
Purification and Characterization of Authentic 30S Ribosomal Precursors Induced by Heat Shock

Abstract
1. Introduction
2. Results
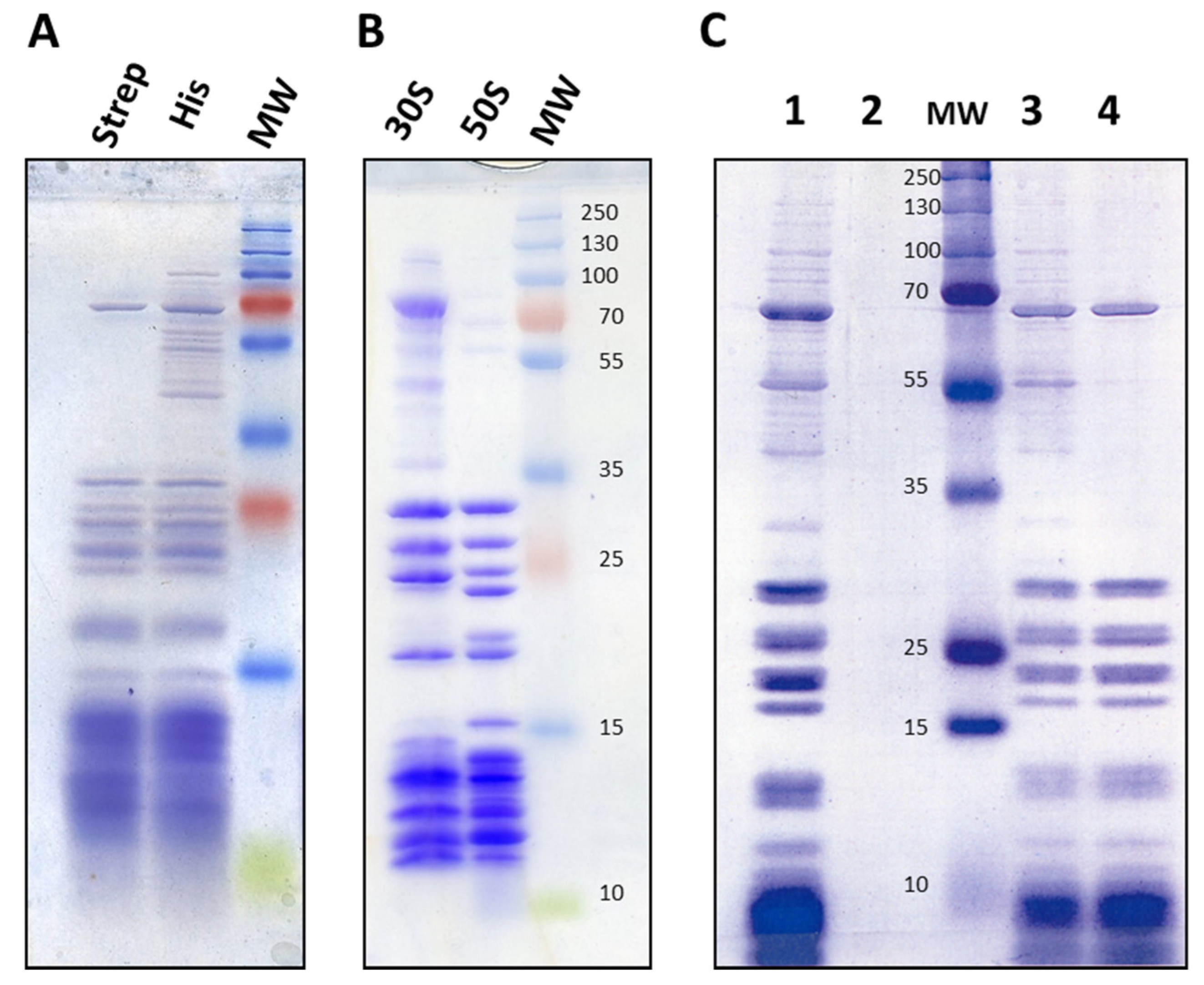
2.1. Introduction of His and Strep Affinity Tags onto 30S Ribosomal Proteins
2.2. Ribosome Synthesis at 45 °C
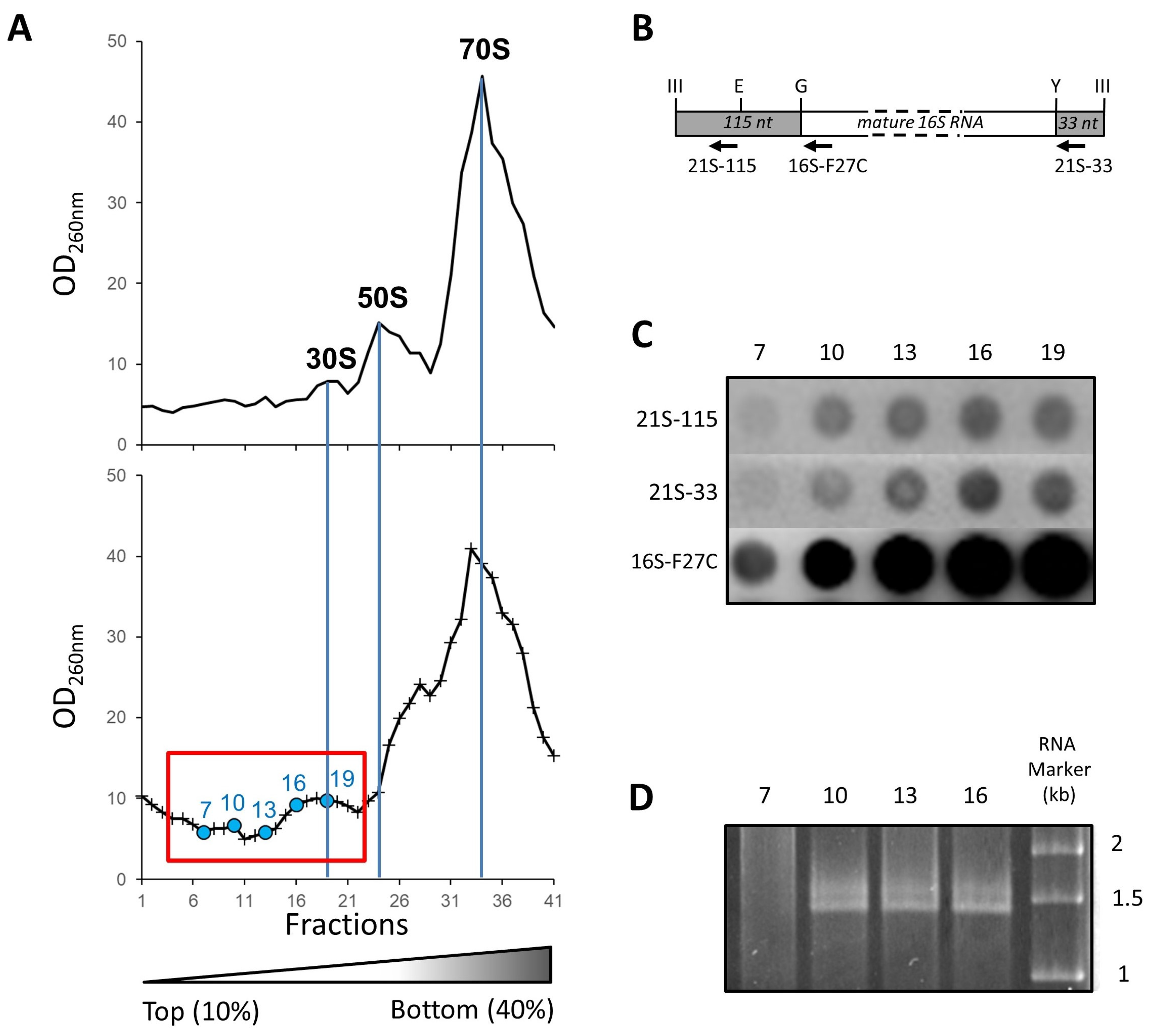
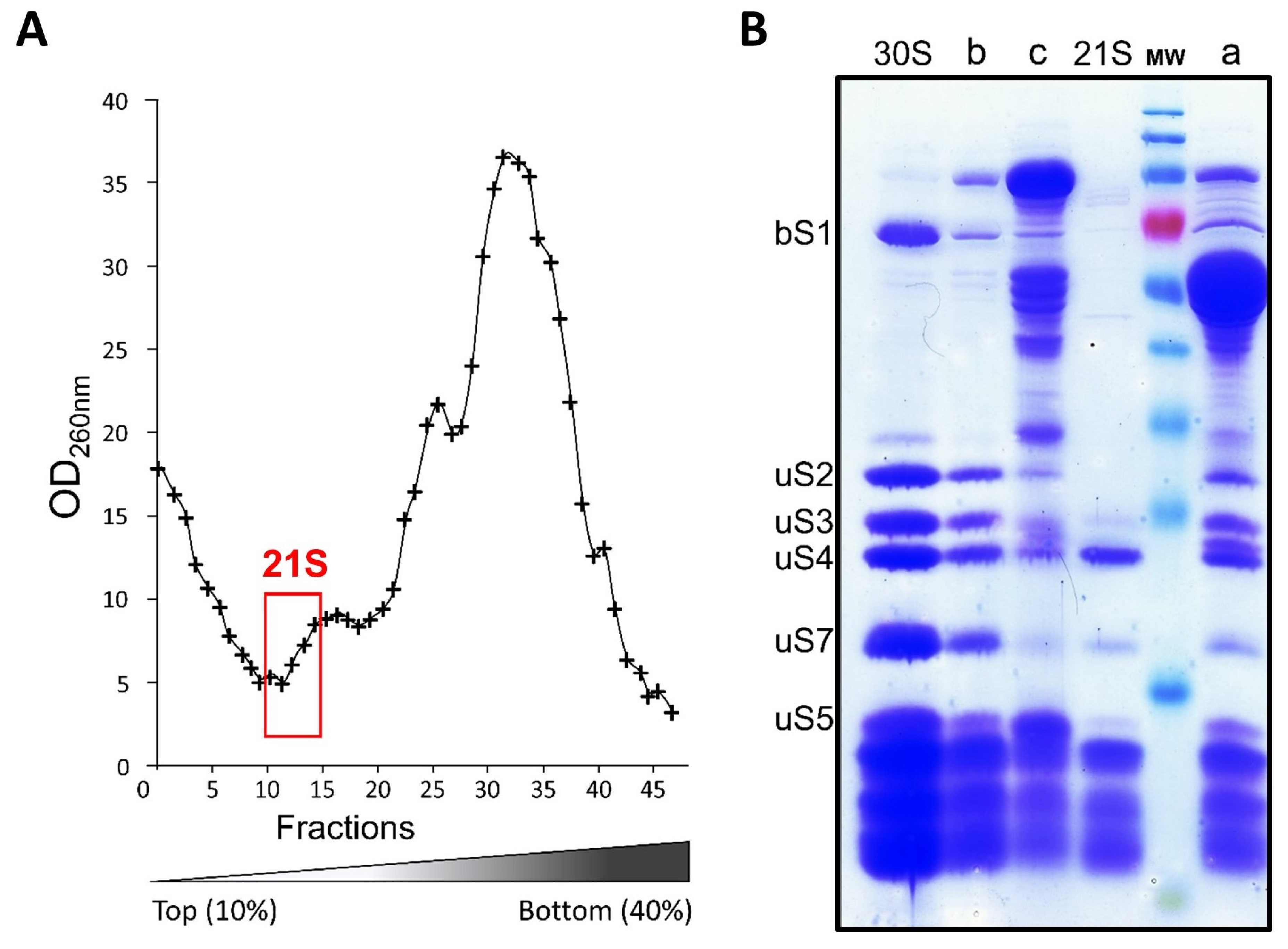
2.3. Isolation and Affinity Purification of Tagged 21S Intermediates
2.4. Composition of 21S Proteins
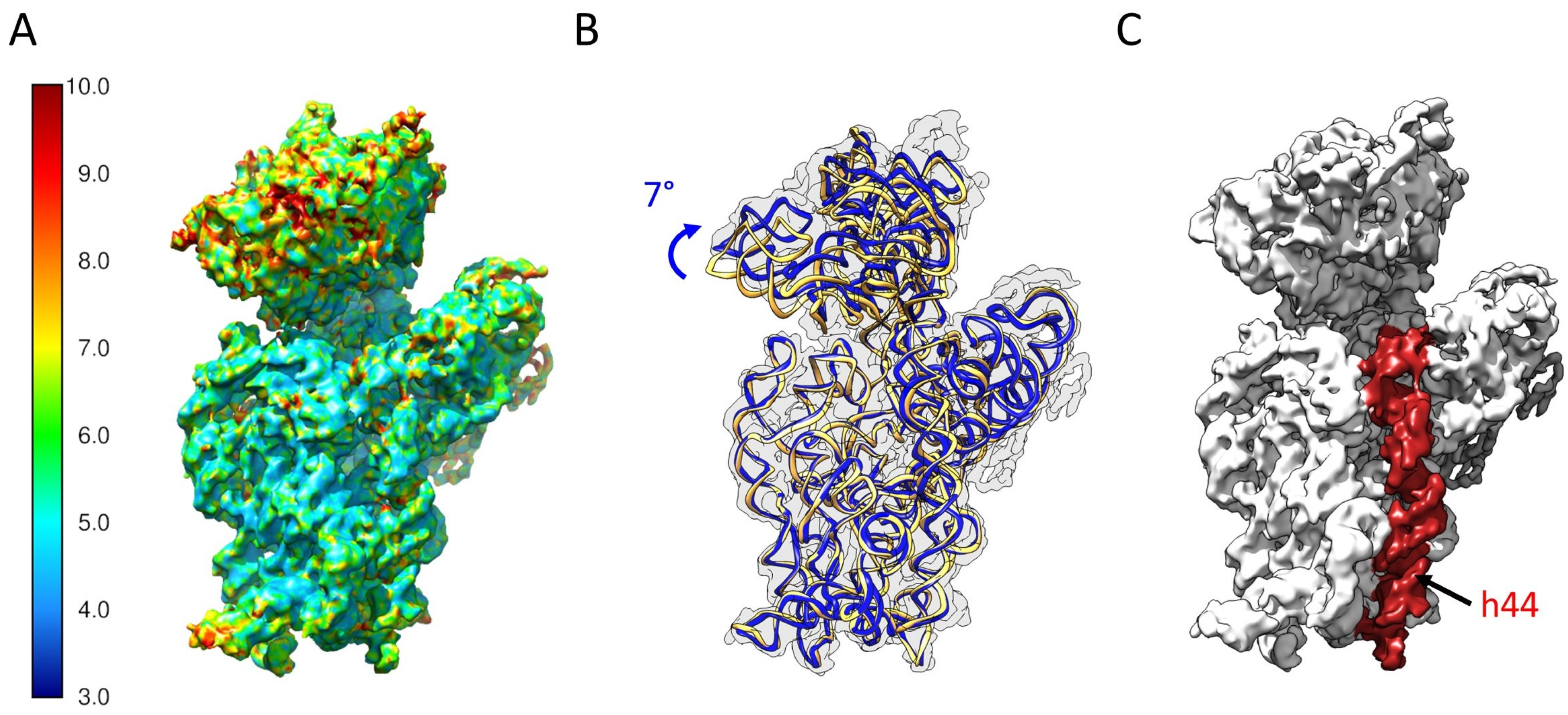
2.5. Determination of the Structure of 21S
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Growth Media, and Genetic Procedures
4.2. Tagging of uS2 and bS20
4.3. Cloning of rpsT:Strep
4.4. Purification of 30S Precursors
4.5. RNA Experiments
4.6. Tandem Mass Spectrometry (MS/MS) Analysis
4.7. Protein Identification Using Liquid Chromatography with Tandem Mass Spectrometry (LC-MS/MS)
4.7.1. Liquid Digestion of Ribosomal Samples
4.7.2. LC-MS/MS Analysis
4.7.3. Cryo-Electron Microscopy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schwarz, E.; Lilie, H.; Rudolph, R. The effect of molecular chaperones on in vivo and in vitro folding processes. Biol. Chem. 1996, 377, 411–416. [Google Scholar] [PubMed]
- Roghanian, M.; Van Nerom, K.; Takada, H.; Caballero-Montes, J.; Tamman, H.; Kudrin, P.; Talavera, A.; Dzhygyr, I.; Ekström, S.; Atkinson, G.C.; et al. (p)ppGpp controls stringent factors by exploiting antagonistic allosteric coupling between catalytic domains. Mol. Cell. 2021, 81, 3310–3322.e6. [Google Scholar] [CrossRef] [PubMed]
- VanBogelen, R.A.; Neidhardt, F.C. Ribosomes as sensors of heat and cold shock in Escherichia coli. Proc. Natl. Acad. Sci. USA 1990, 87, 5589–5593. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, G.C.; Tenson, T.; Hauryliuk, V. The RelA/SpoT Homolog (RSH) Superfamily: Distribution and Functional Evolution of ppGpp Synthetases and Hydrolases across the Tree of Life. PLoS ONE 2011, 6, e23479. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, H.; Beckert, B.; Frese, C.K.; Steinchen, W.; Nuss, A.M.; Beckstette, M.; Hantke, I.; Driller, K.; Sudzinová, P.; Krásný, L.; et al. The alarmones (p)ppGpp are part of the heat shock response of Bacillus subtilis. PLoS Genet. 2020, 16, e1008275. [Google Scholar] [CrossRef]
- Shajani, Z.; Sykes, M.T.; Williamson, J.R. Assembly of Bacterial Ribosomes. Annu. Rev. Biochem. 2011, 80, 501–526. [Google Scholar] [CrossRef]
- Chen, S.S.; Williamson, J.R. Characterization of the ribosome biogenesis landscape in E. coli using quantitative mass spectrometry. J. Mol. Biol. 2013, 425, 767–779. [Google Scholar] [CrossRef]
- Bunner, A.E.; Nord, S.; Wikström, P.M.; Williamson, J.R. The Effect of Ribosome Assembly Cofactors on In Vitro 30S Subunit Reconstitution. J. Mol. Biol. 2010, 398, 1–7. [Google Scholar] [CrossRef]
- Jomaa, A.; Stewart, G.; Martín-Benito, J.; Zielke, R.; Campbell, T.L.; Maddock, J.R.; Brown, E.D.; Ortega, J. Understanding ribosome assembly: The structure of in vivo assembled immature 30S subunits revealed by cryo-electron microscopy. RNA 2011, 17, 697–709. [Google Scholar] [CrossRef]
- Clatterbuck Soper, S.F.; Dator, R.P.; Limbach, P.A.; Woodson, S.A. In vivo X-ray footprinting of pre-30S ribosomes reveals chaperone-dependent remodeling of late assembly intermediates. Mol. Cell. 2013, 52, 506–516. [Google Scholar] [CrossRef]
- Guo, Q.; Goto, S.; Chen, Y.; Feng, B.; Xu, Y.; Muto, A.; Himeno, H.; Deng, H.; Lei, J.; Gao, N. Dissecting the in vivo assembly of the 30S ribosomal subunit reveals the role of RimM and general features of the assembly process. Nucleic Acids Res. 2013, 41, 2609–2620. [Google Scholar] [CrossRef]
- Leong, V.; Kent, M.; Jomaa, A.; Ortega, J. Escherichia coli rimM and yjeQ null strains accumulate immature 30S subunits of similar structure and protein complement. RNA 2013, 19, 789–802. [Google Scholar] [CrossRef]
- Yang, Z.; Guo, Q.; Goto, S.; Chen, Y.; Li, N.; Yan, K.; Zhang, Y.; Muto, A.; Deng, H.; Himeno, H.; et al. Structural insights into the assembly of the 30S ribosomal subunit in vivo: Functional role of S5 and location of the 17S rRNA precursor sequence. Protein Cell 2014, 5, 394–407. [Google Scholar] [CrossRef]
- Razi, A.; Davis, J.H.; Hao, Y.; Jahagirdar, D.; Thurlow, B.; Basu, K.; Jain, N.; Gomez-Blanco, J.; A Britton, R.; Vargas, J.; et al. Role of Era in assembly and homeostasis of the ribosomal small subunit. Nucleic Acids Res. 2019, 47, 8301–8317. [Google Scholar] [CrossRef]
- Maksimova, E.M.; Korepanov, A.P.; Kravchenko, O.V.; Baymukhametov, T.N.; Myasnikov, A.G.; Vassilenko, K.S.; Afonina, Z.A.; Stolboushkina, E.A. RbfA Is Involved in Two Important Stages of 30S Subunit Assembly: Formation of the Central Pseudoknot and Docking of Helix 44 to the Decoding Center. Int. J. Mol. Sci. 2021, 22, 6140. [Google Scholar] [CrossRef]
- Thurlow, B.; Davis, J.; Leong, V.; Moraes, T.F.; Williamson, J.; Ortega, J. Binding properties of YjeQ (RsgA), RbfA, RimM and Era to assembly intermediates of the 30S subunit. Nucleic Acids Res. 2016, 44, 9918–9932. [Google Scholar] [CrossRef]
- Gupta, N.; Culver, G.M. Multiple in vivo pathways for Escherichia coli small ribosomal subunit assembly occur on one pre-rRNA. Nat. Struct. Mol. Biol. 2014, 21, 937–943. [Google Scholar] [CrossRef]
- Maki, J.A.; Schnobrich, D.J.; Culver, G.M. The DnaK Chaperone System Facilitates 30S Ribosomal Subunit Assembly. Mol. Cell 2002, 10, 129–138. [Google Scholar] [CrossRef]
- Maki, J.A.; Southworth, D.R.; Culver, G.M. Demonstration of the role of the DnaK chaperone system in assembly of 30S ribosomal subunits using a purified in vitro system. RNA 2003, 9, 1418–1421. [Google Scholar] [CrossRef]
- Al Refaii, A.; Alix, J.H. Ribosome biogenesis is temperature-dependent and delayed in Escherichia coli lacking the chaperones DnaK or DnaJ. Mol. Microbiol. 2009, 71, 748–762. [Google Scholar] [CrossRef]
- El Hage, A.; Sbaï, M.; Alix, J.-H. The chaperonin GroEL and other heat-shock proteins, besides DnaK, participate in ribosome biogenesis in Escherichia coli. Mol. Gen. Genet. 2001, 264, 796–808. [Google Scholar] [CrossRef] [PubMed]
- René, O.; Alix, J.-H. Late steps of ribosome assembly in E. coli are sensitive to a severe heat stress but are assisted by the HSP70 chaperone machine. Nucleic Acids Res. 2011, 39, 1855–1867. [Google Scholar] [CrossRef] [PubMed]
- Nierhaus, K. The assembly of prokaryotic ribosomes. Biochimie 1991, 73, 739–755. [Google Scholar] [CrossRef] [PubMed]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [PubMed]
- Parsons, G.D.; A Mackie, G. Expression of the gene for ribosomal protein S20: Effects of gene dosage. J. Bacteriol. 1983, 154, 152–160. [Google Scholar] [CrossRef]
- Ratkowsky, D.A.; Lowry, R.K.; A McMeekin, T.; Stokes, A.N.; E Chandler, R. Model for bacterial culture growth rate throughout the entire biokinetic temperature range. J. Bacteriol. 1983, 154, 1222–1226. [Google Scholar] [CrossRef]
- Jacob, A.I.; Köhrer, C.; Davies, B.W.; RajBhandary, U.L.; Walker, G.C. Conserved Bacterial RNase YbeY Plays Key Roles in 70S Ribosome Quality Control and 16S rRNA Maturation. Mol. Cell 2013, 49, 427–438. [Google Scholar] [CrossRef]
- Delvillani, F.; Papiani, G.; Dehò, G.; Briani, F. S1 ribosomal protein and the interplay between translation and mRNA decay. Nucleic Acids Res. 2011, 39, 7702–7715. [Google Scholar] [CrossRef]
- Rabuck-Gibbons, J.N.; Popova, A.M.; Greene, E.M.; Cervantes, C.F.; Lyumkis, D.; Williamson, J.R. SrmB Rescues Trapped Ribosome Assembly Intermediates. J. Mol. Biol. 2020, 432, 978–990. [Google Scholar] [CrossRef]
- Napper, N.; Culver, G.M. Analysis of r-protein and RNA conformation of 30S subunit intermediates in bacteria. RNA 2015, 21, 1323–1334. [Google Scholar] [CrossRef]
- Duss, O.; Stepanyuk, G.A.; Puglisi, J.D.; Williamson, J.R. Transient Protein-RNA Interactions Guide Nascent Ribosomal RNA Folding. Cell 2019, 179, 1357–1369.e16. [Google Scholar] [CrossRef]
- Guo, Q.; Yuan, Y.; Xu, Y.; Feng, B.; Liu, L.; Chen, K.; Sun, M.; Yang, Z.; Lei, J.; Gao, N. Structural basis for the function of a small GTPase RsgA on the 30S ribosomal subunit maturation revealed by cryoelectron microscopy. Proc. Natl. Acad. Sci. USA 2011, 108, 13100–13105. [Google Scholar] [CrossRef]
- López-Alonso, J.P.; Kaminishi, T.; Kikuchi, T.; Hirata, Y.; Iturrioz, I.; Dhimole, N.; Schedlbauer, A.; Hase, Y.; Goto, S.; Kurita, D.; et al. RsgA couples the maturation state of the 30S ribosomal decoding center to activation of its GTPase pocket. Nucleic Acids Res. 2017, 45, 6945–6959. [Google Scholar] [CrossRef]
- Razi, A.; Guarné, A.; Ortega, J. The cryo-EM structure of YjeQ bound to the 30S subunit suggests a fidelity checkpoint function for this protein in ribosome assembly. Proc. Natl. Acad. Sci. USA 2017, 114, E3396–E3403. [Google Scholar] [CrossRef]
- Bergholz, T.M.; Wick, L.M.; Qi, W.; Riordan, J.T.; Ouellette, L.M.; Whittam, T.S. Global transcriptional response of Escherichia coli O157:H7 to growth transitions in glucose minimal medium. BMC Microbiol. 2007, 7, 97. [Google Scholar] [CrossRef]
- Cho, B.-K.; Zengler, K.; Qiu, Y.; Park, Y.S.; Knight, E.M.; Barrett, C.L.; Gao, Y.; Palsson, B. The transcription unit architecture of the Escherichia coli genome. Nat. Biotechnol. 2009, 27, 1043–1049. [Google Scholar] [CrossRef]
- Jozefczuk, S.; Klie, S.; Catchpole, G.; Szymanski, J.; Cuadros-Inostroza, A.; Steinhauser, D.; Selbig, J.; Willmitzer, L. Metabolomic and transcriptomic stress response of Escherichia coli. Mol. Syst. Biol. 2010, 6, 364. [Google Scholar] [CrossRef]
- Murata, M.; Fujimoto, H.; Nishimura, K.; Charoensuk, K.; Nagamitsu, H.; Raina, S.; Kosaka, T.; Oshima, T.; Ogasawara, N.; Yamada, M. Molecular Strategy for Survival at a Critical High Temperature in Eschierichia coli. PLoS ONE 2011, 6, e20063. [Google Scholar] [CrossRef]
- Lemke, J.J.; Sanchez-Vazquez, P.; Burgos, H.L.; Hedberg, G.; Ross, W.; Gourse, R.L. Direct regulation of Escherichia coli ribosomal protein promoters by the transcription factors ppGpp and DksA. Proc. Natl. Acad. Sci. USA 2011, 108, 5712–5717. [Google Scholar] [CrossRef]
- Duin, J.; Wijnands, R. The Function of Ribosomal Protein S21 in Protein Synthesis. JBIC J. Biol. Inorg. Chem. 1981, 118, 615–619. [Google Scholar] [CrossRef]
- El Hage, A.; Alix, J.-H. Authentic precursors to ribosomal subunits accumulate in Escherichia coli in the absence of functional DnaK chaperone. Mol. Microbiol. 2004, 51, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Mogk, A.; Homuth, G.; Scholz, C.; Kim, L.; Schmid, F.X.; Schumann, W. The GroE chaperonin machine is a major modulator of the CIRCE heat shock regulon of Bacillus subtilis. EMBO J. 1997, 16, 4579–4590. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.L.; Wilson, A.C.; Tan, M. A Chlamydia-Specific C-Terminal Region of the Stress Response Regulator HrcA Modulates Its Repressor Activity. J. Bacteriol. 2011, 193, 6733–6741. [Google Scholar] [CrossRef]
- Bandyopadhyay, B.; Das Gupta, T.; Roy, D.; Das Gupta, S.K. DnaK Dependence of the Mycobacterial Stress-Responsive Regulator HspR Is Mediated through Its Hydrophobic C-Terminal Tail. J. Bacteriol. 2012, 194, 4688–4697. [Google Scholar] [CrossRef] [PubMed]
- Tomoyasu, T.; Ogura, T.; Tatsuta, T.; Bukau, B. Levels of DnaK and DnaJ provide tight control of heat shock gene expression and protein repair in Escherichia coli. Mol. Microbiol. 1998, 30, 567–581. [Google Scholar] [CrossRef]
- Miller, J.H. Experiments in Molecular Genetics; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1972. [Google Scholar]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Jensen, K.F. The Escherichia coli K-12 “wild types” W3110 and MG1655 have an rph frameshift mutation that leads to pyrimidine starvation due to low pyrE expression levels. J. Bacteriol. 1993, 175, 3401–3407. [Google Scholar] [CrossRef]
- Guzman, L.M.; Belin, D.; Carson, M.J.; Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995, 177, 4121–4130. [Google Scholar] [CrossRef]
- Punjani, A.; Rubinstein, J.L.; Fleet, D.J.; A Brubaker, M. cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 2017, 14, 290–296. [Google Scholar] [CrossRef]
- Zivanov, J.; Nakane, T.; Forsberg, B.O.; Kimanius, D.; Hagen, W.J.; Lindahl, E.; Scheres, S.H. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 2018, 7, e42166. [Google Scholar] [CrossRef]
- Rohou, A.; Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015, 192, 216–221. [Google Scholar] [CrossRef]
- Schuwirth, B.S.; Borovinskaya, M.A.; Hau, C.W.; Zhang, W.; Vila-Sanjurjo, A.; Holton, J.M.; Cate, J.H.D. Structures of the Bacterial Ribosome at 3.5 Å Resolution. Science 2005, 310, 827–834. [Google Scholar] [CrossRef]
- Mulder, A.M.; Yoshioka, C.; Beck, A.H.; Bunner, A.E.; Milligan, R.A.; Potter, C.S.; Carragher, B.; Williamson, J.R. Visualizing Ribosome Biogenesis: Parallel Assembly Pathways for the 30 S Subunit. Science 2010, 330, 673–677. [Google Scholar] [CrossRef]
- Sashital, D.G.; A Greeman, C.; Lyumkis, D.; Potter, C.S.; Carragher, B.; Williamson, J.R. A combined quantitative mass spectrometry and electron microscopy analysis of ribosomal 30S subunit assembly in E. coli. eLife 2014, 3, e04491. [Google Scholar] [CrossRef]
- Kucukelbir, A.; Sigworth, F.J.; Tagare, H.D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 2014, 11, 63–65. [Google Scholar] [CrossRef]
- Nakane, T.; Scheres, S.H.W. Multi-body Refinement of Cryo-EM Images in RELION. Methods Mol. Biol. 2021, 2215, 145–160. [Google Scholar] [CrossRef]
- Afonine, P.V.; Poon, B.K.; Read, R.J.; Sobolev, O.V.; Terwilliger, T.C.; Urzhumtsev, A.; Adams, P.D. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 2018, 74, 531–544. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Noeske, J.; Wasserman, M.R.; Terry, D.S.; Altman, R.B.; Blanchard, S.C.; Cate, J.H. High-resolution structure of the Escherichia coli ribosome. Nat Struct Mol Biol. 2015, 2, 336–341. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | ||
|---|---|---|
| MG1655 | F− λ− rph 1 | CGSC 6300 [48] |
| E5990 | MG1655 rpsB:his-KanR | This study |
| E6001 | MG1655 rpsB:his | This study |
| E5998 | MG1655 rpsT:strep-kanR | This study |
| E6004 | MG1655 rpsT:strep | This study |
| E6006 | MG1655 rpsB his rpsT strep | This study |
| Plasmids | ||
| pKD46 | repA101(Ts) araBp-gam-bet-exo ori101 AmpR araC | [24] |
| pKD4 | oriR6Kgamma AmpR rgnB KanR flanked by FRT | [24] |
| pCP20 | Rep Ts CI857 AmpR CmR FLP | [24] |
| pBAD24 | oriColE1 araC araBp | [49] |
| pE7867 | pBAD24 rpsT:strep | This study |
| Type | Sequence | Experiment |
|---|---|---|
| rpsTREPF | GCACGTCATAAGGCTAACCTGACTGCACAGATCAACAAACTGGCTTGGAGCCACCCGCAGTTCGAAAAGTAAGTGTAGGCTGGAGCTGCTTC | rpsT strep construction |
| rpsTREPR | AACCCGCTTGCGCGGGCTTTTTCACAAAGCTTCAGCAAATTGGCGAATGGGAATTAGCCATGGTCC | |
| rpsBHISF | CAGGATCTGGCTTCCCAGGCGGAAGAAAGCTTCGTAGAAGCTGAGCACCACCACCACCACCACTAA GTGTAGGCTGGAGCTGCTTC | rpsB his construction |
| rpsFR | GCCTTTCTGCAACTCGAACTATTTTGGGGGAGTTATCAAGCCTTAATGGGAATTAGCCATGGTCC | |
| pkd4F | AAGCCATCCAGTTTACTTTGC | KanR insertion verification |
| pkd4D | CGCATCGCCTTCTATCGCC | |
| rpsBD | CGTTCTCAGGATCTGGCTTC | rpsT mutation Verification |
| rpsBF | CTCGGAGATGTGATCTGCC | |
| rpsTD | GTAAGCACAACGCAAGCCG | rpsT mutation Verification |
| rpsTF | ACAGAAGCCACTGGAGCAC | |
| rpsTA | TTTTCCATGGCTAATATCAAATCAGCTAAGAAGCGC | cloning of rpsT ORF |
| 21S-115 | 5′ Biotin-TEG CATTTTTCGTGTTGCGACG | detection of 17S |
| 21S-33 | 5′ Biotin-TEG CAAAGAACGCTTCTTTAAG | |
| 16S-F27C | 5′ Biotin-TEG CTGAGCCATGATCAAACTCT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giudice, E.; Georgeault, S.; Lavigne, R.; Pineau, C.; Trautwetter, A.; Ermel, G.; Blanco, C.; Gillet, R. Purification and Characterization of Authentic 30S Ribosomal Precursors Induced by Heat Shock. Int. J. Mol. Sci. 2023, 24, 3491. https://doi.org/10.3390/ijms24043491
Giudice E, Georgeault S, Lavigne R, Pineau C, Trautwetter A, Ermel G, Blanco C, Gillet R. Purification and Characterization of Authentic 30S Ribosomal Precursors Induced by Heat Shock. International Journal of Molecular Sciences. 2023; 24(4):3491. https://doi.org/10.3390/ijms24043491
Chicago/Turabian StyleGiudice, Emmanuel, Sylvie Georgeault, Régis Lavigne, Charles Pineau, Annie Trautwetter, Gwennola Ermel, Carlos Blanco, and Reynald Gillet. 2023. "Purification and Characterization of Authentic 30S Ribosomal Precursors Induced by Heat Shock" International Journal of Molecular Sciences 24, no. 4: 3491. https://doi.org/10.3390/ijms24043491
APA StyleGiudice, E., Georgeault, S., Lavigne, R., Pineau, C., Trautwetter, A., Ermel, G., Blanco, C., & Gillet, R. (2023). Purification and Characterization of Authentic 30S Ribosomal Precursors Induced by Heat Shock. International Journal of Molecular Sciences, 24(4), 3491. https://doi.org/10.3390/ijms24043491