
Insulin Metabolism in Polycystic Ovary Syndrome: Secretion, Signaling, and Clearance

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Role of Insulin Resistance in PCOS
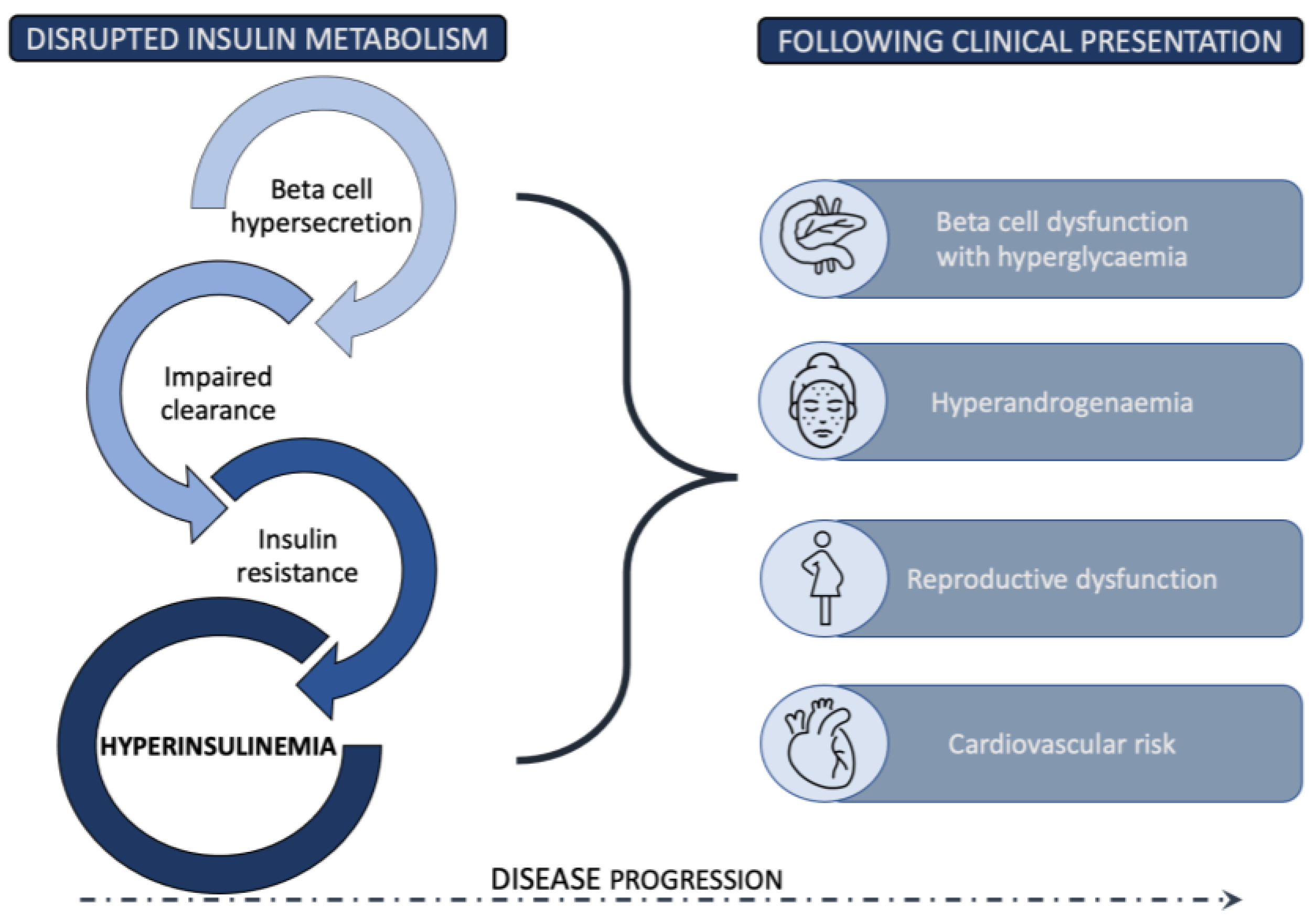
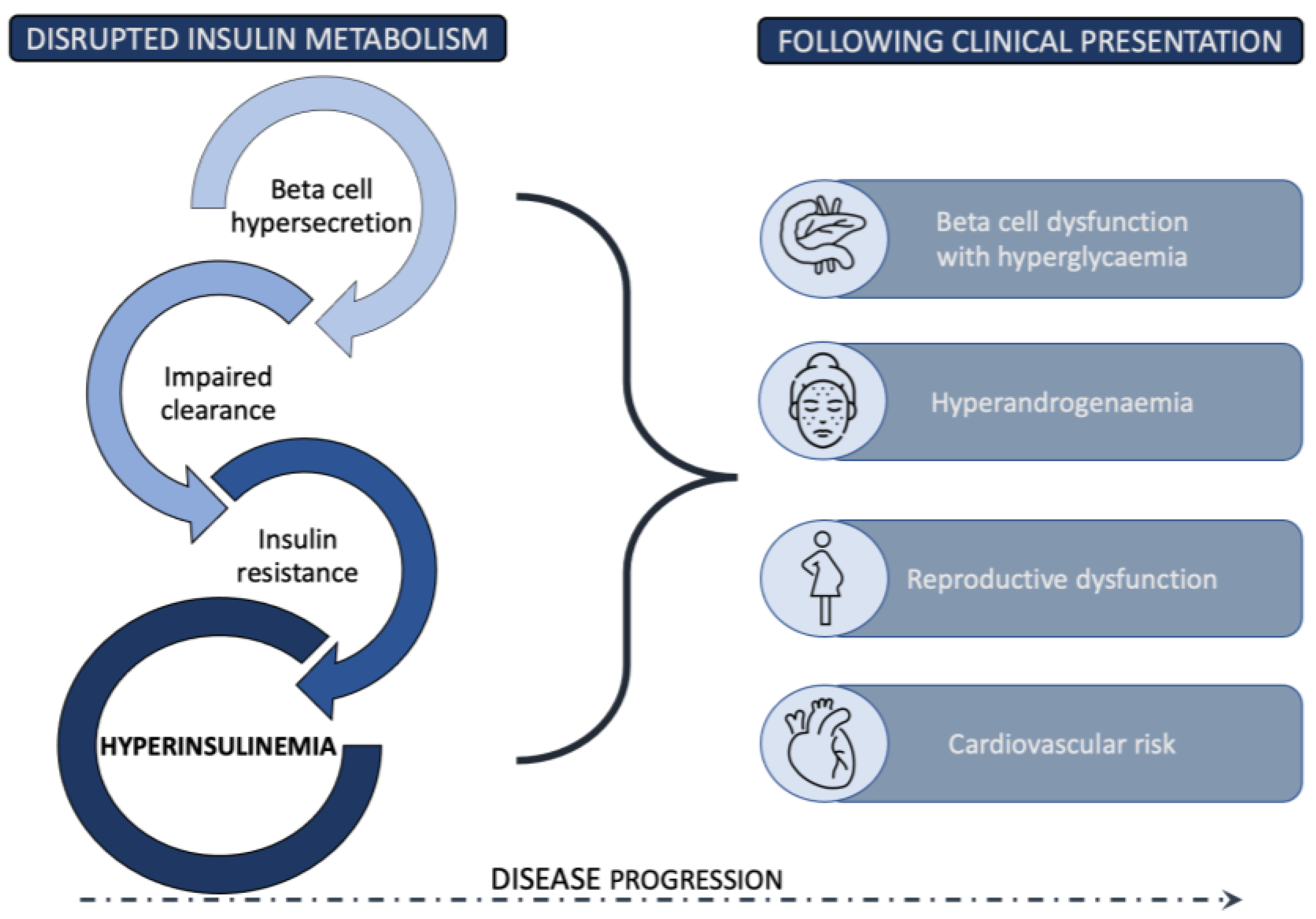
3. Pancreatic Beta Cell Function and PCOS
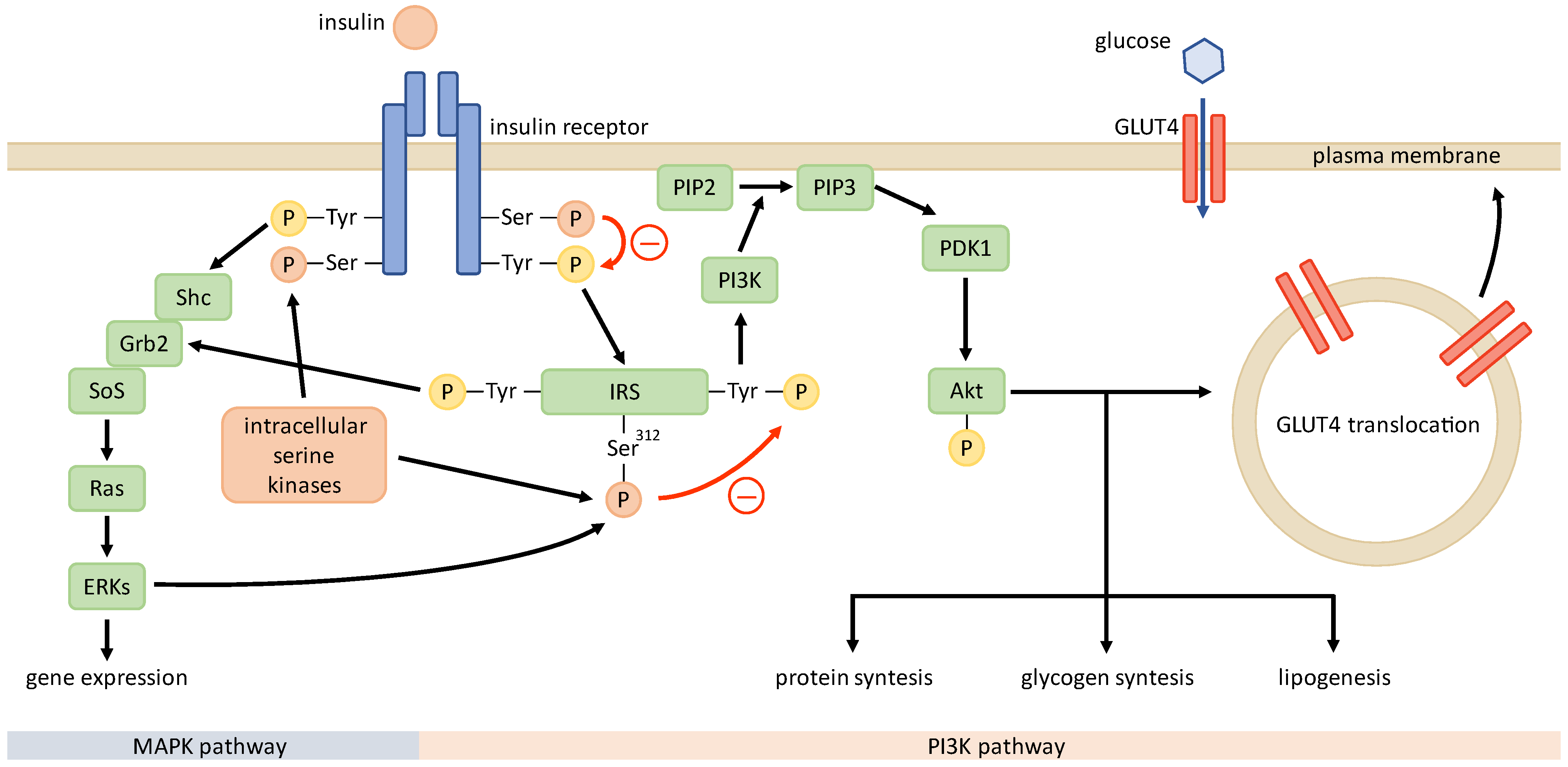
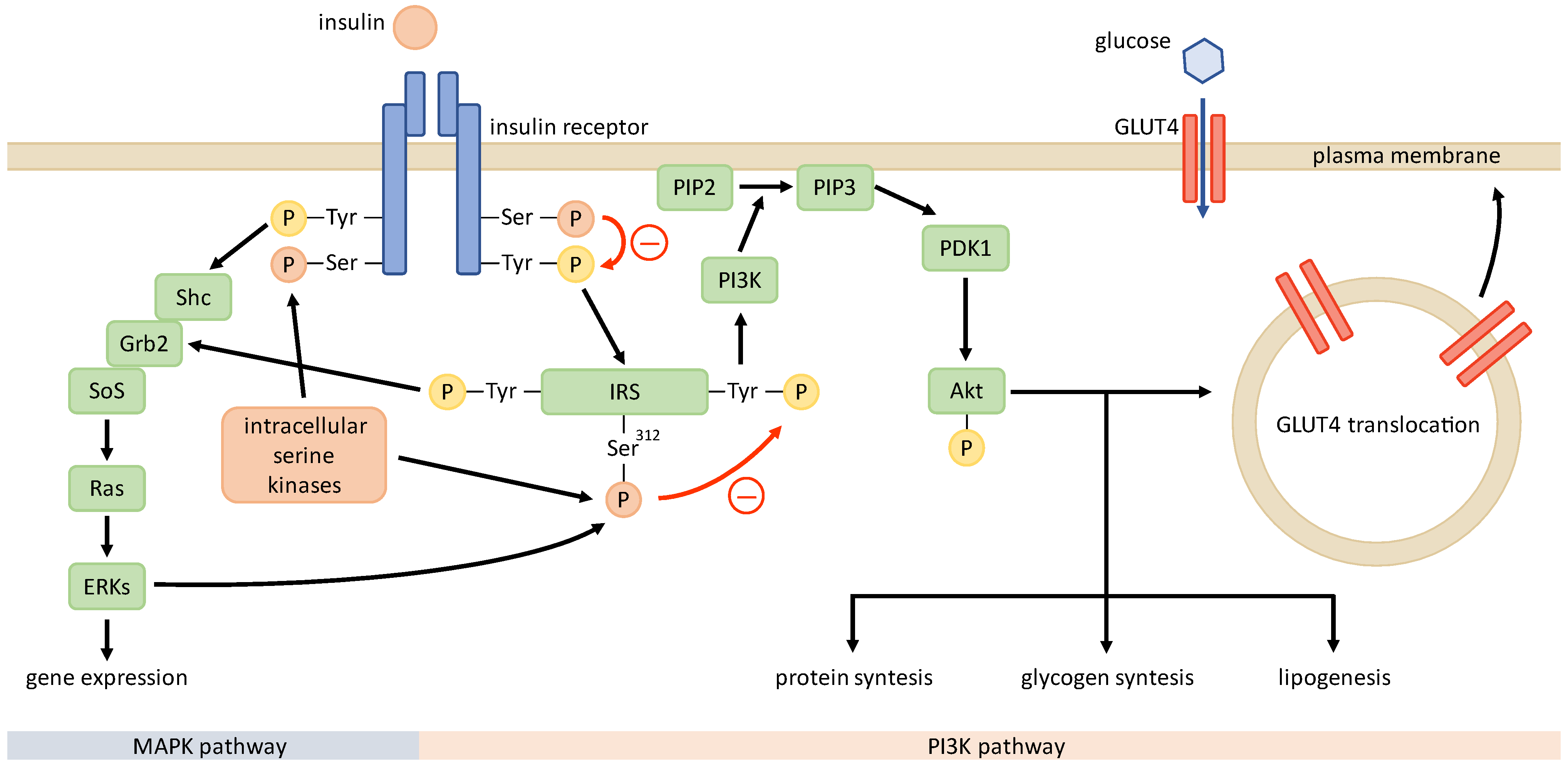
4. Mechanisms of Insulin Resistance
5. Insulin Clearance in PCOS
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Deswal, R.; Narwal, V.; Dang, A.; Pundir, C.S. The Prevalence of Polycystic Ovary Syndrome: A Brief Systematic Review. J. Hum. Reprod. Sci. 2020, 13, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Jensterle, M.; Herman, R.; Janež, A. Therapeutic Potential of Glucagon-like Peptide-1 Agonists in Polycystic Ovary Syndrome: From Current Clinical Evidence to Future Perspectives. Biomedicines 2022, 10, 1989. [Google Scholar] [CrossRef] [PubMed]
- Mumusoglu, S.; Yildiz, B.O. Polycystic Ovary Syndrome Phenotypes and Prevalence: Differential Impact of Diagnostic Criteria and Clinical versus Unselected Population. Curr. Opin. Endocr. Metab. Res. 2020, 12, 66–71. [Google Scholar] [CrossRef]
- Wang, J.; Wu, D.; Guo, H.; Li, M. Hyperandrogenemia and Insulin Resistance: The Chief Culprit of Polycystic Ovary Syndrome. Life Sci. 2019, 236, 116940. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Qiao, J. Association of Insulin Resistance and Elevated Androgen Levels with Polycystic Ovarian Syndrome (PCOS): A Review of Literature. J. Healthc. Eng. 2022, 2022, e9240569. [Google Scholar] [CrossRef] [PubMed]
- Giampaolino, P.; Foreste, V.; Di Filippo, C.; Gallo, A.; Mercorio, A.; Serafino, P.; Improda, F.P.; Verrazzo, P.; Zara, G.; Buonfantino, C.; et al. Microbiome and PCOS: State-of-Art and Future Aspects. Int. J. Mol. Sci. 2021, 22, 2048. [Google Scholar] [CrossRef]
- Burghen, G.A.; Givens, J.R.; Kitabchi, A.E. Correlation of Hyperandrogenism with Hyperinsulinism in Polycystic Ovarian Disease. J. Clin. Endocrinol. Metab. 1980, 50, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Jialal, I.; Naiker, P.; Reddi, K.; Moodley, J.; Joubert, S.M. Evidence for Insulin Resistance in Nonobese Patients with Polycystic Ovarian Disease. J. Clin. Endocrinol. Metab. 1987, 64, 1066–1069. [Google Scholar] [CrossRef]
- Barber, T.M.; Dimitriadis, G.K.; Andreou, A.; Franks, S. Polycystic Ovary Syndrome: Insight into Pathogenesis and a Common Association with Insulin Resistance. Clin. Med. Lond. Engl. 2015, 15 (Suppl. 6), s72–s76. [Google Scholar] [CrossRef]
- Azziz, R.; Carmina, E.; Chen, Z.; Dunaif, A.; Laven, J.S.E.; Legro, R.S.; Lizneva, D.; Natterson-Horowtiz, B.; Teede, H.J.; Yildiz, B.O. Polycystic Ovary Syndrome. Nat. Rev. Dis. Primer 2016, 2, 16057. [Google Scholar] [CrossRef]
- Dahan, M.H.; Reaven, G. Relationship among Obesity, Insulin Resistance, and Hyperinsulinemia in the Polycystic Ovary Syndrome. Endocrine 2019, 64, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Dunaif, A.; Segal, K.R.; Futterweit, W.; Dobrjansky, A. Profound Peripheral Insulin Resistance, Independent of Obesity, in Polycystic Ovary Syndrome. Diabetes 1989, 38, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Toprak, S.; Yönem, A.; Cakir, B.; Güler, S.; Azal, O.; Ozata, M.; Corakçi, A. Insulin Resistance in Nonobese Patients with Polycystic Ovary Syndrome. Horm. Res. 2001, 55, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Ovesen, P.; Moller, J.; Ingerslev, H.J.; Jørgensen, J.O.; Mengel, A.; Schmitz, O.; Alberti, K.G.; Moller, N. Normal Basal and Insulin-Stimulated Fuel Metabolism in Lean Women with the Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 1993, 77, 1636–1640. [Google Scholar] [CrossRef]
- Holte, J.; Bergh, T.; Berne, C.; Berglund, L.; Lithell, H. Enhanced Early Insulin Response to Glucose in Relation to Insulin Resistance in Women with Polycystic Ovary Syndrome and Normal Glucose Tolerance. J. Clin. Endocrinol. Metab. 1994, 78, 1052–1058. [Google Scholar] [CrossRef]
- Vrbíková, J.; Cibula, D.; Dvoráková, K.; Stanická, S.; Sindelka, G.; Hill, M.; Fanta, M.; Vondra, K.; Skrha, J. Insulin Sensitivity in Women with Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2004, 89, 2942–2945. [Google Scholar] [CrossRef]
- Morin-Papunen, L.C.; Vauhkonen, I.; Koivunen, R.M.; Ruokonen, A.; Tapanainen, J.S. Insulin Sensitivity, Insulin Secretion, and Metabolic and Hormonal Parameters in Healthy Women and Women with Polycystic Ovarian Syndrome. Hum. Reprod. 2000, 15, 1266–1274. [Google Scholar] [CrossRef]
- Willis, D.S.; Watson, H.; Mason, H.D.; Galea, R.; Brincat, M.; Franks, S. Premature Response to Luteinizing Hormone of Granulosa Cells from Anovulatory Women with Polycystic Ovary Syndrome: Relevance to Mechanism of Anovulation. J. Clin. Endocrinol. Metab. 1998, 83, 3984–3991. [Google Scholar] [CrossRef]
- Dunaif, A. Insulin Resistance and the Polycystic Ovary Syndrome: Mechanism and Implications for Pathogenesis. Endocr. Rev. 1997, 18, 774–800. [Google Scholar] [CrossRef]
- Yki-Järvinen, H.; Mäkimattila, S.; Utriainen, T.; Rutanen, E.M. Portal Insulin Concentrations Rather than Insulin Sensitivity Regulate Serum Sex Hormone-Binding Globulin and Insulin-like Growth Factor Binding Protein 1 in Vivo. J. Clin. Endocrinol. Metab. 1995, 80, 3227–3232. [Google Scholar] [CrossRef]
- Moghetti, P.; Tosi, F. Insulin Resistance and PCOS: Chicken or Egg? J. Endocrinol. Investig. 2021, 44, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, J.; Yan, Z.; Liu, D.; Ma, J.; Tong, N. Improvement of Insulin Sensitivity Increases Pregnancy Rate in Infertile PCOS Women: A Systemic Review. Front. Endocrinol. 2021, 12, 657889. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.C.; Dunaif, A. All Women With PCOS Should Be Treated For Insulin Resistance. Fertil. Steril. 2012, 97, 18–22. [Google Scholar] [CrossRef]
- Toosy, S.; Sodi, R.; Pappachan, J.M. Lean Polycystic Ovary Syndrome (PCOS): An Evidence-Based Practical Approach. J. Diabetes Metab. Disord. 2018, 17, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Nolan, C.J.; Ruderman, N.B.; Kahn, S.E.; Pedersen, O.; Prentki, M. Insulin Resistance as a Physiological Defense against Metabolic Stress: Implications for the Management of Subsets of Type 2 Diabetes. Diabetes 2015, 64, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Taegtmeyer, H.; Beauloye, C.; Harmancey, R.; Hue, L. Insulin Resistance Protects the Heart from Fuel Overload in Dysregulated Metabolic States. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1693–H1697. [Google Scholar] [CrossRef] [PubMed]
- Connor, T.; Martin, S.D.; Howlett, K.F.; McGee, S.L. Metabolic Remodelling in Obesity and Type 2 Diabetes: Pathological or Protective Mechanisms in Response to Nutrient Excess? Clin. Exp. Pharmacol. Physiol. 2015, 42, 109–115. [Google Scholar] [CrossRef]
- Nolan, C.J.; Ruderman, N.B.; Prentki, M. Intensive Insulin for Type 2 Diabetes: The Risk of Causing Harm. Lancet Diabetes Endocrinol. 2013, 1, 9–10. [Google Scholar] [CrossRef]
- Nolan, C.J.; Prentki, M. Insulin Resistance and Insulin Hypersecretion in the Metabolic Syndrome and Type 2 Diabetes: Time for a Conceptual Framework Shift. Diabetes Vasc. Dis. Res. 2019, 16, 118–127. [Google Scholar] [CrossRef]
- Ye, J. Mechanism of Insulin Resistance in Obesity: A Role of ATP. Front. Med. 2021, 15, 372–382. [Google Scholar] [CrossRef]
- Amato, M.C.; Vesco, R.; Vigneri, E.; Ciresi, A.; Giordano, C. Hyperinsulinism and Polycystic Ovary Syndrome (PCOS): Role of Insulin Clearance. J. Endocrinol. Investig. 2015, 38, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Vrbíková, J.; Bendlová, B.; Hill, M.; Vanková, M.; Vondra, K.; Stárka, L. Insulin Sensitivity and Beta-Cell Function in Women with Polycystic Ovary Syndrome. Diabetes Care 2002, 25, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- O’Meara, N.M.; Blackman, J.D.; Ehrmann, D.A.; Barnes, R.B.; Jaspan, J.B.; Rosenfield, R.L.; Polonsky, K.S. Defects in Beta-Cell Function in Functional Ovarian Hyperandrogenism. J. Clin. Endocrinol. Metab. 1993, 76, 1241–1247. [Google Scholar] [CrossRef]
- Manco, M.; Castagneto-Gissey, L.; Arrighi, E.; Carnicelli, A.; Brufani, C.; Luciano, R.; Mingrone, G. Insulin Dynamics in Young Women with Polycystic Ovary Syndrome and Normal Glucose Tolerance across Categories of Body Mass Index. PLoS ONE 2014, 9, e92995. [Google Scholar] [CrossRef] [PubMed]
- Amato, M.C.; Guarnotta, V.; Ciresi, A.; Modica, R.; Pantò, F.; Giordano, C. No Phenotypic Differences for Polycystic Ovary Syndrome (PCOS) between Women with and without Type 1 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2014, 99, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Thong, E.P.; Milat, F.; Joham, A.E.; Mishra, G.D.; Teede, H. Obesity, Menstrual Irregularity and Polycystic Ovary Syndrome in Young Women with Type 1 Diabetes: A Population-Based Study. Clin. Endocrinol. 2020, 93, 564–571. [Google Scholar] [CrossRef]
- Escobar-Morreale, H.F.; Roldán-Martín, M.B. Type 1 Diabetes and Polycystic Ovary Syndrome: Systematic Review and Meta-Analysis. Diabetes Care 2016, 39, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Polderman, K.H.; Gooren, L.J.; Asscheman, H.; Bakker, A.; Heine, R.J. Induction of Insulin Resistance by Androgens and Estrogens. J. Clin. Endocrinol. Metab. 1994, 79, 265–271. [Google Scholar] [CrossRef]
- Diamond, M.P.; Grainger, D.; Diamond, M.C.; Sherwin, R.S.; Defronzo, R.A. Effects of Methyltestosterone on Insulin Secretion and Sensitivity in Women. J. Clin. Endocrinol. Metab. 1998, 83, 4420–4425. [Google Scholar] [CrossRef]
- Moghetti, P.; Tosi, F.; Castello, R.; Magnani, C.M.; Negri, C.; Brun, E.; Furlani, L.; Caputo, M.; Muggeo, M. The Insulin Resistance in Women with Hyperandrogenism Is Partially Reversed by Antiandrogen Treatment: Evidence That Androgens Impair Insulin Action in Women. J. Clin. Endocrinol. Metab. 1996, 81, 952–960. [Google Scholar] [CrossRef] [Green Version]
- Elkind-Hirsch, K.E.; Valdes, C.T.; Russell Malinak, L. Insulin Resistance Improves in Hyperandrogenic Women Treated with Lupron. Fertil. Steril. 1993, 60, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhang, C.; Shi, Y.; Zhang, F.; Li, L.; Wang, X.; Ling, Y.; Fu, H.; Dong, W.; Shen, J.; et al. Dysbiosis of Gut Microbiota Associated with Clinical Parameters in Polycystic Ovary Syndrome. Front. Microbiol. 2017, 8, 324. [Google Scholar] [CrossRef] [PubMed]
- Torres, P.J.; Siakowska, M.; Banaszewska, B.; Pawelczyk, L.; Duleba, A.J.; Kelley, S.T.; Thackray, V.G. Gut Microbial Diversity in Women With Polycystic Ovary Syndrome Correlates With Hyperandrogenism. J. Clin. Endocrinol. Metab. 2018, 103, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Insenser, M.; Murri, M.; Del Campo, R.; Martínez-García, M.Á.; Fernández-Durán, E.; Escobar-Morreale, H.F. Gut Microbiota and the Polycystic Ovary Syndrome: Influence of Sex, Sex Hormones, and Obesity. J. Clin. Endocrinol. Metab. 2018, 103, 2552–2562. [Google Scholar] [CrossRef] [PubMed]
- Thursby, E.; Juge, N. Introduction to the Human Gut Microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- He, F.-F.; Li, Y.-M. Role of Gut Microbiota in the Development of Insulin Resistance and the Mechanism Underlying Polycystic Ovary Syndrome: A Review. J. Ovarian Res. 2020, 13, 73. [Google Scholar] [CrossRef]
- Pande, A.R.; Guleria, A.K.; Singh, S.D.; Shukla, M.; Dabadghao, P. β Cell Function and Insulin Resistance in Lean Cases with Polycystic Ovary Syndrome. Gynecol. Endocrinol. Off. J. Int. Soc. Gynecol. Endocrinol. 2017, 33, 877–881. [Google Scholar] [CrossRef]
- Song, D.K.; Hong, Y.S.; Sung, Y.-A.; Lee, H. Insulin Resistance According to β-Cell Function in Women with Polycystic Ovary Syndrome and Normal Glucose Tolerance. PLoS ONE 2017, 12, e0178120. [Google Scholar] [CrossRef]
- Gennarelli, G.; Rovei, V.; Novi, R.F.; Holte, J.; Bongioanni, F.; Revelli, A.; Pacini, G.; Cavallo-Perin, P.; Massobrio, M. Preserved Insulin Sensitivity and β-Cell Activity, but Decreased Glucose Effectiveness in Normal-Weight Women with the Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2005, 90, 3381–3386. [Google Scholar] [CrossRef]
- Ehrmann, D.A.; Sturis, J.; Byrne, M.M.; Rosenfield, R.L.; Polonsky, K.S. Beta-Cell Function in Polycystic Ovary Syndrome. In Polycystic Ovary Syndrome; Chang, R.J., Ed.; Serono Symposia USA; Springer: New York, NY, USA, 1996; pp. 126–141. [Google Scholar] [CrossRef]
- Tao, T.; Li, S.; Zhao, A.; Mao, X.; Liu, W. Early Impaired β-Cell Function in Chinese Women with Polycystic Ovary Syndrome. Int. J. Clin. Exp. Pathol. 2012, 5, 777–786. [Google Scholar]
- Colilla, S.; Cox, N.J.; Ehrmann, D.A. Heritability of Insulin Secretion and Insulin Action in Women with Polycystic Ovary Syndrome and Their First Degree Relatives. J. Clin. Endocrinol. Metab. 2001, 86, 2027–2031. [Google Scholar] [CrossRef]
- Sam, S.; Sung, Y.-A.; Legro, R.S.; Dunaif, A. Evidence for Pancreatic Beta-Cell Dysfunction in Brothers of Women with Polycystic Ovary Syndrome. Metabolism 2008, 57, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, M.O.; Erickson, S.; Port, S.C.; Jennrich, R.I.; Korenman, S.G. Beta-Cell Function: A Key Pathological Determinant in Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2005, 90, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, S.; Grace, C.; Mattei, A.A.; Siemienowicz, K.; Brownlee, W.; MacCallum, J.; McNeilly, A.S.; Duncan, W.C.; Rae, M.T. Developmental Programming of Polycystic Ovary Syndrome (PCOS): Prenatal Androgens Establish Pancreatic Islet α/β Cell Ratio and Subsequent Insulin Secretion. Sci. Rep. 2016, 6, 27408. [Google Scholar] [CrossRef] [PubMed]
- Rae, M.; Grace, C.; Hogg, K.; Wilson, L.M.; McHaffie, S.L.; Ramaswamy, S.; MacCallum, J.; Connolly, F.; McNeilly, A.S.; Duncan, C. The Pancreas Is Altered by in Utero Androgen Exposure: Implications for Clinical Conditions Such as Polycystic Ovary Syndrome (PCOS). PLoS ONE 2013, 8, e56263. [Google Scholar] [CrossRef]
- Nicol, L.E.; O’Brien, T.D.; Dumesic, D.A.; Grogan, T.; Tarantal, A.F.; Abbott, D.H. Abnormal Infant Islet Morphology Precedes Insulin Resistance in PCOS-like Monkeys. PLoS ONE 2014, 9, e106527. [Google Scholar] [CrossRef]
- Højlund, K. Metabolism and insulin signaling in common metabolic disorders and inherited insulin resistance. Dan. Med. J. 2014, 61, B4890. [Google Scholar] [PubMed]
- James, D.E.; Stöckli, J.; Birnbaum, M.J. The Aetiology and Molecular Landscape of Insulin Resistance. Nat. Rev. Mol. Cell Biol. 2021, 22, 751–771. [Google Scholar] [CrossRef]
- Dunaif, A.; Xia, J.; Book, C.B.; Schenker, E.; Tang, Z. Excessive Insulin Receptor Serine Phosphorylation in Cultured Fibroblasts and in Skeletal Muscle. A Potential Mechanism for Insulin Resistance in the Polycystic Ovary Syndrome. J. Clin. Investig. 1995, 96, 801–810. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [Green Version]
- Corbould, A.; Zhao, H.; Mirzoeva, S.; Aird, F.; Dunaif, A. Enhanced Mitogenic Signaling in Skeletal Muscle of Women With Polycystic Ovary Syndrome. Diabetes 2006, 55, 751–759. [Google Scholar] [CrossRef]
- Rajkhowa, M.; Brett, S.; Cuthbertson, D.J.; Lipina, C.; Ruiz-Alcaraz, A.J.; Thomas, G.E.; Logie, L.; Petrie, J.R.; Sutherland, C. Insulin Resistance in Polycystic Ovary Syndrome Is Associated with Defective Regulation of ERK1/2 by Insulin in Skeletal Muscle in Vivo. Biochem. J. 2009, 418, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Diamanti-Kandarakis, E.; Dunaif, A. Insulin Resistance and the Polycystic Ovary Syndrome Revisited: An Update on Mechanisms and Implications. Endocr. Rev. 2012, 33, 981–1030. [Google Scholar] [CrossRef]
- Macut, D.; Bjekić-Macut, J.; Rahelić, D.; Doknić, M. Insulin and the Polycystic Ovary Syndrome. Diabetes Res. Clin. Pract. 2017, 130, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Dunaif, A.; Segal, K.R.; Shelley, D.R.; Green, G.; Dobrjansky, A.; Licholai, T. Evidence for distinctive and intrinsic defects in insulin action in polycystic ovary syndrome. Diabetes 1992, 41, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Corbould, A.; Kim, Y.-B.; Youngren, J.F.; Pender, C.; Kahn, B.B.; Lee, A.; Dunaif, A. Insulin Resistance in the Skeletal Muscle of Women with PCOS Involves Intrinsic and Acquired Defects in Insulin Signaling. Am. J. Physiol.-Endocrinol. Metab. 2005, 288, E1047–E1054. [Google Scholar] [CrossRef]
- Takayama, S.; White, M.F.; Kahn, C.R. Phorbol Ester-Induced Serine Phosphorylation of the Insulin Receptor Decreases Its Tyrosine Kinase Activity. J. Biol. Chem. 1988, 263, 3440–3447. [Google Scholar] [CrossRef]
- Li, M.; Youngren, J.F.; Dunaif, A.; Goldfine, I.D.; Maddux, B.A.; Zhang, B.B.; Evans, J.L. Decreased Insulin Receptor (IR) Autophosphorylation in Fibroblasts from Patients with PCOS: Effects of Serine Kinase Inhibitors and IR Activators. J. Clin. Endocrinol. Metab. 2002, 87, 4088–4093. [Google Scholar] [CrossRef]
- Dunaif, A.; Wu, X.; Lee, A.; Diamanti-Kandarakis, E. Defects in Insulin Receptor Signaling in Vivo in the Polycystic Ovary Syndrome (PCOS). Am. J. Physiol.-Endocrinol. Metab. 2001, 281, E392–E399. [Google Scholar] [CrossRef]
- Lin, Q.; Zhang, H.; Zhao, J.; Wang, Z. Expression and Contribution of Insulin Signaling Pathway to the Development of Polycystic Ovary Syndrome. In Polycystic Ovarian Syndrome; Wang, Z., Ed.; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Bao, Y.; Zhou, X.; Zheng, L. Polycystic Ovary Syndrome and Mitochondrial Dysfunction. Reprod. Biol. Endocrinol. 2019, 17, 67. [Google Scholar] [CrossRef]
- Skov, V.; Glintborg, D.; Knudsen, S.; Jensen, T.; Kruse, T.A.; Tan, Q.; Brusgaard, K.; Beck-Nielsen, H.; Højlund, K. Reduced Expression of Nuclear-Encoded Genes Involved in Mitochondrial Oxidative Metabolism in Skeletal Muscle of Insulin-Resistant Women With Polycystic Ovary Syndrome. Diabetes 2007, 56, 2349–2355. [Google Scholar] [CrossRef]
- Skov, V.; Glintborg, D.; Knudsen, S.; Tan, Q.; Jensen, T.; Kruse, T.A.; Beck-Nielsen, H.; Højlund, K. Pioglitazone Enhances Mitochondrial Biogenesis and Ribosomal Protein Biosynthesis in Skeletal Muscle in Polycystic Ovary Syndrome. PLoS ONE 2008, 3, e2466. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.L.; Maddux, B.A.; Goldfine, I.D. The Molecular Basis for Oxidative Stress-Induced Insulin Resistance. Antioxid. Redox Signal. 2005, 7, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Diao, F.; Xu, M.; Hu, Y.; Li, J.; Xu, Z.; Lin, M.; Wang, L.; Zhou, Y.; Zhou, Z.; Liu, J.; et al. The Molecular Characteristics of Polycystic Ovary Syndrome (PCOS) Ovary Defined by Human Ovary CDNA Microarray. J. Mol. Endocrinol. 2004, 33, 59–72. [Google Scholar] [CrossRef]
- Lee, H.; Oh, J.-Y.; Sung, Y.-A.; Chung, H.; Kim, H.-L.; Kim, G.S.; Cho, Y.S.; Kim, J.T. Genome-Wide Association Study Identified New Susceptibility Loci for Polycystic Ovary Syndrome. Hum. Reprod. 2015, 30, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-J.; Zhao, H.; He, L.; Shi, Y.; Qin, Y.; Shi, Y.; Li, Z.; You, L.; Zhao, J.; Liu, J.; et al. Genome-Wide Association Study Identifies Susceptibility Loci for Polycystic Ovary Syndrome on Chromosome 2p16.3, 2p21 and 9q33.3. Nat. Genet. 2011, 43, 55–59. [Google Scholar] [CrossRef]
- Shi, Y.; Zhao, H.; Shi, Y.; Cao, Y.; Yang, D.; Li, Z.; Zhang, B.; Liang, X.; Li, T.; Chen, J.; et al. Genome-Wide Association Study Identifies Eight New Risk Loci for Polycystic Ovary Syndrome. Nat. Genet. 2012, 44, 1020–1025. [Google Scholar] [CrossRef]
- Goodarzi, M.O.; Jones, M.R.; Li, X.; Chua, A.K.; Garcia, O.A.; Chen, Y.-D.I.; Krauss, R.M.; Rotter, J.I.; Ankener, W.; Legro, R.S.; et al. Replication of Association of DENND1A and THADA Variants with Polycystic Ovary Syndrome in European Cohorts. J. Med. Genet. 2012, 49, 90–95. [Google Scholar] [CrossRef]
- Hayes, M.G.; Urbanek, M.; Ehrmann, D.A.; Armstrong, L.L.; Lee, J.Y.; Sisk, R.; Karaderi, T.; Barber, T.M.; McCarthy, M.I.; Franks, S.; et al. Genome-Wide Association of Polycystic Ovary Syndrome Implicates Alterations in Gonadotropin Secretion in European Ancestry Populations. Nat. Commun. 2015, 6, 7502. [Google Scholar] [CrossRef] [Green Version]
- Louwers, Y.V.; Stolk, L.; Uitterlinden, A.G.; Laven, J.S.E. Cross-Ethnic Meta-Analysis of Genetic Variants for Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2013, 98, E2006–E2012. [Google Scholar] [CrossRef]
- Day, F.; Karaderi, T.; Jones, M.R.; Meun, C.; He, C.; Drong, A.; Kraft, P.; Lin, N.; Huang, H.; Broer, L.; et al. Large-Scale Genome-Wide Meta-Analysis of Polycystic Ovary Syndrome Suggests Shared Genetic Architecture for Different Diagnosis Criteria. PLoS Genet. 2018, 14, e1007813. [Google Scholar] [CrossRef]
- Duckworth, W.C.; Bennett, R.G.; Hamel, F.G. Insulin Degradation: Progress and Potential. Endocr. Rev. 1998, 19, 608–624. [Google Scholar] [CrossRef]
- Tosi, F.; Dal Molin, F.; Zamboni, F.; Saggiorato, E.; Salvagno, G.L.; Fiers, T.; Kaufman, J.-M.; Bonora, E.; Moghetti, P. Serum Androgens Are Independent Predictors of Insulin Clearance but Not of Insulin Secretion in Women With PCOS. J. Clin. Endocrinol. Metab. 2020, 105, dgaa095. [Google Scholar] [CrossRef]
- Wahlqvist, M.L.; Kaijser, L.; Lassers, B.W.; Löw, H.; Carlson, L.A. Release of Immunoreactive Insulin from the Human Heart. Eur. J. Clin. Investig. 1972, 2, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Hennes, M.M.; Dua, A.; Kissebah, A.H. Effects of Free Fatty Acids and Glucose on Splanchnic Insulin Dynamics. Diabetes 1997, 46, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Pagano, C.; Rizzato, M.; Lombardi, A.M.; Fabris, R.; Favaro, A.; Federspil, G.; Vettor, R. Effect of Lactate on Hepatic Insulin Clearance in Perfused Rat Liver. Am. J. Physiol. 1996, 270 Pt 2, R682–R687. [Google Scholar] [CrossRef] [PubMed]
- Svedberg, J.; Björntorp, P.; Smith, U.; Lönnroth, P. Free-Fatty Acid Inhibition of Insulin Binding, Degradation, and Action in Isolated Rat Hepatocytes. Diabetes 1990, 39, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Kupfer, S.R.; Marschke, K.B.; Wilson, E.M.; French, F.S. Receptor Accessory Factor Enhances Specific DNA Binding of Androgen and Glucocorticoid Receptors. J. Biol. Chem. 1993, 268, 17519–17527. [Google Scholar] [CrossRef]
- Kupfer, S.R.; Wilson, E.M.; French, F.S. Androgen and Glucocorticoid Receptors Interact with Insulin Degrading Enzyme. J. Biol. Chem. 1994, 269, 20622–20628. [Google Scholar] [CrossRef]
- Buffington, C.K.; Kitabchi, A.E. Evidence for a Defect in Insulin Metabolism in Hyperandrogenic Women with Polycystic Ovarian Syndrome. Metabolism 1994, 43, 1367–1372. [Google Scholar] [CrossRef]
- Ciampelli, M.; Fulghesu, A.M.; Cucinelli, F.; Pavone, V.; Caruso, A.; Mancuso, S.; Lanzone, A. Heterogeneity in Beta Cell Activity, Hepatic Insulin Clearance and Peripheral Insulin Sensitivity in Women with Polycystic Ovary Syndrome. Hum. Reprod. 1997, 12, 1897–1901. [Google Scholar] [CrossRef] [PubMed]
- Stassek, J.; Erdmann, J.; Ohnolz, F.; Berg, F.D.; Kiechle, M.; Seifert-Klauss, V. C-Peptide, Baseline and Postprandial Insulin Resistance after a Carbohydrate-Rich Test Meal—Evidence for an Increased Insulin Clearance in PCOS Patients? Geburtshilfe Frauenheilkd. 2017, 77, 59–65. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herman, R.; Sikonja, J.; Jensterle, M.; Janez, A.; Dolzan, V. Insulin Metabolism in Polycystic Ovary Syndrome: Secretion, Signaling, and Clearance. Int. J. Mol. Sci. 2023, 24, 3140. https://doi.org/10.3390/ijms24043140
Herman R, Sikonja J, Jensterle M, Janez A, Dolzan V. Insulin Metabolism in Polycystic Ovary Syndrome: Secretion, Signaling, and Clearance. International Journal of Molecular Sciences. 2023; 24(4):3140. https://doi.org/10.3390/ijms24043140
Chicago/Turabian StyleHerman, Rok, Jaka Sikonja, Mojca Jensterle, Andrej Janez, and Vita Dolzan. 2023. "Insulin Metabolism in Polycystic Ovary Syndrome: Secretion, Signaling, and Clearance" International Journal of Molecular Sciences 24, no. 4: 3140. https://doi.org/10.3390/ijms24043140