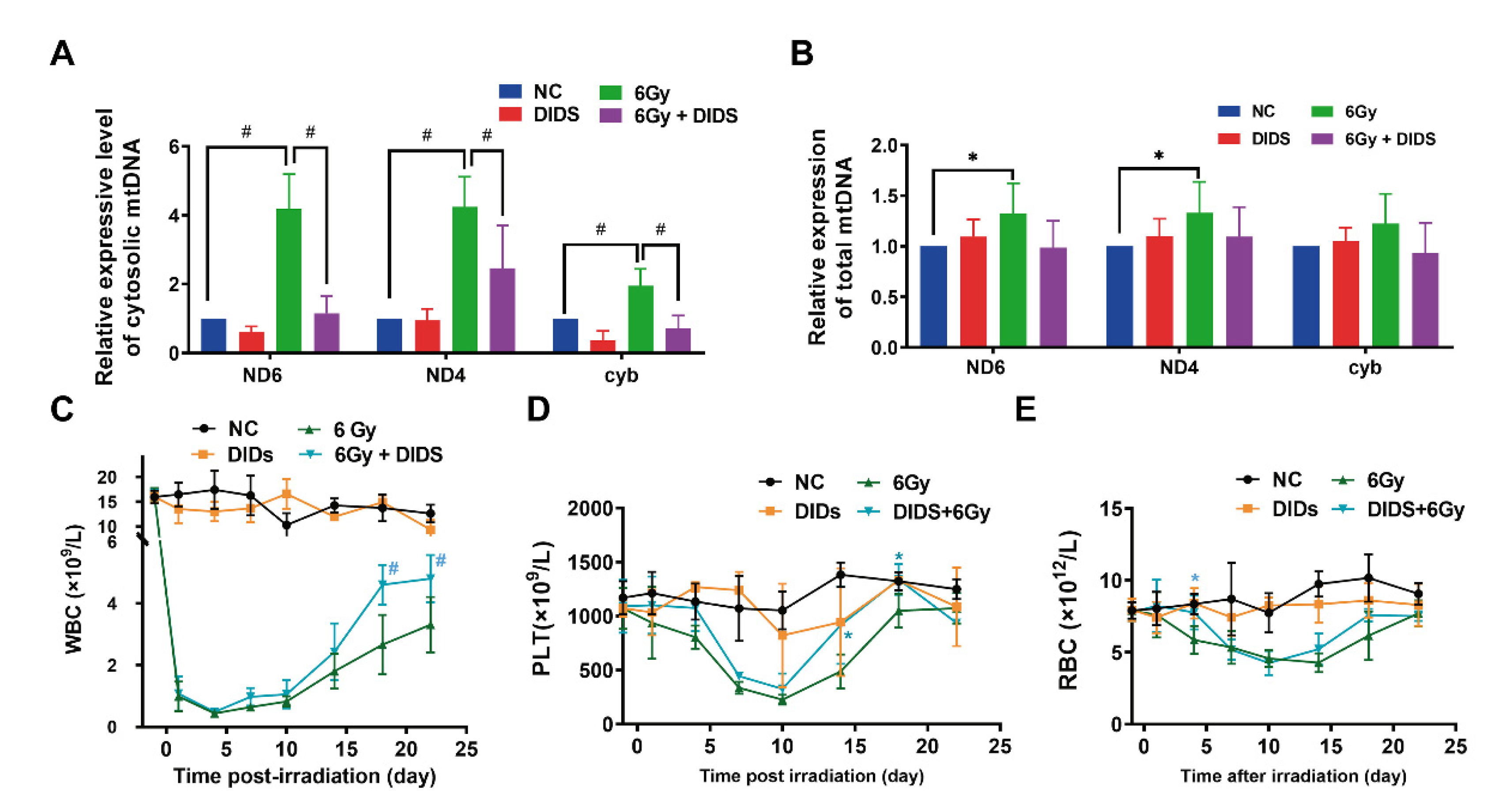
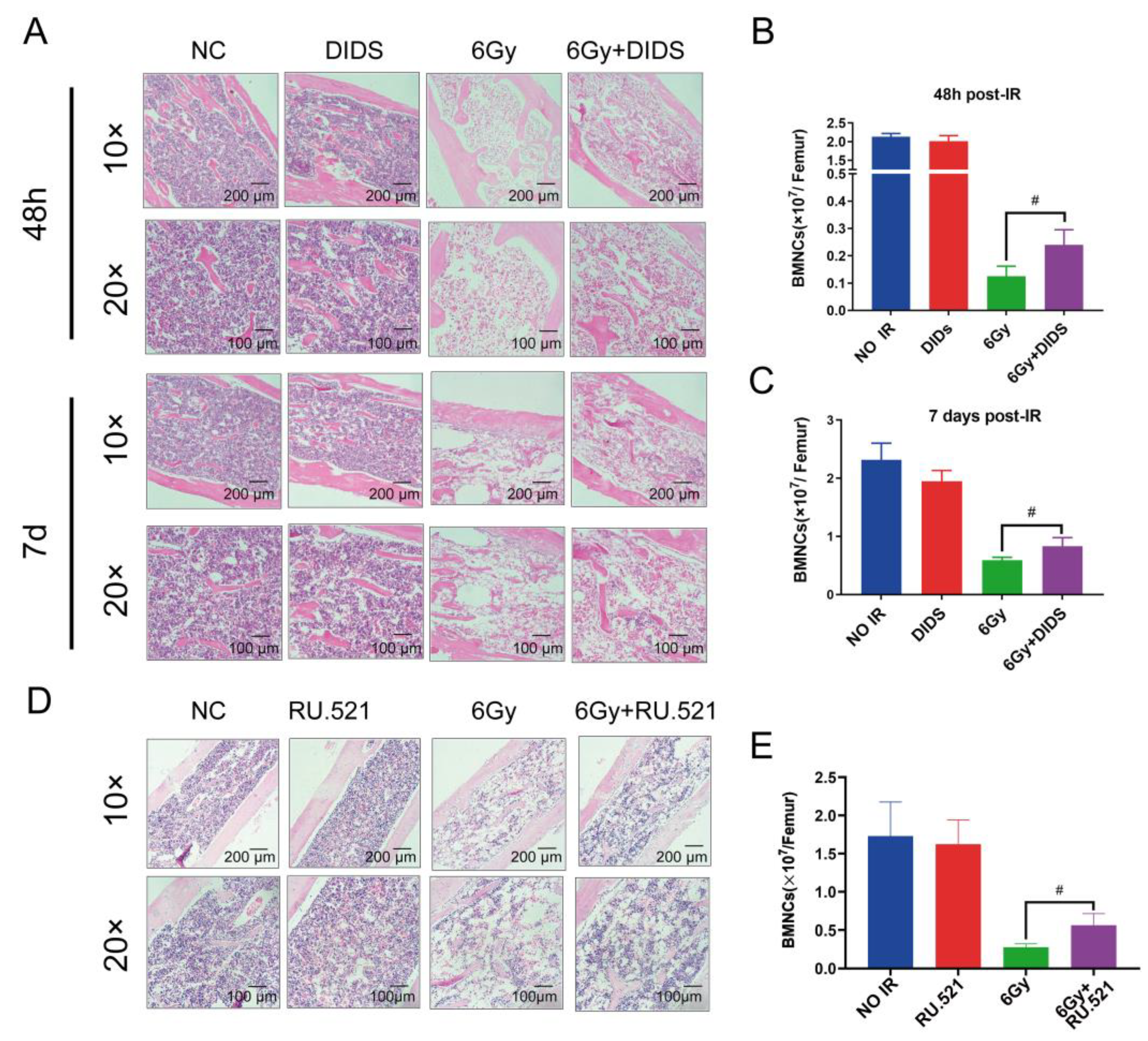
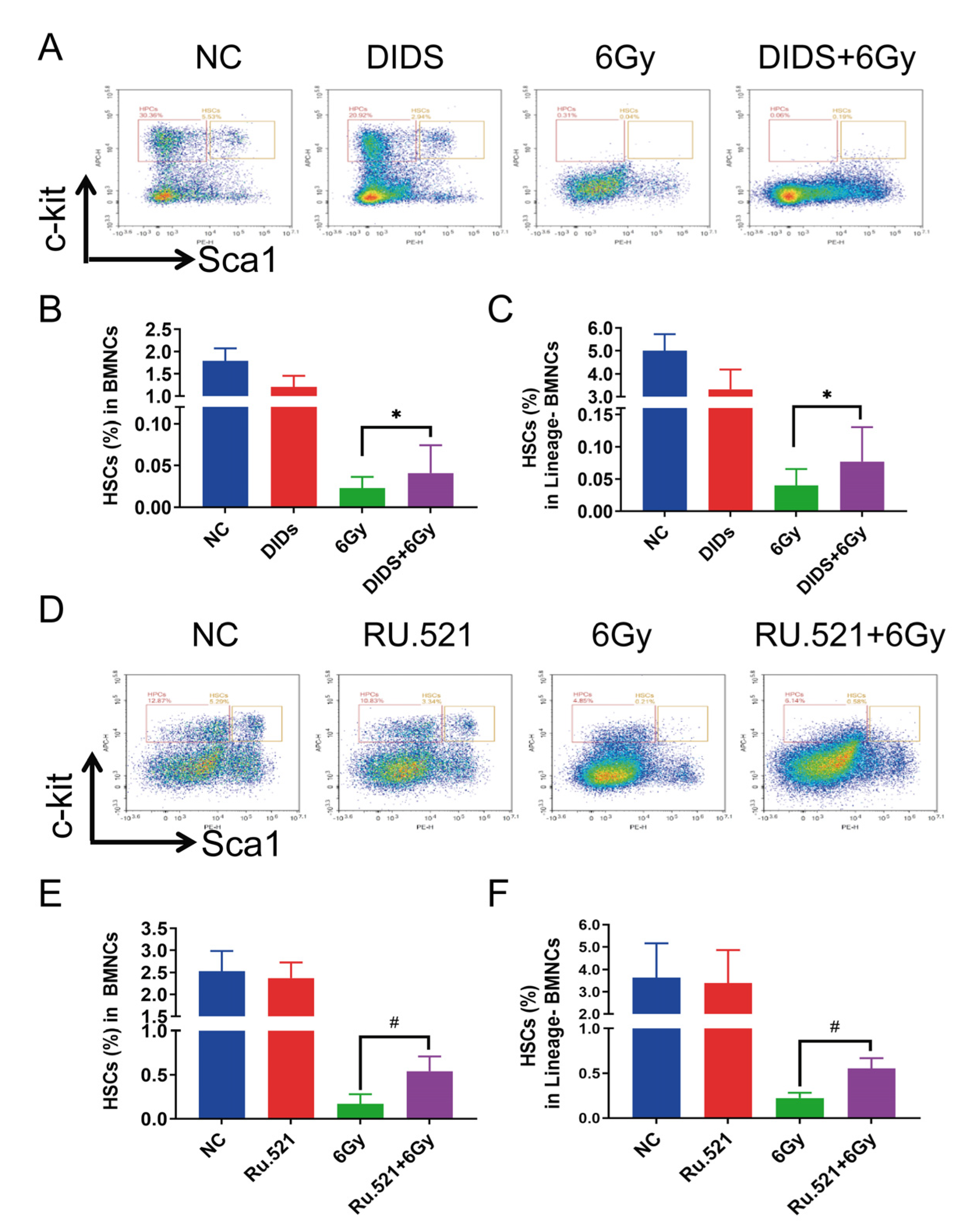
2.4. Cytosolic Release of Mitochondrial DNA Involved in Bone Marrow Tissue Injury of γ-ray Irradiated Mice
2.5. Radioprotection of Targeting Cytosolic mtDNA-cGAS Signaling Pathway Hematopoietic Cell Injury in Mice by Regulation
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Mice and Irradiation
4.3. Western Blot
4.4. Subcellular Fractionation and Quantification of Mitochondrial DNA
4.5. RNA Extraction and Real-Time PCR
4.6. Peripheral Blood Cell Counts
4.7. Analysis of the Surface Markers of Mouse Bone Marrow Cells by Flow Cytometry
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Authors on behalf of ICRP; Stewart, F.A.; Akleyev, A.V.; Hauer-Jensen, M.; Hendry, J.H.; Kleiman, N.J.; Macvittie, T.J.; Aleman, B.M.; Edgar, A.B.; Mabuchi, K.; et al. ICRP publication 118: ICRP statement on tissue reactions and early and late effects of radiation in normal tissues and organs—Threshold doses for tissue reactions in a radiation protection context. Ann. ICRP 2012, 41, 1–322. [Google Scholar] [CrossRef]
- Kuntz, L.; Noel, G. Pelvic irradiation and hematopoietic toxicity: A review of the literature. Cancer Radiother. 2021, 25, 77–91. [Google Scholar] [CrossRef]
- Morgan, W.F. Is there a common mechanism underlying genomic instability, bystander effects and other nontargeted effects of exposure to ionizing radiation? Oncogene 2003, 22, 7094–7099. [Google Scholar] [CrossRef]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef]
- Marín, A.; Martín, M.; Liñán, O.; Alvarenga, F.; López, M.; Fernández, L.; Büchser, D.; Cerezo, L. Bystander effects and radiotherapy. Rep. Pract. Oncol. Radiother. J. Greatpoland Cancer Cent. Pozn. Pol. Soc. Radiat. Oncol. 2015, 20, 12–21. [Google Scholar] [CrossRef]
- Mothersill, C.; Rusin, A.; Fernandez-Palomo, C.; Seymour, C. History of bystander effects research 1905-present; what is in a name? Int. J. Radiat. Biol. 2018, 94, 696–707. [Google Scholar] [CrossRef]
- Mo, L.J.; Song, M.; Huang, Q.H.; Guan, H.; Liu, X.D.; Xie, D.F.; Huang, B.; Huang, R.X.; Zhou, P.K. Exosome-packaged miR-1246 contributes to bystander DNA damage by targeting LIG4. Br. J. Cancer 2018, 119, 492–502. [Google Scholar] [CrossRef]
- Huang, B.; Shang, Z.F.; Li, B.; Wang, Y.; Liu, X.D.; Zhang, S.M.; Guan, H.; Rang, W.Q.; Hu, J.A.; Zhou, P.K. DNA-PKcs associates with PLK1 and is involved in proper chromosome segregation and cytokinesis. J. Cell. Biochem. 2014, 115, 1077–1088. [Google Scholar] [CrossRef]
- Lippert, T.P.; Greenberg, R.A. The abscopal effect: A sense of DNA damage is in the air. J. Clin. Investig. 2021, 131, e148274. [Google Scholar] [CrossRef]
- Miranda, S.; Correia, M.; Dias, A.G.; Pestana, A.; Soares, P.; Nunes, J.; Lima, J.; Máximo, V.; Boaventura, P. Evaluation of the role of mitochondria in the non-targeted effects of ionizing radiation using cybrid cellular models. Sci. Rep. 2020, 10, 6131. [Google Scholar] [CrossRef]
- Wang, H.; Yu, K.N.; Hou, J.; Liu, Q.; Han, W. Radiation-induced bystander effect: Early process and rapid assessment. Cancer Lett. 2015, 356, 137–144. [Google Scholar] [CrossRef]
- Fan, P.C.; Zhang, Y.; Wang, Y.; Wei, W.; Zhou, Y.X.; Xie, Y.; Wang, X.; Qi, Y.Z.; Chang, L.; Jia, Z.P.; et al. Quantitative proteomics reveals mitochondrial respiratory chain as a dominant target for carbon ion radiation: Delayed reactive oxygen species generation caused DNA damage. Free. Radic. Biol. Med. 2019, 130, 436–445. [Google Scholar] [CrossRef]
- Vignard, J.; Mirey, G.; Salles, B. Ionizing-radiation induced DNA double-strand breaks: A direct and indirect lighting up. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2013, 108, 362–369. [Google Scholar] [CrossRef]
- Morales, A.; Miranda, M.; Sánchez-Reyes, A.; Biete, A.; Fernández-Checa, J.C. Oxidative damage of mitochondrial and nuclear DNA induced by ionizing radiation in human hepatoblastoma cells. Int. J. Radiat. Oncol. Biol. Phys. 1998, 42, 191–203. [Google Scholar] [CrossRef]
- Narayanan, P.K.; Goodwin, E.H.; Lehnert, B.E. Alpha particles initiate biological production of superoxide anions and hydrogen peroxide in human cells. Cancer Res. 1997, 57, 3963–3971. [Google Scholar]
- Guliaeva, N.A.; Abdullaev, S.A.; Malakhova, L.V.; Antipova, V.N.; Bezlepkin, V.G.; Gaziev, A.I. Reduction of the number of mutant copies of mitochondrial DNA in tissues of irradiated mice in the postradiation period. Genetika 2009, 45, 949–956. [Google Scholar] [CrossRef]
- Rogounovitch, T.I.; Saenko, V.A.; Shimizu-Yoshida, Y.; Abrosimov, A.Y.; Lushnikov, E.F.; Roumiantsev, P.O.; Ohtsuru, A.; Namba, H.; Tsyb, A.F.; Yamashita, S. Large deletions in mitochondrial DNA in radiation-associated human thyroid tumors. Cancer Res. 2002, 62, 7031–7041. [Google Scholar]
- Singh, G.; Hauswirth, W.W.; Ross, W.E.; Neims, A.H. A method for assessing damage to mitochondrial DNA caused by radiation and epichlorohydrin. Mol. Pharmacol. 1985, 27, 167–170. [Google Scholar]
- Meyer, J.N.; Leuthner, T.C.; Luz, A.L. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 2017, 391, 42–53. [Google Scholar] [CrossRef]
- Aarreberg, L.D.; Esser-Nobis, K.; Driscoll, C.; Shuvarikov, A.; Roby, J.A.; Gale, M., Jr. Interleukin-1β Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol. Cell 2019, 74, 801–815.e6. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, S.; Guan, H.; Zhou, P.K. Radiation-induced non-targeted effect of immunity provoked by mitochondrial DNA damage triggered cGAS/ AIM2 pathways. Radiat. Med. Prot. 2022, 3, e148274. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, J.; Alu, A.; Han, X.; Wei, Y.; Wei, X. cGAS-STING pathway in cancer biotherapy. Mol. Cancer 2020, 19, 136. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Chen, P.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; Ye, L.; He, Y.; et al. cGAS-STING, an important pathway in cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 81. [Google Scholar] [CrossRef]
- Sun, B.; Sundström, K.B.; Chew, J.J.; Bist, P.; Gan, E.S.; Tan, H.C.; Goh, K.C.; Chawla, T.; Tang, C.K.; Ooi, E.E. Dengue virus activates cGAS through the release of mitochondrial DNA. Sci. Rep. 2017, 7, 3594. [Google Scholar] [CrossRef]
- Xia, P.; Wang, S.; Ye, B.; Du, Y.; Li, C.; Xiong, Z.; Qu, Y.; Fan, Z. A Circular RNA Protects Dormant Hematopoietic Stem Cells from DNA Sensor cGAS-Mediated Exhaustion. Immunity 2018, 48, 688–701.e7. [Google Scholar] [CrossRef]
- Sato, T.; Onai, N.; Yoshihara, H.; Arai, F.; Suda, T.; Ohteki, T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon–dependent exhaustion. Nat. Med. 2009, 15, 696–700. [Google Scholar] [CrossRef]
- Smith, J.N.P.; Zhang, Y.; Li, J.J.; McCabe, A.; Jo, H.J.; Maloney, J.; MacNamara, K.C. Type I IFNs drive hematopoietic stem and progenitor cell collapse via impaired proliferation and increased RIPK1-dependent cell death during shock-like ehrlichial infection. PLoS Pathog. 2018, 14, e1007234. [Google Scholar] [CrossRef]
- Singh, V.K.; Garcia, M.; Seed, T.M. A review of radiation countermeasures focusing on injury-specific medicinals and regulatory approval status: Part II. Countermeasures for limited indications, internalized radionuclides, emesis, late effects, and agents demonstrating efficacy in large animals with or without FDA IND status. Int. J. Radiat. Biol. 2017, 93, 870–884. [Google Scholar]
- Richter, C.; Park, J.W.; Ames, B.N. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467. [Google Scholar] [CrossRef]
- May, A.; Bohr, V.A. Gene-specific repair of gamma-ray-induced DNA strand breaks in colon cancer cells: No coupling to transcription and no removal from the mitochondrial genome. Biochem. Biophys. Res. Commun. 2000, 269, 433–437. [Google Scholar] [CrossRef]
- Yoshida, T.; Goto, S.; Kawakatsu, M.; Urata, Y.; Li, T.S. Mitochondrial dysfunction, a probable cause of persistent oxidative stress after exposure to ionizing radiation. Free. Radic. Res. 2012, 46, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, M.; Hanawalt, P.C. Efficient protection against oxidative DNA damage in chromatin. Mol. Carcinog. 1992, 5, 264–269. [Google Scholar] [CrossRef]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liao, W.; Chen, J.; Wu, Y.; Liu, C.; Chen, S.; Xu, Y.; Wang, S.; Su, Y.; Du, C.; et al. Caffeic acid attenuates irradiation-induced hematopoietic stem cell apoptosis through inhibiting mitochondrial damage. Exp. Cell Res. 2021, 409, 112934. [Google Scholar] [CrossRef]
- Ortega, M.T.; Pecaut, M.J.; Gridley, D.S.; Stodieck, L.S.; Ferguson, V.; Chapes, S.K. Shifts in bone marrow cell phenotypes caused by spaceflight. J. Appl. Physiol. 2009, 106, 548–555. [Google Scholar] [CrossRef]
- Shan, S.; Liu, Z.; Wang, S.; Liu, Z.; Huang, Z.; Yang, Y.; Zhang, C.; Song, F. Drp1-mediated mitochondrial fission promotes carbon tetrachloride-induced hepatic fibrogenesis in mice. Toxicol. Res. 2022, 11, 486–497. [Google Scholar] [CrossRef]
- Wang, H.X.; Zhang, R.; Li, Z.; Wang, L.S.; Yu, Y.; Wang, Q.; Ding, Z.; Zhang, J.P.; Zhang, M.R.; Xu, L.C. Cypermethrin induces Sertoli cell apoptosis through mitochondrial pathway associated with calcium. Toxicol. Res. 2021, 10, 742–750. [Google Scholar] [CrossRef]
- Hu, S.; Gao, Y.; Zhou, H.; Kong, F.; Xiao, F.; Zhou, P.; Chen, Y. New insight into mitochondrial changes in vascular endothelial cells irradiated by gamma ray. Int. J. Radiat. Biol. 2017, 93, 470–476. [Google Scholar] [CrossRef]
- Mohammadnejad, L.; Soltaninejad, K.; Seyedabadi, M.; Ghasem Pouri, S.K.; Shokrzadeh, M.; Mohammadi, H. Evaluation of mitochondrial dysfunction due to oxidative stress in therapeutic, toxic and lethal concentrations of tramadol. Toxicol. Res. 2021, 10, 1162–1170. [Google Scholar] [CrossRef]
- Zhou, P.K.; Huang, R.X. Targeting of the respiratory chain by toxicants: Beyond the toxicities to mitochondrial morphology. Toxicol. Res. 2018, 7, 1008–1011. [Google Scholar] [CrossRef] [PubMed]
- Al-Kafaji, G.; Golbahar, J. High glucose-induced oxidative stress increases the copy number of mitochondrial DNA in human mesangial cells. BioMed Res. Int. 2013, 2013, 754946. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Cui, R.; Geng, X.; Li, J.; Zhou, Y.; He, L.; Cao, C.; Zhang, C.; Chen, Z.; Ying, S. LPS-induced mitochondrial DNA synthesis and release facilitate RAD50-dependent acute lung injury. Signal Transduct. Target. Ther. 2021, 6, 103. [Google Scholar] [CrossRef]
- García, N.; Chávez, E. Mitochondrial DNA fragments released through the permeability transition pore correspond to specific gene size. Life Sci. 2007, 81, 1160–1166. [Google Scholar] [CrossRef]
- García, N.; García, J.J.; Correa, F.; Chávez, E. The permeability transition pore as a pathway for the release of mitochondrial DNA. Life Sci. 2005, 76, 2873–2880. [Google Scholar] [CrossRef] [PubMed]
- Patrushev, M.; Kasymov, V.; Patrusheva, V.; Ushakova, T.; Gogvadze, V.; Gaziev, A.I. Release of mitochondrial DNA fragments from brain mitochondria of irradiated mice. Mitochondrion 2006, 6, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhou, P.K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 254. [Google Scholar] [CrossRef]
- Li, Y.; Chen, H.; Yang, Q.; Wan, L.; Zhao, J.; Wu, Y.; Wang, J.; Yang, Y.; Niu, M.; Liu, H.; et al. Increased Drp1 promotes autophagy and ESCC progression by mtDNA stress mediated cGAS-STING pathway. J. Exp. Clin. Cancer Res. 2022, 41, 76. [Google Scholar] [CrossRef]
- Yamazaki, T.; Galluzzi, L. Mitochondrial control of innate immune signaling by irradiated cancer cells. Oncoimmunology 2020, 9, 1797292. [Google Scholar] [CrossRef]
- Guan, J.; Lu, C.; Jin, Q.; Lu, H.; Chen, X.; Tian, L.; Zhang, Y.; Ortega, J.; Zhang, J.; Siteni, S.; et al. MLH1 Deficiency-Triggered DNA Hyperexcision by Exonuclease 1 Activates the cGAS-STING Pathway. Cancer Cell. 2021, 39, 109–121.e105. [Google Scholar] [CrossRef]
- Feng, X.; Tubbs, A.; Zhang, C.; Tang, M.; Sridharan, S.; Wang, C.; Jiang, D.; Su, D.; Zhang, H.; Chen, Z.; et al. ATR inhibition potentiates ionizing radiation-induced interferon response via cytosolic nucleic acid-sensing pathways. EMBO J. 2020, 39, e104036. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Shao, L.; Chang, J.; Feng, W.; Liu, Y.L.; Cottler-Fox, M.H.; Emanuel, P.D.; Hauer-Jensen, M.; Bernstein, I.D.; Liu, L.; et al. M1 and M2 macrophages differentially regulate hematopoietic stem cell self-renewal and ex vivo expansion. Blood Adv. 2018, 2, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Yan, Z.; Wang, P.; Liu, Y.; Ao, X.; Liu, Z.; Wang, D.; Liu, X.; Zhu, M.; Gao, S.; et al. Irradiation Activates MZF1 to Inhibit miR-541-5p Expression and Promote Epithelial-Mesenchymal Transition (EMT) in Radiation-Induced Pulmonary Fibrosis (RIPF) by Upregulating Slug. Int. J. Mol. Sci. 2021, 22, 11309. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Manufacturer | NO. | Dilution Ratio |
|---|---|---|---|
| anti-VDAC | CST | #4866 | 1:1000 |
| anti-pIRF3(Ser396) | CST | #29037 | 1:1000 |
| anti-IRF3 | Santa | sc-33641 | 1:1000 |
| anti-TBK1 | Santa | Sc-52957 | 1:1000 |
| Anti-pTBK1(ser172) | CST | #5483 | 1:1000 |
| anti-COXIV | Proteintech | 11242-1-AP | 1:1000 |
| anti-AKT | Proteintech | 10176-2-AP | 1:1000 |
| anti-Lamin B1 | Proteintech | 12987-1-AP | 1:1000 |
| Name | Primer Sequence (F) | Primer Sequence (R) |
|---|---|---|
| mmt-ND6 | TTAGCATTAAAGCCTTCACC | CCAACAAACCCACTAACAAT |
| mmt-ND4 | AACGGATCCACAGCCGTA | AGTCCTCGGGCCATGATT |
| mmt-cyb | GGCTACGTCCTTCCATGAGG | TCGGGTCAAGGTTGCTTTGT |
| GAPDH(mouse) | AGGAGCGAGACCCCACTAACA | AGGGGGGCTAAGCAGTTGGT |
| Name | Primer Sequence (F) | Primer Sequence (R) |
|---|---|---|
| Ifna (mouse) | ACCATCCCTGTCCTCCATGA | GAGGGTCTCATCCCAAGCAG |
| Ifnb (mouse) | TGACTATGGTCCAGGCACAG | TTGTTGAGAACCTCCTGGCT |
| Actin (mouse) | CACTGTCGAGTCGCGTC | GTCATCCATGGCGAACTGGT |
| GAPDH (mouse) | AGGAGCGAGACCCCACTAACA | AGGGGGGCTAAGCAGTTGGT |
| Name | Manufacturer | NO. |
|---|---|---|
| FITC anti-mouse Lineage Cocktail with | Biolegend | 133302 |
| APC anti-mouse CD117 (c-Kit) Antibody | Biolegend | 105811 |
| PE anti-mouse Ly-6A/E (Sca-1) Antibody | Biolegend | 108107 |
| PE anti-mouse Ly-6C | Biolegend | 128007 |
| F4/80 Monoclonal Antibody (BM8),FITC | eBioscience | 11-4801-85 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, H.; Zhang, W.; Xie, D.; Nie, Y.; Chen, S.; Sun, X.; Zhao, H.; Liu, X.; Wang, H.; Huang, X.; et al. Cytosolic Release of Mitochondrial DNA and Associated cGAS Signaling Mediates Radiation-Induced Hematopoietic Injury of Mice. Int. J. Mol. Sci. 2023, 24, 4020. https://doi.org/10.3390/ijms24044020
Guan H, Zhang W, Xie D, Nie Y, Chen S, Sun X, Zhao H, Liu X, Wang H, Huang X, et al. Cytosolic Release of Mitochondrial DNA and Associated cGAS Signaling Mediates Radiation-Induced Hematopoietic Injury of Mice. International Journal of Molecular Sciences. 2023; 24(4):4020. https://doi.org/10.3390/ijms24044020
Chicago/Turabian StyleGuan, Hua, Wen Zhang, Dafei Xie, Yuehua Nie, Shi Chen, Xiaoya Sun, Hongling Zhao, Xiaochang Liu, Hua Wang, Xin Huang, and et al. 2023. "Cytosolic Release of Mitochondrial DNA and Associated cGAS Signaling Mediates Radiation-Induced Hematopoietic Injury of Mice" International Journal of Molecular Sciences 24, no. 4: 4020. https://doi.org/10.3390/ijms24044020
APA StyleGuan, H., Zhang, W., Xie, D., Nie, Y., Chen, S., Sun, X., Zhao, H., Liu, X., Wang, H., Huang, X., Bai, C., Huang, B., Zhou, P., & Gao, S. (2023). Cytosolic Release of Mitochondrial DNA and Associated cGAS Signaling Mediates Radiation-Induced Hematopoietic Injury of Mice. International Journal of Molecular Sciences, 24(4), 4020. https://doi.org/10.3390/ijms24044020