
Thymosin Beta 4 Inhibits LPS and ATP-Induced Hepatic Stellate Cells via the Regulation of Multiple Signaling Pathways


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Inhibitory Effect of Tβ4 on Cellular Viability of RAW264.7 and LX-2 Cells
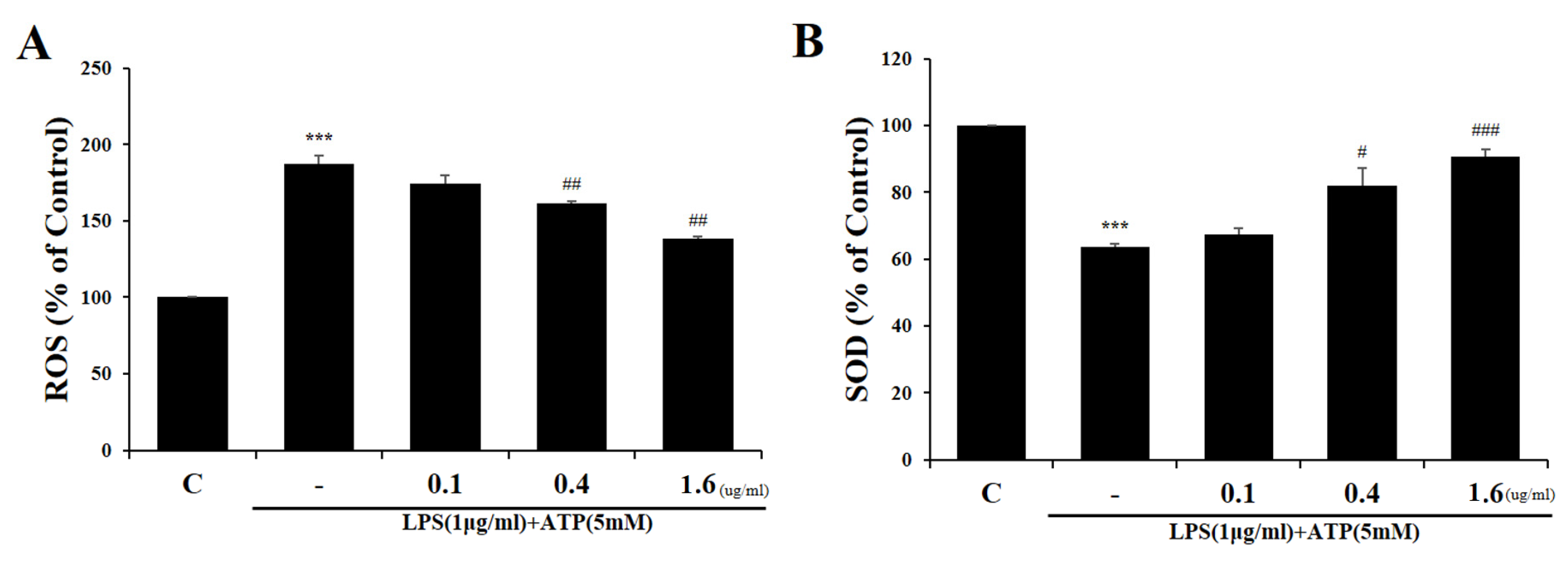
2.2. Tβ4 Suppresses ROS Prudction Induced by Activation Signals
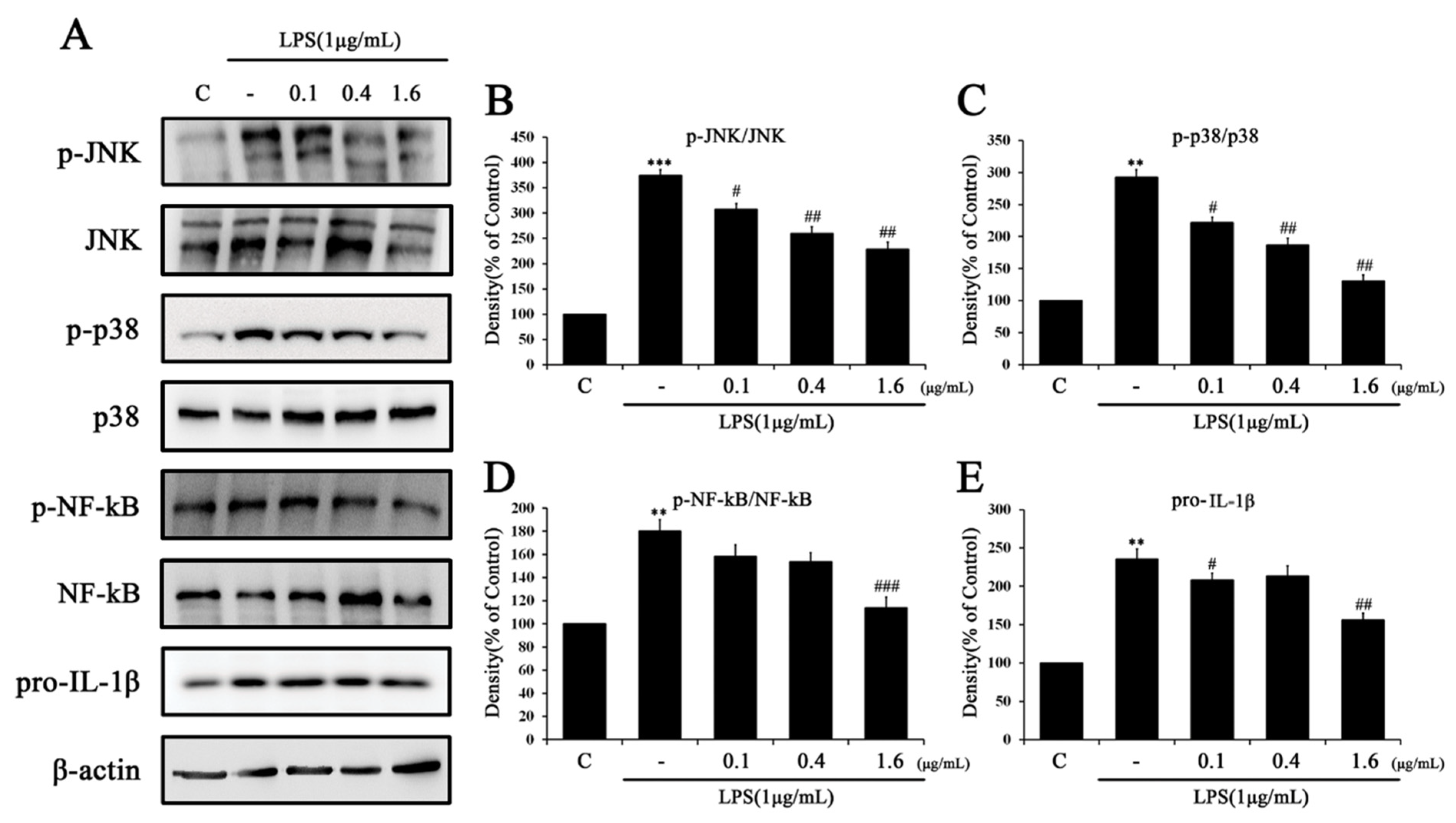
2.3. Tβ4 Regulates the Priming Signal of p38 MAPK Signaling Pathway
2.4. Tβ4 Improves Autophagy Associated with NLRP3 Inflammasome Inhibition
2.5. Tβ4 Regulates the Activation Signal of the NLRP3 Inflammasome
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture and Treatment
4.3. Measurement of Cell Viability
4.4. Intracellular ROS Assay
4.5. Superoxide Dismutase (SOD) Activity Assay
4.6. Preparation of Cellular Extracts and Western Blot Analysis
4.7. IL-1β Concentration Analysis in RAW264.7 Cells
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, L.; Wang, Y.; Quan, J. Exosomal miR-223 derived from natural killer cells inhibits hepatic stellate cell activation by suppressing autophagy. Mol. Med. 2020, 26, 81. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Aspects Med. 2019, 65, 37–55. [Google Scholar] [CrossRef]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis-A common pathway to organ injury and failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, F.; Xiong, X.; Lu, C.; Lian, N.; Lu, Y.; Zheng, S. Etramethylpyrazine reduces inflammation in liver fibrosis and inhibits inflammatory cytokine expression in hepatic stellate cells by modulating NLRP3 inflammasome pathway. IUBMB Life 2015, 67, 312–321. [Google Scholar] [CrossRef]
- Czaja, A.J.; Carpenter, H.A. Progressive fibrosis during corticosteroid therapy of autoimmune hepatitis. Hepatology 2004, 39, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Friedman, S.L. Hepatic fibrogenesis. Semin. Liver Dis. 2007, 27, 413–426. [Google Scholar] [CrossRef]
- Sasaki, R.; Devhare, P.B.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus-induced CCL5 secretion from macrophages activates hepatic stellate cells. Hepatology 2017, 66, 746–757. [Google Scholar] [CrossRef]
- Shrivastava, S.; Mukherjee, A.; Ray, R.; Ray, R.B. Hepatitis C virus induces interleukin-1β (IL-1β)/IL-18 in circulatory and resident liver macrophage. J. Virol. 2013, 87, 12284–12290. [Google Scholar] [CrossRef]
- Negash, A.A.; Ramos, H.J.; Crochet, N.; Lau, D.T.Y.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; Hagedorn, C.H.; et al. IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013, 9, e1003330. [Google Scholar] [CrossRef]
- Bansal, R.; Baarlen, J.V.; Storm, G.; Prakash, J. The interplay of the Notch signaling in hepatic stellate cells and macrophages determines the fate of liver fibrogenesis. Sci. Rep. 2015, 14, 18272. [Google Scholar] [CrossRef]
- Sica, A.; Invernizzi, P.; Mantovani, A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 2014, 59, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Shi, Z.; Zhang, M.; Dong, X.; Zheng, L.; Li, G.; Han, X.; Yao, Z.; Han, T.; Hong, H. Silencing incRNA Lfar1 alleviates the classical activation and pyoptosis of macrophage in hepatic fibrosis. Cell Death Dis. 2020, 11, 132. [Google Scholar] [CrossRef]
- Yu, Z.; Xie, X.; Su, X.; Lv, H.; Song, S.; Liu, C.; You, Y.; Tian, M.; Zhu, L.; Wang, L.; et al. TRA-meidated-crosstalk between stellate cells and Kupffer cells inhibits autophagy and promotes NLRP3 activation in acute liver injury. Cell Signal. 2022, 93, 110304. [Google Scholar] [CrossRef]
- Cai, X.; Wang, J.; Wang, J.; Zhou, Q.; Yang, B.; He, Q.; Weng, Q. Intercellular crosstalk of hepatic stellate cells in liver fibrosis: New sights into therapy. Pharacol. Res. 2020, 155, 104720. [Google Scholar] [CrossRef]
- Gaul, S.; Leszczynska, A.; Alegre, F.; Kaufmann, B.; Johnson, C.D.; Adams, L.A.; Wree, A.; Damm, G.; Seehofer, D.; Calvente, C.J.; et al. Hepatocyte pyroptosis and release of inflammasome particles induced stellate cell activation and liver fibrosis. J. Hepatol. 2021, 74, 156–167. [Google Scholar] [CrossRef]
- Yu, H.; Finlay, B. The caspase-1 inflammasome: A pilot of innate immune response. Cell Host Microbe 2008, 4, 198–208. [Google Scholar] [CrossRef]
- De-Nardo, D.; De-Nardo, C.; Latz, E. New insights into mechanisms controlling the NLRP3 inflammasome and its role in lug diseases. Am. J. Pathol. 2014, 184, 42–54. [Google Scholar] [CrossRef]
- Dinarello, C. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Bauernfeind, F.; Ablasser, A.; Bartok, E.; Kim, S.; Schmid-Burgk, J.; Cavlar, T.; Hornung, V. Inflammasomes: Current understading and open questions. Cell Mol. Life Sci. 2011, 68, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Thomas, C.J.; Guarda, G.; Tschopp, J. The inflammasome: An intergrated view. Immunol. Rev. 2011, 243, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Rooyen, D.M.V.; Haczeyni, F.; Teoh, N.C.H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Reiter, F.P.; Wimmer, R.; Wottke, L.; Artmann, R.; Nagel, J.M.; Carranza, M.O.; Mayr, D.; Rust, C.; Rickert, P.; Trauner, M.; et al. Role of interleukin-1 and its antagonism of hepatic stellate cell proliferation and liver fibrosis in the Abcb4(−/−) mouse model. World J. Hepatol. 2016, 8, 401–410. [Google Scholar] [CrossRef]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradiation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay between NLRP3 inflammasome and autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef] [PubMed]
- Toshima, T.; Shirabe, K.; Fukuhara, T.; Ikegami, T.; Yoshizumi, T.; Seojima, Y.; Ikeda, T.; Okano, S.; Maehara, Y. Suppression of autophagy during liver regeneration impairs energy charge and hepatocyte senescence in mice. Hepatology 2014, 60, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Lodder, J.; Denaës, T.; Chobert, M.N.; Wan, J.H.; El-Benna, J.; Pawlotsky, J.M.; Lotersztajn, S.; Teixeira-Clerc, F. Macrophage autophagy protects against liver fibrosis in mice. Autophagy 2015, 11, 1280–1290. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Gea, V.; Hilscher, M.; Rozenfeld, R.; Lim, M.P.; Nieto, N.; Werner, S.; Devi, L.A.; Friedman, S.L. Endeplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J. Hepatol. 2013, 59, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Mallat, A.; Lodder, J.; Teixeira-Clerc, F.; Moreau, R.; Codogno, P.; Lotersztajn, S. Autophagy: A multifaceted partner in liver fibrosis. Biomed. Res. Int. 2014, 2014, 869390. [Google Scholar] [CrossRef]
- Kumar, S.; Gupta, S. Thymosin beta 4 prevents oxidative stress by targeting antioxidant and anti-apoptotic genes in cardiac fibroblasts. PLoS ONE 2011, 6, e26912. [Google Scholar] [CrossRef]
- Philp, D.; Goldstein, A.L.; Kleinman, H.K. Thymosin beta4 promotes angiogenesis, wound healing, and hair follicle development. Mech. Ageing Dev. 2004, 125, 113–115. [Google Scholar] [CrossRef]
- Sosne, G.; Chan, C.C.; Thai, K.; Kennedy, M.; Szliter, E.A.; Hazlett, L.D.; Kleinman, H.K. Thymosin beta 4 promotes corneal wound healing and modulates inflammatory mediators in vivo. Exp. Eye Res. 2001, 72, 605–608. [Google Scholar] [CrossRef]
- Han, H.J.; Kim, S.; Kwon, J. Thymosin beta 4-induced autophagy increases cholinergic signaling in PrP (106–126)-treated HT22 cells. Neurotox. Res. 2019, 36, 58–65. [Google Scholar] [CrossRef]
- Xiao, Y.; Qu, C.; Ge, W.; Wang, B.; Wu, J.; Xu, L.; Chen, Y. Depletion of thymosin β4 promotes the proliferation, migration, and activation of human hepatic stellate cells. Cell Physiol. Biochem. 2014, 34, 356–367. [Google Scholar] [CrossRef]
- Kim, J.; Wang, S.; Hyun, J.; Choi, S.S.; Cha, H.; Ock, M.; Jung, Y. Hepatic stellate cells express thymosin beta 4 in chronically damaged liver. PLoS ONE 2015, 10, e0122758. [Google Scholar] [CrossRef]
- Zhu, L.; Cheng, M.; Liu, Y.; Yao, Y.; Zhu, Z.; Zhang, B.; Mou, Q.; Cheng, Y. Thymosin-β4 inhibits proliferation and induces apoptosis of hepatic stellate cells through PI3K/AKT pathway. Oncotarget 2017, 8, 68847–68853. [Google Scholar] [CrossRef] [PubMed]
- Kma, L.; Baruah, T.J. The interplay of ROS and the PI3K/Akt pathway in autophagy regulation. Biotechnol. Appl. Biochem. 2022, 69, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, X.; Ding, Y.; Zhou, W.; Tao, L.; Lu, P.; Wang, Y.; Hu, R. Nuclear factor E2-related factor-2 negatively regulates NLRP3 inflammasome activity inhibiting reactive oxygen species-induced NLRP-3 Priming. Antioxid. Redox Signal. 2017, 26, 28–43. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, Y.; Guan, X.; Kang, X.; Wang, X.; Liu, Q.; Li, D.; Xu, H.; Tao, Z.; Deng, Y. Atorvastatin inhibits renal inflammatory response induced by calcium oxalate crystals via inhibiting the activation of TLR4/NF-κB and NLRP3 inflammasome. IUBMB Life 2020, 72, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, J.; Zhang, Q.; Zhang, D.; Xiang, F.; Jia, J.; Wei, P.; Zhang, J.; Hu, J.; Huang, Y. High glucose suppresses keratinocyte migration through the inhibition of p38 MAPK/Autophagy pathway. Front. Physiol. 2019, 10, 24. [Google Scholar] [CrossRef]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zhang, Y.; Zheng, J.H.; Li, X.; Yao, Y.L.; Wu, Y.L.; Song, S.Z.; Sun, P.; Nan, J.X.; Lian, L.H. Potentiation of hepatic stellate cell activation by extracellular ATP is dependent on P2X7R-mediated NLRP3 inflammasome activation. Pharmacol. Res. 2017, 117, 82–93. [Google Scholar] [CrossRef]
- Bein, K.; Giuseppe, M.D.; Mischler, S.E.; Ortiz, L.A.; Leikauf, G.D. LPS-treated macrophage cytokines repress surfactant protein-B in lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 2013, 49, 306–315. [Google Scholar] [CrossRef]
- Dong, Z.; Zhuang, Q.; Ning, M.; Wu, S.; Lu, L.; Wan, X. Palmitic acid stimulates NLRP3 inflammasome activation through TLR4-NF-κB signal pathway in hepatic stellate cells. Ann. Transl. Med. 2020, 8, 168. [Google Scholar] [CrossRef]
- Sziksz, E.; Pap, D.; Lippai, R.; Béres, N.J.; Szabó, A.J.; Vannay, Á. Fibrosis related inflammatory mediators: Role of the IL-10 cytokine family. Mediat. Inflamm. 2015, 2015, 764641. [Google Scholar] [CrossRef]
- Tacke, F. Functional role of intrahepatic monocyte subsets for the progression of liver inflammation and liver fibrosis in vivo. Fibrogenesis Tissue Repair 2012, 5, S27. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Lin, C.J.; Chen, K.H.; Wu, J.C.; Huang, S.H.; Wang, S.M. Macrophage activation increases the invasive properties of hepatoma cells by destabilization of the adherens junction. FEBS Lett. 2006, 580, 3042–3050. [Google Scholar] [CrossRef] [PubMed]
- Dranoff, J.A.; Ogawa, M.; Kruglov, E.A.; Gaça, M.D.A.; Sévigny, J.; Robson, S.C.; Wells, R.G. Expression of P2Y nucleotide receptors and ectonucleotidases in quiescent and activated rat hepatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G417–G424. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, R.; Tacke, F. Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg. Nutr. 2014, 3, 344–363. [Google Scholar] [PubMed]
- Chang, J.; Hisamatsu, T.; Shimamura, K.; Yoneno, K.; Adachi, M.; Naruse, H.; Igarashi, T.; Higuchi, H.; Matsuoka, K.; Kitazume, M.T.; et al. Activated hepatic stellate cells mediate the differentiation of macrophage. Hepatol. Res. 2013, 43, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jung, Y. Potential role of thymosin beta 4 in liver fibrosis. Int. J. Mol. Sci. 2015, 16, 10624–10635. [Google Scholar] [CrossRef]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and fucntions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Komatsu, M.; Ichimura, Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 2010, 584, 1374–1378. [Google Scholar] [CrossRef]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef]
- Allaire, M.; Rautou, P.E.; Codogno, P.; Lotersztajin, S. Autophagy in liver diseases: Time for translation? J. Hepatol. 2019, 70, 985–998. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhou, Y.; Li, R.; Deng, S. Macrophages produce PGE2 to promote hepatic stellate cell autophagy and liver fibrosis. Autophagy Rep. 2022, 1, 389–392. [Google Scholar] [CrossRef]
- Singh, B.N.; Kumar, D.; Shanker, S.; Srivastava, R.K. Rottlerin induces autophagy which leads to apoptotic cell death through inhibition of PI3K/Akt/mTOR pathway in human pancreatic cancer stem cells. Biochem. Pharmacol. 2012, 84, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Ritzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptor license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Saitoh, T.; Akira, S. Regulation of inflammasomes by autophagy. J. Allergy Clin. Immunol. 2016, 138, 28–36. [Google Scholar] [CrossRef]
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, NLRP3 inflammasome and auto-inflammatory/immune diseases. Clin. Exp. Rheumatol. 2016, 34, 12–16. [Google Scholar]
- Sosne, G.; Qiu, P.; Christopherson, P.L.; Wheater, M.K. Thymosin beta 4 suppression of corneal NFkappaB: A potential anti-inflammaoty pathway. Exp. Eye Res. 2007, 84, 663–669. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.; Cho, Y.; Choi, H.; Lee, S.; Han, H.; Lee, J.; Kwon, J. Thymosin Beta 4 Inhibits LPS and ATP-Induced Hepatic Stellate Cells via the Regulation of Multiple Signaling Pathways. Int. J. Mol. Sci. 2023, 24, 3439. https://doi.org/10.3390/ijms24043439
Choi J, Cho Y, Choi H, Lee S, Han H, Lee J, Kwon J. Thymosin Beta 4 Inhibits LPS and ATP-Induced Hepatic Stellate Cells via the Regulation of Multiple Signaling Pathways. International Journal of Molecular Sciences. 2023; 24(4):3439. https://doi.org/10.3390/ijms24043439
Chicago/Turabian StyleChoi, Jihye, Yunsang Cho, Hwal Choi, Sangmin Lee, Hyeju Han, Jeonghyeon Lee, and Jungkee Kwon. 2023. "Thymosin Beta 4 Inhibits LPS and ATP-Induced Hepatic Stellate Cells via the Regulation of Multiple Signaling Pathways" International Journal of Molecular Sciences 24, no. 4: 3439. https://doi.org/10.3390/ijms24043439
APA StyleChoi, J., Cho, Y., Choi, H., Lee, S., Han, H., Lee, J., & Kwon, J. (2023). Thymosin Beta 4 Inhibits LPS and ATP-Induced Hepatic Stellate Cells via the Regulation of Multiple Signaling Pathways. International Journal of Molecular Sciences, 24(4), 3439. https://doi.org/10.3390/ijms24043439