
Bioinorganic Modulators of Ferroptosis: A Review of Recent Findings

Abstract
1. Introduction
2. Concise Mechanism of Ferroptotic Progression
2.1. The Key Role of Iron
2.2. Increased Formation of Reactive Oxygen Species (ROS)
2.3. Compromised Cellular Defense against ROS
2.4. Target of ROS: PUFA Metabolism
3. Gallium Salts, Complexes and Nanoparticles
4. Chalcogens: Sulphur, Selenium, Tellurium
5. Transition Metals
6. Poisons and Toxicants
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Seiler, A.; Schneider, M.; Forster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Radmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef]
- DIXON, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in cancer therapy: A novel approach to reversing drug resistance. Mol. Cancer 2022, 21, 47. [Google Scholar] [CrossRef]
- Diepstraten, S.T.; Anderson, M.A.; Czabotar, P.E.; Lessene, G.; Strasser, A.; Kelly, G.L. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat. Rev. Cancer 2022, 22, 45–64. [Google Scholar] [CrossRef]
- Zhang, J. The osteoprotective effects of artemisinin compounds and the possible mechanisms associated with intracellular iron: A review of in vivo and in vitro studies. Environ. Toxicol. Pharmacol. 2020, 76, 103358. [Google Scholar] [CrossRef]
- Duan, J.Y.; Lin, X.; Xu, F.; Shan, S.K.; Guo, B.; Li, F.X.; Wang, Y.; Zheng, M.H.; Xu, Q.S.; Lei, L.M.; et al. Ferroptosis and Its Potential Role in Metabolic Diseases: A Curse or Revitalization? Front. Cell Dev. Biol. 2021, 9, 701788. [Google Scholar] [CrossRef]
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple cell death modalities and their key features (Review). World Acad. Sci. J. 2020, 2, 39–48. [Google Scholar] [CrossRef]
- Abe, C.; Miyazawa, T.; Miyazawa, T. Current Use of Fenton Reaction in Drugs and Food. Molecules 2022, 27, 5451. [Google Scholar] [CrossRef]
- Li, Z. Imaging of hydrogen peroxide (H2O2) during the ferroptosis process in living cancer cells with a practical fluorescence probe. Talanta 2020, 212, 120804. [Google Scholar] [CrossRef]
- Park, E.; Chung, S.W. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019, 10, 822. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef]
- Rafael, O. A History of the Fenton Reactions (Fenton Chemistry for Beginners). In Reactive Oxygen Species; Rizwan, A., Ed.; IntechOpen: Rijeka, Croatia, 2022. [Google Scholar] [CrossRef]
- You, H.; Wang, L.; Bu, F.; Meng, H.; Huang, C.; Fang, G.; Li, J. Ferroptosis: Shedding Light on Mechanisms and Therapeutic Opportunities in Liver Diseases. Cells 2022, 11, 3301. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Kajiwara, K.; Beharier, O.; Chng, C.-P.; Goff, J.P.; Ouyang, Y.; St Croix, C.M.; Huang, C.; Kagan, V.E.; Hsia, K.J.; Sadovsky, Y. Ferroptosis induces membrane blebbing in placental trophoblasts. J. Cell Sci. 2021, 135, jcs255737. [Google Scholar] [CrossRef]
- Graceffa, V. Therapeutic Potential of Reactive Oxygen Species: State of the Art and Recent Advances. SLAS TECHNOLOGY: Transl. Life Sci. Innov. 2020, 26, 140–158. [Google Scholar] [CrossRef]
- Ren, X.; Liu, H.; Wu, X.; Weng, W.; Wang, X.; Su, J. Reactive Oxygen Species (ROS)-Responsive Biomaterials for the Treatment of Bone-Related Diseases. Front. Bioeng. Biotechnol. 2022, 9, 820468. [Google Scholar] [CrossRef]
- Shields, H.J.; Traa, A.; Van Raamsdonk, J.M. Beneficial and Detrimental Effects of Reactive Oxygen Species on Lifespan: A Comprehensive Review of Comparative and Experimental Studies. Front. Cell Dev. Biol. 2021, 9, 628157. [Google Scholar] [CrossRef]
- Eljebbawi, A.; Guerrero, Y.d.C.R.; Dunand, C.; Estevez, J.M. Highlighting reactive oxygen species as multitaskers in root development. iScience 2021, 24, 101978. [Google Scholar] [CrossRef]
- Villalpando-Rodriguez, G.E.; Gibson, S.B. Reactive Oxygen Species (ROS) Regulates Different Types of Cell Death by Acting as a Rheostat. Oxidative Med. Cell. Longev. 2021, 2021, 9912436. [Google Scholar] [CrossRef]
- Nimse, S.B.; Pal, D. Free radicals, natural antioxidants, and their reaction mechanisms. RSC Adv. 2015, 5, 27986–28006. [Google Scholar] [CrossRef]
- Atika, E.; Naouel, E. Endogenous Enzymatic Antioxidant Defense and Pathologies. In Antioxidants; Viduranga, W., Ed.; IntechOpen: Rijeka, Croatia, 2021. [Google Scholar] [CrossRef]
- Herb, M.; Gluschko, A.; Schramm, M. Reactive Oxygen Species: Not Omnipresent but Important in Many Locations. Front. Cell Dev. Biol. 2021, 9, 716406. [Google Scholar] [CrossRef]
- Zhao, Z. Iron and oxidizing species in oxidative stress and Alzheimer’s disease. Aging Med. 2019, 2, 82–87. [Google Scholar] [CrossRef]
- Mailloux, R.J. Mitochondrial Antioxidants and the Maintenance of Cellular Hydrogen Peroxide Levels. Oxidative Med. Cell. Longev. 2018, 2018, 7857251. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Jo, D.S.; Park, N.Y.; Cho, D.-H. Peroxisome quality control and dysregulated lipid metabolism in neurodegenerative diseases. Exp. Mol. Med. 2020, 52, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Bou-Fakhredin, R.; Dia, B.; Ghadieh, H.E.; Rivella, S.; Cappellini, M.D.; Eid, A.A.; Taher, A.T. CYP450 Mediates Reactive Oxygen Species Production in a Mouse Model of β-Thalassemia through an Increase in 20-HETE Activity. Int. J. Mol. Sci. 2021, 22, 1106. [Google Scholar] [CrossRef] [PubMed]
- Sinenko, S.A.; Starkova, T.Y.; Kuzmin, A.A.; Tomilin, A.N. Physiological Signaling Functions of Reactive Oxygen Species in Stem Cells: From Flies to Man. Front. Cell Dev. Biol. 2021, 9, 714370. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Tang, L.J.; Luo, X.J.; Ai, K.L.; Peng, J. Insights into the novel function of system Xc- in regulated cell death. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 1650–1662. [Google Scholar] [CrossRef]
- Feng, Z.; Qin, Y.; Huo, F.; Jian, Z.; Li, X.; Geng, J.; Li, Y.; Wu, J. NMN recruits GSH to enhance GPX4-mediated ferroptosis defense in UV irradiation induced skin injury. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2022, 1868, 166287. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Xu, C.; Sun, S.; Johnson, T.; Qi, R.; Zhang, S.; Zhang, J.; Yang, K. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep. 2021, 35, 109235. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxidative Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef] [PubMed]
- Li, F.-J.; Long, H.-Z.; Zhou, Z.-W.; Luo, H.-Y.; Xu, S.-G.; Gao, L.-C. System Xc−/GSH/GPX4 axis: An important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front. Pharmacol. 2022, 13, 910292. [Google Scholar] [CrossRef]
- Pohl, E.E.; Jovanovic, O. The Role of Phosphatidylethanolamine Adducts in Modification of the Activity of Membrane Proteins under Oxidative Stress. Molecules 2019, 24, 4545. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Chen, Y.; Xue, F.; Liu, K.; Zhu, B.; Gao, J.; Yin, J.; Zhang, C.; Li, G. Contribution of ferroptosis and GPX4’s dual functions to osteoarthritis progression. eBioMedicine 2022, 76, 103847. [Google Scholar] [CrossRef] [PubMed]
- Fei, W.; Chen, D.; Tang, H.; Li, C.; Zheng, W.; Chen, F.; Song, Q.; Zhao, Y.; Zou, Y.; Zheng, C. Targeted GSH-exhausting and hydroxyl radical self-producing manganese-silica nanomissiles for MRI guided ferroptotic cancer therapy. Nanoscale 2020, 12, 16738–16754. [Google Scholar] [CrossRef]
- Yan, B.; Ai, Y.; Sun, Q.; Ma, Y.; Cao, Y.; Wang, J.; Zhang, Z.; Wang, X. Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol. Cell 2021, 81, 355–369.e310. [Google Scholar] [CrossRef]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef]
- Gan, B. ACSL4, PUFA, and ferroptosis: New arsenal in anti-tumor immunity. Signal Transduct. Target. Ther. 2022, 7, 128. [Google Scholar] [CrossRef]
- Li, D.; Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct. Target. Ther. 2020, 5, 108. [Google Scholar] [CrossRef]
- Nai, A.; Lidonnici, M.R.; Rausa, M.; Mandelli, G.; Pagani, A.; Silvestri, L.; Ferrari, G.; Camaschella, C. The second transferrin receptor regulates red blood cell production in mice. Blood 2015, 125, 1170–1179. [Google Scholar] [CrossRef]
- Zeng, X.; An, H.; Yu, F.; Wang, K.; Zheng, L.; Zhou, W.; Bao, Y.; Yang, J.; Shen, N.; Huang, D. Benefits of Iron Chelators in the Treatment of Parkinson’s Disease. Neurochem. Res. 2021, 46, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Liu, S.; Li, C.; Ai, Z.; Shen, W.; Ren, W.; Yang, X. Discovery of a novel ferroptosis inducer-talaroconvolutin A—Killing colorectal cancer cells in vitro and in vivo. Cell Death Dis. 2020, 11, 988. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mo, J.; Dai, J.; Ye, C.; Cen, W.; Zheng, X.; Jiang, L.; Ye, L. Cetuximab promotes RSL3-induced ferroptosis by suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant colorectal cancer. Cell Death Dis. 2021, 12, 1079. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Xu, B.; Xiong, Q.; Feng, Y.; Du, H. Erastin-induced ferroptosis causes physiological and pathological changes in healthy tissues of mice. Mol. Med. Rep. 2021, 24, 713. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef]
- Ghoochani, A.; Hsu, E.-C.; Aslan, M.; Rice, M.A.; Nguyen, H.M.; Brooks, J.D.; Corey, E.; Paulmurugan, R.; Stoyanova, T. Ferroptosis Inducers Are a Novel Therapeutic Approach for Advanced Prostate Cancer. Cancer Res. 2021, 81, 1583–1594. [Google Scholar] [CrossRef]
- Li, B.; Yang, L.; Peng, X.; Fan, Q.; Wei, S.; Yang, S.; Li, X.; Jin, H.; Wu, B.; Huang, M.; et al. Emerging mechanisms and applications of ferroptosis in the treatment of resistant cancers. Biomed. Pharmacother. 2020, 130, 110710. [Google Scholar] [CrossRef]
- Liang, C.; Zhang, X.; Yang, M.; Dong, X. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv. Mater. 2019, 31, 1904197. [Google Scholar] [CrossRef]
- Yan, H.-f.; Zou, T.; Tuo, Q.-z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and links with diseases. Signal Transduct. Target. Ther. 2021, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Qiao, Y.; Wang, D.; Tang, C.; Yan, G. Ferritinophagy and ferroptosis in cardiovascular disease: Mechanisms and potential applications. Biomed. Pharmacother. 2021, 141, 111872. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Berdun, F.; Bartoli, C.; Steelheart, C.; Alegre, M.; Bayir, H.; Tyurina, Y.Y.; Kagan, V.E.; Salerno, G.; Pagnussat, G.; et al. C-ferroptosis is an iron-dependent form of regulated cell death in cyanobacteria. J. Cell Biol. 2022, 221, e201911005. [Google Scholar] [CrossRef] [PubMed]
- Kurtuldu, F.; Mutlu, N.; Boccaccini, A.R.; Galusek, D. Gallium containing bioactive materials: A review of anticancer, antibacterial, and osteogenic properties. Bioact. Mater. 2022, 17, 125–146. [Google Scholar] [CrossRef]
- Chitambar, C.R.; Al-Gizawiy, M.M.; Alhajala, H.S.; Pechman, K.R.; Wereley, J.P.; Wujek, R.; Clark, P.A.; Kuo, J.S.; Antholine, W.E.; Schmainda, K.M. Gallium Maltolate Disrupts Tumor Iron Metabolism and Retards the Growth of Glioblastoma by Inhibiting Mitochondrial Function and Ribonucleotide Reductase. Mol. Cancer 2018, 17, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Awais, M.; Aizaz, A.; Nazneen, A.; Bhatti, Q.; Akhtar, M.; Wadood, A.; Ur Rehman, M.A. A Review on the Recent Advancements on Therapeutic Effects of Ions in the Physiological Environments. Prosthesis 2022, 4, 263–316. [Google Scholar] [CrossRef]
- Guo, Q.; Li, L.; Hou, S.; Yuan, Z.; Li, C.; Zhang, W.; Zheng, L.; Li, X. The Role of Iron in Cancer Progression. Front. Oncol. 2021, 11, 778492. [Google Scholar] [CrossRef]
- Li, F.; Liu, F.; Huang, K.; Yang, S. Advancement of Gallium and Gallium-Based Compounds as Antimicrobial Agents. Front. Bioeng. Biotechnol. 2022, 10, 827960. [Google Scholar] [CrossRef]
- Chitambar, C.R. Gallium Complexes as Anticancer Drugs. Met. Ions Life Sci. 2018, 18, 281–301. [Google Scholar] [CrossRef]
- Ellahioui, Y.; Prashar, S.; Gómez-Ruiz, S. Anticancer Applications and Recent Investigations of Metallodrugs Based on Gallium, Tin and Titanium. Inorganics 2017, 5, 4. [Google Scholar] [CrossRef]
- Chitambar, C.R. Gallium-containing anticancer compounds. Future Med. Chem. 2012, 4, 1257–1272. [Google Scholar] [CrossRef]
- Chitambar, C.; Wereley, J. Resistance to the Antitumor Agent Gallium Nitrate in Human Leukemic Cells Is Associated with Decreased Gallium/Iron Uptake, Increased Activity of Iron Regulatory Protein1, and Decreased Ferritin Production. J. Biol. Chem. 1997, 272, 12151–12157. [Google Scholar] [CrossRef]
- Bernstein, L.; Tanner, T.; Godfrey, C.; Noll, B. Chemistry and Pharmacokinetics of Gallium Maltolate, a Compound With High Oral Gallium Bioavailability. Met.-Based Drugs 2000, 7, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Timerbaev, A.R. Advances in developing tris(8-quinolinolato)gallium(iii) as an anticancer drug: Critical appraisal and prospects. Metallomics 2009, 1, 193–198. [Google Scholar] [CrossRef]
- Moawed, F.S.; El-Sonbaty, S.M.; Mansour, S.Z. Gallium nanoparticles along with low-dose gamma radiation modulate TGF-β/MMP-9 expression in hepatocellular carcinogenesis in rats. Tumor Biol. 2019, 41, 1010428319834856. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.M.; Cheung, P.C.K. Gallic Acid Triggers Iron-Dependent Cell Death with Apoptotic, Ferroptotic, and Necroptotic Features. Toxins 2019, 11, 492. [Google Scholar] [CrossRef]
- Mostafa, N.; Salem, A.; Mansour, S.Z.; El-Sonbaty, S.M.; Moawed, F.S.M.; Kandil, E.I. Rationale for Tailoring an Alternative Oncology Trial Using a Novel Gallium-Based Nanocomplex: Mechanistic Insights and Preclinical Challenges. Technol. Cancer Res. Treat. 2022, 21, 15330338221085376. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef]
- Chen, M.; Zhu, J.-Y.; Mu, W.-J.; Guo, L. Cysteine dioxygenase type 1 (CDO1): Its functional role in physiological and pathophysiological processes. Genes Dis. 2022; in press. [Google Scholar] [CrossRef]
- Kang, R.; Tang, D. Heat Shock Proteins: Endogenous Modulators of Ferroptosis. Ferroptosis Health Dis. 2019, 61–81. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015, 34, 5617–5625. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Tang, L.; Zhang, Q.; Shi, C.; Huang, Z.; Nijiati, S.; Chen, X.; Zhou, Z. Coordinating the Mechanisms of Action of Ferroptosis and the Photothermal Effect for Cancer Theranostics. Angew. Chem. Int. Ed. 2022, 61, e202112925. [Google Scholar] [CrossRef]
- Cao, C.; Yang, N.; Su, Y.; Zhang, Z.; Wang, C.; Song, X.; Chen, P.; Wang, W.; Dong, X. Starvation, Ferroptosis, and Prodrug Therapy Synergistically Enabled by a Cytochrome c Oxidase like Nanozyme. Adv. Mater. 2022, 34, 2203236. [Google Scholar] [CrossRef]
- El-sonbaty, S.M.; Moawed, F.S.; Kandil, E.I.; Tamamm, A.M. Antitumor and Antibacterial Efficacy of Gallium Nanoparticles Coated by Ellagic Acid. Dose-Response 2022, 20, 15593258211068998. [Google Scholar] [CrossRef]
- Wu, J.; Wang, Y.; Jiang, R.; Xue, R.; Yin, X.; Wu, M.; Meng, Q. Ferroptosis in liver disease: New insights into disease mechanisms. Cell Death Discov. 2021, 7, 276. [Google Scholar] [CrossRef]
- Fujii, J.; Homma, T.; Osaki, T. Superoxide Radicals in the Execution of Cell Death. Antioxidants 2022, 11, 501. [Google Scholar] [CrossRef]
- Liang, J.; Shen, Y.; Wang, Y.; Huang, Y.; Wang, J.; Zhu, Q.; Tong, G.; Yu, K.; Cao, W.; Wang, Q.; et al. Ferroptosis participates in neuron damage in experimental cerebral malaria and is partially induced by activated CD8+ T cells. Mol. Brain 2022, 15, 57. [Google Scholar] [CrossRef]
- Xie, X.; Zhang, Y.; Su, X.; Wang, J.; Yao, X.; Lv, D.; Zhou, Q.; Mao, J.; Chen, J.; Han, F.; et al. Targeting iron metabolism using gallium nanoparticles to suppress ferroptosis and effectively mitigate acute kidney injury. Nano Res. 2022, 15, 6315–6327. [Google Scholar] [CrossRef]
- Hreusova, M.; Novohradsky, V.; Markova, L.; Kostrhunova, H.; Potocnak, I.; Brabec, V.; Kasparkova, J. Gallium(III) Complex with Cloxyquin Ligands Induces Ferroptosis in Cancer Cells and Is a Potent Agent against Both Differentiated and Tumorigenic Cancer Stem Rhabdomyosarcoma Cells. Bioinorg. Chem. Appl. 2022, 2022, 3095749. [Google Scholar] [CrossRef]
- Xie, W.; Allioux, F.-M.; Ou, J.Z.; Miyako, E.; Tang, S.-Y.; Kalantar-Zadeh, K. Gallium-Based Liquid Metal Particles for Therapeutics. Trends Biotechnol. 2021, 39, 624–640. [Google Scholar] [CrossRef]
- Leu, J.I.; Murphy, M.E.; George, D.L. Functional interplay among thiol-based redox signaling, metabolism, and ferroptosis unveiled by a genetic variant of TP53. Proc. Natl. Acad. Sci. USA 2020, 117, 26804–26811. [Google Scholar] [CrossRef]
- Wu, Z.; Khodade, V.S.; Chauvin, J.-P.R.; Rodriguez, D.; Toscano, J.P.; Pratt, D.A. Hydropersulfides Inhibit Lipid Peroxidation and Protect Cells from Ferroptosis. J. Am. Chem. Soc. 2022, 144, 15825–15837. [Google Scholar] [CrossRef]
- Toyokuni, S.; Ito, F.; Yamashita, K.; Okazaki, Y.; Akatsuka, S. Iron and thiol redox signaling in cancer: An exquisite balance to escape ferroptosis. Free Radic. Biol. Med. 2017, 108, 610–626. [Google Scholar] [CrossRef] [PubMed]
- Barayeu, U.; Schilling, D.; Eid, M.; Xavier da Silva, T.N.; Schlicker, L.; Mitreska, N.; Zapp, C.; Grater, F.; Miller, A.K.; Kappl, R.; et al. Hydropersulfides inhibit lipid peroxidation and ferroptosis by scavenging radicals. Nat. Chem. Biol. 2023, 19, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Guo, Z.; Liu, X.; Wang, Y.; Xiao, C. A glutathione-responsive sulfur dioxide polymer prodrug selectively induces ferroptosis in gastric cancer therapy. Biomater. Sci. 2022, 10, 4184–4192. [Google Scholar] [CrossRef]
- Xiang, N.; Zhao, R.; Zhong, W. Sodium selenite induces apoptosis by generation of superoxide via the mitochondrial-dependent pathway in human prostate cancer cells. Cancer Chemother. Pharmacol. 2009, 63, 351–362. [Google Scholar] [CrossRef]
- Johnstone, M.A.; Nelson, S.J.; O’Leary, C.; Self, W.T. Exploring the selenium-over-sulfur substrate specificity and kinetics of a bacterial selenocysteine lyase. Biochimie 2021, 182, 166–176. [Google Scholar] [CrossRef]
- Olm, E.; Fernandes, A.P.; Hebert, C.; Rundlöf, A.-K.; Larsen, E.H.; Danielsson, O.; Björnstedt, M. Extracellular thiol-assisted selenium uptake dependent on the xc cystine transporter explains the cancer-specific cytotoxicity of selenite. Proc. Natl. Acad. Sci. 2009, 106, 11400–11405. [Google Scholar] [CrossRef]
- Radomska, D.; Czarnomysy, R.; Radomski, D.; Bielawski, K. Selenium Compounds as Novel Potential Anticancer Agents. Int. J. Mol. Sci. 2021, 22, 1009. [Google Scholar] [CrossRef]
- Subburayan, K.; Thayyullathil, F.; Pallichankandy, S.; Cheratta, A.R.; Galadari, S. Superoxide-mediated ferroptosis in human cancer cells induced by sodium selenite. Transl. Oncol. 2020, 13, 100843. [Google Scholar] [CrossRef] [PubMed]
- Vinceti, M.; Filippini, T.; Jablonska, E.; Saito, Y.; Wise, L.A. Safety of selenium exposure and limitations of selenoprotein maximization: Molecular and epidemiologic perspectives. Environ. Res. 2022, 211, 113092. [Google Scholar] [CrossRef]
- Vinceti, M.; Vicentini, M.; Wise, L.A.; Sacchettini, C.; Malagoli, C.; Ballotari, P.; Filippini, T.; Malavolti, M.; Rossi, P.G. Cancer incidence following long-term consumption of drinking water with high inorganic selenium content. Sci. Total Environ. 2018, 635, 390–396. [Google Scholar] [CrossRef]
- Ramakrishnan, M.; Arivalagan, J.; Satish, L.; Mohan, M.; Samuel Selvan Christyraj, J.R.; Chandran, S.A.; Ju, H.-J.; Ramesh, T.; Ignacimuthu, S.; Kalishwaralal, K. Selenium: A potent regulator of ferroptosis and biomass production. Chemosphere 2022, 306, 135531. [Google Scholar] [CrossRef]
- Gao, M.; Hu, J.; Zhu, Y.; Wang, X.; Zeng, S.; Hong, Y.; Zhao, G. Ferroptosis and Apoptosis Are Involved in the Formation of L-Selenomethionine-Induced Ocular Defects in Zebrafish Embryos. Int. J. Mol. Sci. 2022, 23, 4783. [Google Scholar] [CrossRef] [PubMed]
- Stathopoulou, M.G.; Kanoni, S.; Papanikolaou, G.; Antonopoulou, S.; Nomikos, T.; Dedoussis, G. Mineral Intake. In Progress in Molecular Biology and Translational Science; Bouchard, C., Ordovas, J.M., Eds.; Academic Press: Cambridge, MA, USA, 2012; Volume 108, pp. 201–236. [Google Scholar]
- Tuo, Q.-Z.; Masaldan, S.; Southon, A.; Mawal, C.; Ayton, S.; Bush, A.I.; Lei, P.; Belaidi, A.A. Characterization of Selenium Compounds for Anti-ferroptotic Activity in Neuronal Cells and after Cerebral Ischemia–Reperfusion Injury. Neurotherapeutics 2021, 18, 2682–2691. [Google Scholar] [CrossRef] [PubMed]
- Alim, I.; Caulfield, J.T.; Chen, Y.; Swarup, V.; Geschwind, D.H.; Ivanova, E.; Seravalli, J.; Ai, Y.; Sansing, L.H.; Ste.Marie, E.J.; et al. Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell 2019, 177, 1262–1279.e1225. [Google Scholar] [CrossRef]
- Chen, Y.-X.; Zuliyaer, T.; Liu, B.; Guo, S.; Yang, D.-G.; Gao, F.; Yu, Y.; Yang, M.-L.; Du, L.-J.; Li, J.-J. Sodium selenite promotes neurological function recovery after spinal cord injury by inhibiting ferroptosis. Neural Regen. Res. 2022, 17, 2702–2709. [Google Scholar] [CrossRef]
- Jiang, X.; Guo, S.; Xu, M.; Ma, B.; Liu, R.; Xu, Y.; Zhang, Y. TFAP2C-Mediated lncRNA PCAT1 Inhibits Ferroptosis in Docetaxel-Resistant Prostate Cancer Through c-Myc/miR-25-3p/SLC7A11 Signaling. Front. Oncol. 2022, 12, 862015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Hu, Y.; Hu, J.-E.; Ding, Y.; Shen, Y.; Xu, H.; Chen, H.; Wu, N. Sp1-mediated upregulation of Prdx6 expression prevents podocyte injury in diabetic nephropathy via mitigation of oxidative stress and ferroptosis. Life Sci. 2021, 278, 119529. [Google Scholar] [CrossRef]
- Li, Y.; Feng, D.; Wang, Z.; Zhao, Y.; Sun, R.; Tian, D.; Liu, D.; Zhang, F.; Ning, S.; Yao, J.; et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019, 26, 2284–2299. [Google Scholar] [CrossRef]
- Liu, S.; Tang, Y.; Liu, L.; Yang, L.; Li, P.; Liu, X.; Yin, H. Proteomic analysis reveals that ACSL4 activation during reflux esophagitis contributes to ferroptosis-mediated esophageal mucosal damage. Eur. J. Pharmacol. 2022, 931, 175175. [Google Scholar] [CrossRef]
- Yi, L.; Hu, Y.; Wu, Z.; Li, Y.; Kong, M.; Kang, Z.; Zuoyuan, B.; Yang, Z. TFRC upregulation promotes ferroptosis in CVB3 infection via nucleus recruitment of Sp1. Cell Death Dis. 2022, 13, 592. [Google Scholar] [CrossRef]
- Liu, N.; Liang, Y.; Wei, T.; Zou, L.; Huang, X.; Kong, L.; Tang, M.; Zhang, T. The role of ferroptosis mediated by NRF2/ERK-regulated ferritinophagy in CdTe QDs-induced inflammation in macrophage. J. Hazard. Mater. 2022, 436, 129043. [Google Scholar] [CrossRef]
- Song, X.; Long, D. Nrf2 and Ferroptosis: A New Research Direction for Neurodegenerative Diseases. Front. Neurosci. 2020, 14, 267. [Google Scholar] [CrossRef]
- Wang, D.; Tang, L.; Zhang, Y.; Ge, G.; Jiang, X.; Mo, Y.; Wu, P.; Deng, X.; Li, L.; Zuo, S.; et al. Regulatory pathways and drugs associated with ferroptosis in tumors. Cell Death Dis. 2022, 13, 544. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wu, D.; Duan, J.; Xiao, H.; Zhou, Y.; Zhao, L.; Feng, Y. NRF2 regulates the sensitivity of human NSCLC cells to cystine deprivation-induced ferroptosis via FOCAD-FAK signaling pathway. Redox Biol. 2020, 37, 101702. [Google Scholar] [CrossRef]
- Wang, M.; Liu, Y.; Shi, H.; Li, S.; Chen, S. Yielding hydroxyl radicals in the Fenton-like reaction induced by manganese (II) oxidation determines Cd mobilization upon soil aeration in paddy soil systems. Environ. Pollut. 2022, 292, 118311. [Google Scholar] [CrossRef]
- Wang, P.; Liang, C.; Zhu, J.; Yang, N.; Jiao, A.; Wang, W.; Song, X.; Dong, X. Manganese-Based Nanoplatform As Metal Ion-Enhanced ROS Generator for Combined Chemodynamic/Photodynamic Therapy. ACS Appl. Mater. Interfaces 2019, 11, 41140–41147. [Google Scholar] [CrossRef]
- Lin, L.-S.; Song, J.; Song, L.; Ke, K.; Liu, Y.; Zhou, Z.; Shen, Z.; Li, J.; Yang, Z.; Tang, W.; et al. Simultaneous Fenton-like Ion Delivery and Glutathione Depletion by MnO2-Based Nanoagent to Enhance Chemodynamic Therapy. Angew. Chem. Int. Ed. 2018, 57, 4902–4906. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, F.; Qiao, R.; Hu, X.; Liao, H.; Chen, L.; Wu, J.; Wu, H.; Zhao, M.; Liu, J.; et al. Arginine-Rich Manganese Silicate Nanobubbles as a Ferroptosis-Inducing Agent for Tumor-Targeted Theranostics. ACS Nano 2018, 12, 12380–12392. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhong, X.; Yuan, Y. Does Baking Soda Function as a Magic Bullet for Patients With Cancer? A Mini Review. Integr. Cancer Ther. 2020, 19, 1534735420922579. [Google Scholar] [CrossRef]
- Cheng, J.; Zhu, Y.; Xing, X.; Xiao, J.; Chen, H.; Zhang, H.; Wang, D.; Zhang, Y.; Zhang, G.; Wu, Z.; et al. Manganese-deposited iron oxide promotes tumor-responsive ferroptosis that synergizes the apoptosis of cisplatin. Theranostics 2021, 11, 5418–5429. [Google Scholar] [CrossRef]
- Ameta, R.; Chohadia, A.K.; Jain, A.; Punjabi, P.B. Chapter 3—Fenton and Photo-Fenton Processes. In Advanced Oxidation Processes for Waste Water Treatment; Ameta, S.C., Ameta, R., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 49–87. [Google Scholar] [CrossRef]
- Ma, B.; Wang, S.; Liu, F.; Zhang, S.; Duan, J.; Li, Z.; Kong, Y.; Sang, Y.; Liu, H.; Bu, W.; et al. Self-Assembled Copper–Amino Acid Nanoparticles for in Situ Glutathione “AND” H2O2 Sequentially Triggered Chemodynamic Therapy. J. Am. Chem. Soc. 2019, 141, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, D.; Zhang, S.; Cheng, Y.; Yang, F.; Xing, Y.; Xu, T.; Dong, H.; Zhang, X. Biodegradable Biomimic Copper/Manganese Silicate Nanospheres for Chemodynamic/Photodynamic Synergistic Therapy with Simultaneous Glutathione Depletion and Hypoxia Relief. ACS Nano 2019, 13, 4267–4277. [Google Scholar] [CrossRef] [PubMed]
- Southon, A.; Szostak, K.; Acevedo, K.M.; Dent, K.A.; Volitakis, I.; Belaidi, A.A.; Barnham, K.J.; Crouch, P.J.; Ayton, S.; Donnelly, P.S.; et al. Cu(II) (atsm) inhibits ferroptosis: Implications for treatment of neurodegenerative disease. Br. J. Pharmacol. 2020, 177, 656–667. [Google Scholar] [CrossRef]
- Xue, Q.; Yan, D.; Chen, X.; Li, X.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Chen, X.; Tang, D.; Liu, J. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy, 2023; Online ahead of print. [Google Scholar] [CrossRef]
- Pushpalatha, C.; Suresh, J.; Gayathri, V.; Sowmya, S.; Augustine, D.; Alamoudi, A.; Zidane, B.; Mohammad Albar, N.H.; Patil, S. Zinc Oxide Nanoparticles: A Review on Its Applications in Dentistry. Front. Bioeng. Biotechnol. 2022, 10, 917990. [Google Scholar] [CrossRef]
- Chauhan, R.; Kumar, A.; Tripathi, R.; Kumar, A. Advancing of Zinc Oxide Nanoparticles for Cosmetic Applications. In Handbook of Consumer Nanoproducts; Springer Nature Singapore: Singapore, 2022; pp. 1057–1072. [Google Scholar] [CrossRef]
- Youn, S.-M.; Choi, S.-J. Food Additive Zinc Oxide Nanoparticles: Dissolution, Interaction, Fate, Cytotoxicity, and Oral Toxicity. Int. J. Mol. Sci. 2022, 23, 6074. [Google Scholar] [CrossRef]
- Qin, X.; Zhang, J.; Wang, B.; Xu, G.; Yang, X.; Zou, Z.; Yu, C. Ferritinophagy is involved in the zinc oxide nanoparticles-induced ferroptosis of vascular endothelial cells. Autophagy 2021, 17, 4266–4285. [Google Scholar] [CrossRef]
- Chang, N.C. Autophagy and Stem Cells: Self-Eating for Self-Renewal. Front. Cell Dev. Biol. 2020, 8, 138. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-Z.; Kong, N.; Zhang, G.-Y.; Xu, Q.; Xu, Y.; Ke, P.; Liu, C. The critical role of ferritinophagy in human disease. Front. Pharmacol. 2022, 13. [Google Scholar] [CrossRef]
- Hamaï, A.; Cañeque, T.; Müller, S.; Mai, T.T.; Hienzsch, A.; Ginestier, C.; Charafe-Jauffret, E.; Codogno, P.; Mehrpour, M.; Rodriguez, R. An iron hand over cancer stem cells. Autophagy 2017, 13, 1465–1466. [Google Scholar] [CrossRef]
- Quiles del Rey, M.; Mancias, J.D. NCOA4-Mediated Ferritinophagy: A Potential Link to Neurodegeneration. Front. Neurosci. 2019, 13, 238. [Google Scholar] [CrossRef]
- Bellelli, R.; Federico, G.; Matte’, A.; Colecchia, D.; Iolascon, A.; Chiariello, M.; Santoro, M.; De Franceschi, L.; Carlomagno, F. NCOA4 Deficiency Impairs Systemic Iron Homeostasis. Cell Rep. 2016, 14, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Sindikubwabo, F.; Cañeque, T.; Lafon, A.; Versini, A.; Lombard, B.; Loew, D.; Wu, T.-D.; Ginestier, C.; Charafe-Jauffret, E.; et al. CD44 regulates epigenetic plasticity by mediating iron endocytosis. Nat. Chem. 2020, 12, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Turcu, A.L.; Versini, A.; Khene, N.; Gaillet, C.; Cañeque, T.; Müller, S.; Rodriguez, R. DMT1 Inhibitors Kill Cancer Stem Cells by Blocking Lysosomal Iron Translocation. Chem. Eur. J. 2020, 26, 7369–7373. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Chi, J.T. Unexpected zinc dependency of ferroptosis: What is in a name? Oncotarget 2021, 12, 1126–1127. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Du, T.; Yang, H.; Lei, L.; Guo, M.; Ding, H.F.; Zhang, J.; Wang, H.; Chen, X.; et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc−. Cell Death Differ. 2020, 27, 662–675. [Google Scholar] [CrossRef]
- Yan, R.; Xie, E.; Li, Y.; Li, J.; Zhang, Y.; Chi, X.; Hu, X.; Xu, L.; Hou, T.; Stockwell, B.R.; et al. The structure of erastin-bound xCT–4F2hc complex reveals molecular mechanisms underlying erastin-induced ferroptosis. Cell Res. 2022, 32, 687–690. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, Z.; Zhang, Y.; Ma, L.; Song, E.; Song, Y. “Iron free” zinc oxide nanoparticles with ion-leaking properties disrupt intracellular ROS and iron homeostasis to induce ferroptosis. Cell Death Dis. 2020, 11, 183. [Google Scholar] [CrossRef]
- Han, Z.; Xu, Z.; Chen, L.; Ye, D.; Yu, Y.; Zhang, Y.; Cao, Y.; Djibril, B.; Guo, X.; Gao, X.; et al. Iron overload inhibits self-renewal of human pluripotent stem cells via DNA damage and generation of reactive oxygen species. FEBS Open Bio 2020, 10, 726–733. [Google Scholar] [CrossRef]
- Basuli, D.; Tesfay, L.; Deng, Z.; Paul, B.; Yamamoto, Y.; Ning, G.; Xian, W.; McKeon, F.; Lynch, M.; Crum, C.P.; et al. Iron addiction: A novel therapeutic target in ovarian cancer. Oncogene 2017, 36, 4089–4099. [Google Scholar] [CrossRef]
- Mai, T.T.; Hamaï, A.; Hienzsch, A.; Cañeque, T.; Müller, S.; Wicinski, J.; Cabaud, O.; Leroy, C.; David, A.; Acevedo, V.; et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem. 2017, 9, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Antoszczak, M.; Müller, S.; Cañeque, T.; Colombeau, L.; Dusetti, N.; Santofimia-Castaño, P.; Gaillet, C.; Puisieux, A.; Iovanna, J.L.; Rodriguez, R. Iron-Sensitive Prodrugs That Trigger Active Ferroptosis in Drug-Tolerant Pancreatic Cancer Cells. J. Am. Chem. Soc. 2022, 144, 11536–11545. [Google Scholar] [CrossRef] [PubMed]
- Planeta Kepp, K. Bioinorganic Chemistry of Zinc in Relation to the Immune System. ChemBioChem 2022, 23, e202100554. [Google Scholar] [CrossRef]
- Limmer, M.A.; Seyfferth, A.L. Altering the localization and toxicity of arsenic in rice grain. Sci. Rep. 2022, 12, 5210. [Google Scholar] [CrossRef]
- Xiao, J.; Zhang, S.; Tu, B.; Jiang, X.; Cheng, S.; Tang, Q.; Zhang, J.; Qin, X.; Wang, B.; Zou, Z.; et al. Arsenite induces ferroptosis in the neuronal cells via activation of ferritinophagy. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2021, 151, 112114. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, X.; He, C.; Guo, X.; Cai, H.; Aierken, A.; Hua, J.; Peng, S. Effects of Ferroptosis on Male Reproduction. Int. J. Mol. Sci. 2022, 23, 7139. [Google Scholar] [CrossRef]
- Meng, P.; Zhang, S.; Jiang, X.; Cheng, S.; Zhang, J.; Cao, X.; Qin, X.; Zou, Z.; Chen, C. Arsenite induces testicular oxidative stress in vivo and in vitro leading to ferroptosis. Ecotoxicol. Environ. Saf. 2020, 194, 110360. [Google Scholar] [CrossRef]
- Tirichen, H.; Yaigoub, H.; Xu, W.; Wu, C.; Li, R.; Li, Y. Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression Through Oxidative Stress. Front. Physiol. 2021, 12, 627837. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Lee, D.H.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Ferroptosis-Induced Endoplasmic Reticulum Stress: Cross-talk between Ferroptosis and Apoptosis. Mol. Cancer Res. MCR 2018, 16, 1073–1076. [Google Scholar] [CrossRef] [PubMed]
- Li, M.D.; Fu, L.; Lv, B.B.; Xiang, Y.; Xiang, H.X.; Xu, D.X.; Zhao, H. Arsenic induces ferroptosis and acute lung injury through mtROS-mediated mitochondria-associated endoplasmic reticulum membrane dysfunction. Ecotoxicol. Environ. Saf. 2022, 238, 113595. [Google Scholar] [CrossRef]
- Yang, M.; Li, C.; Yang, S.; Xiao, Y.; Xiong, X.; Chen, W.; Zhao, H.; Zhang, Q.; Han, Y.; Sun, L. Mitochondria-Associated ER Membranes—The Origin Site of Autophagy. Front. Cell Dev. Biol. 2020, 8, 595. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Yamasaki, T.; Hasebe, R.; Suzuki, A.; Horiuchi, M. Enhanced phosphorylation of PERK in primary cultured neurons as an autonomous neuronal response to prion infection. PLoS ONE 2020, 15, e0234147. [Google Scholar] [CrossRef] [PubMed]
- SIOP ABSTRACTS. Pediatr. Blood Cancer 2020, 67, e28742. [CrossRef]
- Feng, C.; Wu, Y.; Chen, Y.; Xiong, X.; Li, P.; Peng, X.; Li, C.; Weng, W.; Zhu, Y.; Zhou, D.; et al. Arsenic trioxide increases apoptosis of SK-N-BE (2) cells partially by inducing GPX4-mediated ferroptosis. Mol. Biol. Rep. 2022, 49, 6573–6580. [Google Scholar] [CrossRef]
- Wang, L.; Liu, S.; Gao, C.; Chen, H.; Li, J.; Lu, J.; Yuan, Y.; Zheng, X.; He, H.; Zhang, X.; et al. Arsenic trioxide-induced cardiotoxicity triggers ferroptosis in cardiomyoblast cells. Hum. Exp. Toxicol. 2022, 41, 09603271211064537. [Google Scholar] [CrossRef]
- Tang, S.; Shen, Y.; Wei, X.; Shen, Z.; Lu, W.; Xu, J. Olaparib synergizes with arsenic trioxide by promoting apoptosis and ferroptosis in platinum-resistant ovarian cancer. Cell Death Dis. 2022, 13, 826. [Google Scholar] [CrossRef]
- Yang, Y.; Zhu, T.; Wang, X.; Xiong, F.; Hu, Z.; Qiao, X.; Yuan, X.; Wang, D. ACSL3 and ACSL4, Distinct Roles in Ferroptosis and Cancers. Cancers 2022, 14, 5896. [Google Scholar] [CrossRef]
- Chu, J.-H.; Li, L.-X.; Gao, P.-C.; Chen, X.-W.; Wang, Z.-Y.; Fan, R.-F. Mercuric chloride induces sequential activation of ferroptosis and necroptosis in chicken embryo kidney cells by triggering ferritinophagy. Free Radic. Biol. Med. 2022, 188, 35–44. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, P.; Li, Y. Gut microbiota-mediated ferroptosis contributes to mercury exposure-induced brain injury in common carp. Metallomics 2022, 14, mfab072. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Yang, B.; Zhang, Y.; Wang, S.; Li, F.; Xing, G.; Farina, M.; Zhang, Y.; Appiah-Kubi, K.; Tinkov, A.A.; et al. Ferroptosis contributes to methylmercury-induced cytotoxicity in rat primary astrocytes and Buffalo rat liver cells. Neurotoxicology 2022, 90, 228–236. [Google Scholar] [CrossRef]
- Wang, W.; Shi, F.; Cui, J.; Pang, S.; Zheng, G.; Zhang, Y. MiR-378a-3p/ SLC7A11 regulate ferroptosis in nerve injury induced by lead exposure. Ecotoxicol. Environ. Saf. 2022, 239, 113639. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.-L.; Chao, S.-C.; Chu, P.-M.; Yu, C.-C. Regulation of Ferroptosis by Non-Coding RNAs in Head and Neck Cancers. Int. J. Mol. Sci. 2022, 23, 3142. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chan, Y.-T.; Tan, H.-Y.; Zhang, C.; Guo, W.; Xu, Y.; Sharma, R.; Chen, Z.-S.; Zheng, Y.-C.; Wang, N.; et al. Epigenetic regulation of ferroptosis via ETS1/miR-23a-3p/ACSL4 axis mediates sorafenib resistance in human hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2022, 41, 3. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhu, D.; Luo, B.; Kou, W.; Cheng, Y.; Zhu, Y. IFNγ enhances ferroptosis by increasing JAK-STAT pathway activation to suppress SLCA711 expression in adrenocortical carcinoma. Oncol. Rep. 2022, 47, 97. [Google Scholar] [CrossRef]
- Wang, H.; Lin, D.; Yu, Q.; Li, Z.; Lenahan, C.; Dong, Y.; Wei, Q.; Shao, A. A Promising Future of Ferroptosis in Tumor Therapy. Front. Cell Dev. Biol. 2021, 9, 629150. [Google Scholar] [CrossRef]


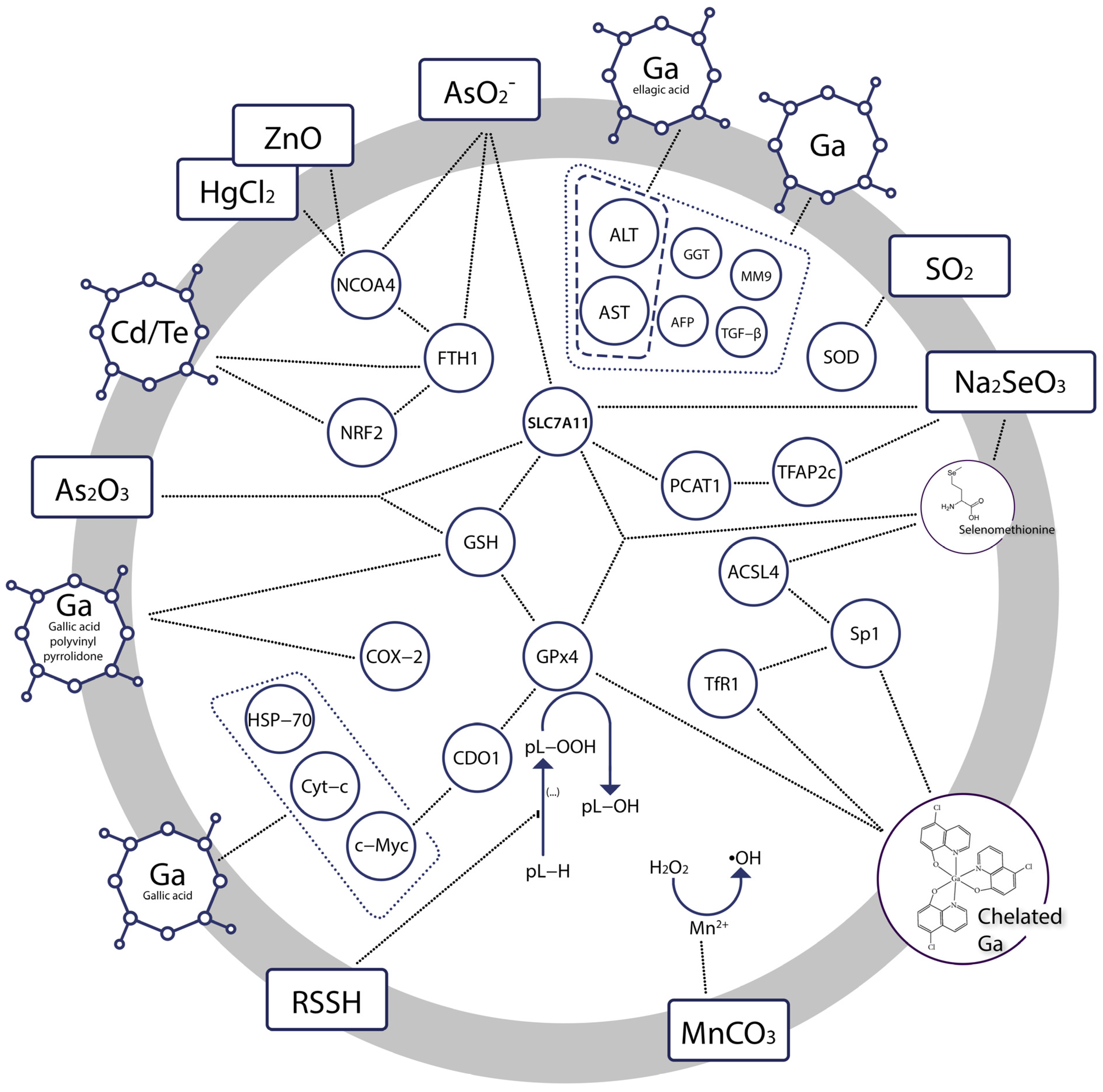{kind=link}
| Bioinorganic Modulator | Research Model | Outcome | Ref. |
|---|---|---|---|
| Gallium nanoparticles +low level of gamma radiation | Diethylnitrosamine (DEN)—induced hepatocellular carcinoma (HCC) in rats Human hepatocellular carcinoma (HepG2) cell line | Reduced serum levels of AST, ALT, AFP, GGT, MM9, TGF-β in HCC Cytotoxic effect in HepG-2, IC50 8.0 μg/mL | [66] |
| Gallium nanoparticles coated by gallic acid | Diethylnitrosamine (DEN)—induced hepatocellular carcinoma (HCC) in rats Human hepatocellular cancer (HepG-2) cell line | Cytotoxic effects at the interface of primary hepatocarcinogenesis, key hallmarks of cancer diminished (normalized liver C-Myc, HSP-70, and Cyt-c mRNA expression, ameliorated iron accumulation), reduced liver function indices ALP, ALT, AST Strong cytotoxic effect in HepG-2, IC50 0.71 ± 0.02 µg/mL | [68] |
| Gallium nanoparticles coated by ellagic acid | 7, 12 dimethyl benz[a] anthracene (DMBA)—induced mammary gland carcinogenesis in rats Human breast cancer (MCF-7) cell line | Reduced serum levels of ALT, AST, urea, and creatinine, decreased MDA, increased SOD, GSH, total iron binding capacity, mild recurrence of normal histopathological appearance Cytotoxic effect in MCF-7, IC50 2.86 ± 0.3 μg/mL | [75] |
| Gallic acid-gallium polyvinyl pyrrolidone nanoparticles | CP—induced and ischemia-reperfusion injury—induced acute kidney injury in mice Cisplatin (CP)—induced ferroptosis in HK-2 cells | Protection against renal ischemia-reperfusion injury in mice Reduced accumulation of intracellular free iron and mitochondrial dysfunction, suppressed parameters of ferroptosis (attenuated iron accumulation, inhibited ferritinophagy, restored GPx4 and NCOA4 expression) | [79] |
| Gallium(III) complex with cloxyquin | Human rhabdomyosarcoma cells | Killed bulk and stem rhabdomyosarcoma cells, IC50 1.3 ± 0.1 and 6–8 ± 2 μM, respectively Induced ferroptosis, significant increase in ROS level, reduced GPx4, stimulated expression of TfR1, potentiated accumulation of lipid peroxides Coincubation with ferrostatin reduced the potency of the compound | [80] |
| Bioinorganic Modulator | Research Model | Outcome | Ref. |
|---|---|---|---|
| In situ generated hydropersulfides from cumyl-SSH | (1S,3R)-RSL3—induced ferroptosis in Pfa1 mouse embryonic fibroblasts | Restored cell viability in a dose-dependent manner, IC50 6.2 μM Ferroptosis previously induced through inactivation of GPx4 or deletion of the gene encoding it was suppressed | [83] |
| mPEG-PLG(DNs), i.e., amphiphilic polymer prodrug of sulfur dioxide | Human gastric cancer cell line MKN-1 Human gastric mucosa cell line GES-1 | GSH depletion and SO2 generation resulting in amplified oxidative stress accelerated and lipid peroxidation in malignant cells, downregulated the expression of GPx4 in malignant cells IC50 240.9 ± 26.3 μg/mL in MKN-1 IC50 440.9 ± 44.7 μg/mL in GES-1 | [86] |
| Sodium selenite | Human cancer cell lines: MCF-7, PC3, U87MG, HT-1080, HeLa, HCT-116 and A172 cells Cytotoxicity testing: normal human fetal glial cells (SVG P12) vs. U87MG human malignant glioma cells | Reduced cell viability Downregulated SLC7A11, GSH, GPx4 Increased iron accumulation and lipid peroxidation Preferentially cytotoxic to malignant cells | [91] |
| Selenate and selenite transformed into selenomethionine | Zebrafish embryos | Disrupted mitochondrial morphology, elevated ROS-induced oxidative stress, significant increase in MDA, decrease in GSH, ferroptosis in embryonic eye cells At 24 h post fertilisation: gpx4a and gpx4b up-regulated, while slc7a11 down-regulated At 96 h post fertilisation: gpx4a and gpx4b decreased, slc7a11 unchanged relative to the controls | [95] |
| Sodium selenite, and sodium selenate | N27 immortalized neuronal cell line | anti-ferroptotic, protective against erastin- and RSL3-induced ferroptosis, increase in GPx1 and GPx4 expression Selenite was the most potent inhibitor of ferroptosis with EC50erastin: 38 nM | [97] |
| Sodium selenite | Rat model of T10 vertebral contusion injury | Decreased iron concentration and levels of the lipid peroxidation, increased the protein and mRNA expression of Sp1 and GPx4, promoted survival of neurons and oligodendrocytes, inhibited proliferation of astrocytes | [99] |
| Sodium selenite | 8–10 week old Male C57BL/6 mice HT22 murine hippocampal cells cultures Human HT-1080 fibrosarcoma cells | Dose-dependent neuroprotective efect against ferroptotic death in transformed and non-transformed cells, induction of selenoproteins (incl. GPx4), activation of the transcription factors i.a. TFAP2c and Sp1 | [98] |
| Cadmium telluride quantum dots | RAW264.7 macrophages | Ferroptotic death as a result of imbalanced oxidative and antioxidant systems, associated with decrease in NRF2, phosphorylation of ERK1/2, activated ferritinophagy | [105] |
| Bioinorganic Modulator | Research Model | Outcome | Ref. |
|---|---|---|---|
| Manganese carbonate nanocubes | HeLa cells Tumor bearing mice | Upregulated intracellular ROS was prompted by Mn2+ ions and contributed to antitumor effect | [111] |
| Manganese dioxide-coated mesoporous silica nanoparticles | U87MG human glioma cells | Decline of antioxidant defense resulting from GSH depletion, ROS generation prompted by Mn2+ ions, dose- and time-dependent toxicity of Mn2+ ions | [112] |
| Arginine-rich manganese silicate nanobubbles | Huh7 liver cancer cells Liver L02 normal cells Huh7 tumor xenograft nude mice | Upregulation of reactive oxygen species, GSH depletion, inactivation of GPx4 | [113] |
| Cisplatin prodrug-loaded manganese-deposited iron oxide nanoplatform | HeLa cells Tumor-bearing BALB/c-Nude mice | Ferroptotic effect through generation of reactive oxygen species, reduced cell viability, high levels of intracellular lipid peroxide, significantly lowered GSH, prominent anti-tumor efficacy in vivo | [115] |
| Copper–amino acid mercaptide nanoparticles | Human cervical carcinoma (HeLa) cells, human prostate cancer (PC-3) cells, human adipose-derived stem cells (hADSCs), human renal tubular epithelial (HK-2) cells, human bone mesenchymal stem cells (hbMSCs), human breast cancer cells (MCF-7), doxorubicin-resistant MCF-7 cells (MCF-7R) | Anti-cancer effect based on GSH depletion, generation of reactive oxygen species, low systematic toxicity, selective cytotoxicity, tumor suppression at ∼72.3% inhibition rate | [117] |
| Copper/Manganese Silicate Nanospheres | MCF-7, A549 and NHDF cells MCF-7 cancer-bearing female Balb/c mice | Upregulation of reactive oxygen species, GSH depletion, disrupted tumor microenvironment through relief of intracellular hypoxia, cancer cells growth significantly inhibited | [118] |
| Cu2+ (atsm) | Primary cortical neurons Immortalised neuronal cell lines, N27 | EC50 ≈ 130 nM Cytoprotective effect against lipid peroxidation and ferroptotic lethality | [119] |
| Copper sulfate | AsPC-1, PANC-1, MIA PaCa-2, and SW 1990 cell lines Mouse xenograft model | Amplified cell death and lipid peroxidation, GPx4 autophagy | [120] |
| Zinc oxide nanoparticles | HUVECs and EA.hy926 cells | Macroautophagy/autophagy-dependent NCOA4-mediated ferroptosis | [124] |
| Bioinorganic Modulator | Research Model | Outcome | Ref. |
|---|---|---|---|
| Arsenite | Neuronal-like PC-12 Adh cells C57BL/6J mice | Decreased levels of ferritin and NCOA4, increased autophagy marker LC3B and lipid peroxidation, elevated iron content, SLC7A11 and GPX4 expression declined Neuronal ferroptotic cell death in the hippocampus via activation of ferritinophagy, pathological changes in the mitochondria | [142] |
| Arsenite | C57BL/6J mice GC-2spd cells | Pathological changes in mouse testis, reduced number of sperm, mitochondrial oxidative damage Reduced cell viability, accumulation of iron, production of reactive oxygen species, lipid peroxidation, reduced GPX4 and SCL7A11 expression | [144] |
| Arsenic trioxide | SK-N-BE(2) neuroblastoma cell line | Inhibited proliferation, reduced viability, ROS and iron accumulation, ferritinophagy, downregulated GPx4 | [151] |
| Arsenic trioxide | Rat cardiomyocyte H9c2 cells | Reduced viability, IC50 20.7 μM production of ROS, loss of the mitochondrial membrane potential, distortion and enlargement of myocardial mitochondria, hyperactive ER stress, impaired autophagy, accumulation of MDA, depletion of GPx-4 | [152] |
| Arsenic trioxide +olaparib | Platinum-resistant A2780-CIS and SKOV3-CIS cell lines BALB/c mice | Suppressed cell proliferation and loss of viability indicating synergetic effects, increased lipid peroxidation, significant reduction of formed colonies, increased MDA, decreased SLC7A11and GPx4 levels, ferroptotic cell death and suppressed tumor growth | [153] |
| Mercuric chloride | Chicken embryo kidney cells | Ferroptosis followed by necroptosis, ferritinophagy, ROS generation, iron overload | [155] |
| Mercuric chloride | Common carp | Mitochondrial defects, increased levels of iron and MDA, depleted GSH, Brain injury and memory loss, gut microbiota disorders | [156] |
| Lead acetate | C57 mice HT22 hippocampal neuronal cell line | Decreased cell viability related to Pb-promoted iron overload, accumulation of ROS and MDA, depleted GSH, downregulated expression of SLC7A11 Ferroptosis reported in brain tissues (hippocampus, cortex, hypothalamus, striatum) | [158] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartos, A.; Sikora, J. Bioinorganic Modulators of Ferroptosis: A Review of Recent Findings. Int. J. Mol. Sci. 2023, 24, 3634. https://doi.org/10.3390/ijms24043634
Bartos A, Sikora J. Bioinorganic Modulators of Ferroptosis: A Review of Recent Findings. International Journal of Molecular Sciences. 2023; 24(4):3634. https://doi.org/10.3390/ijms24043634
Chicago/Turabian StyleBartos, Adrian, and Joanna Sikora. 2023. "Bioinorganic Modulators of Ferroptosis: A Review of Recent Findings" International Journal of Molecular Sciences 24, no. 4: 3634. https://doi.org/10.3390/ijms24043634
APA StyleBartos, A., & Sikora, J. (2023). Bioinorganic Modulators of Ferroptosis: A Review of Recent Findings. International Journal of Molecular Sciences, 24(4), 3634. https://doi.org/10.3390/ijms24043634