
The Protective Role of Glutathione on Zinc-Induced Neuron Death after Brain Injuries

 ,
, 
Abstract
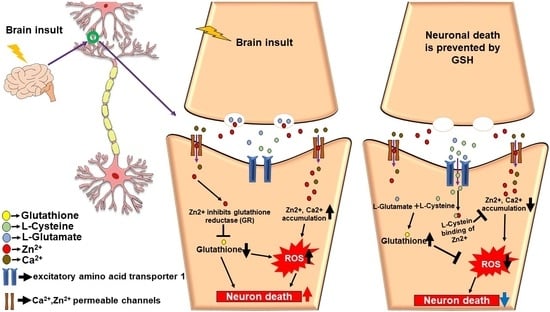
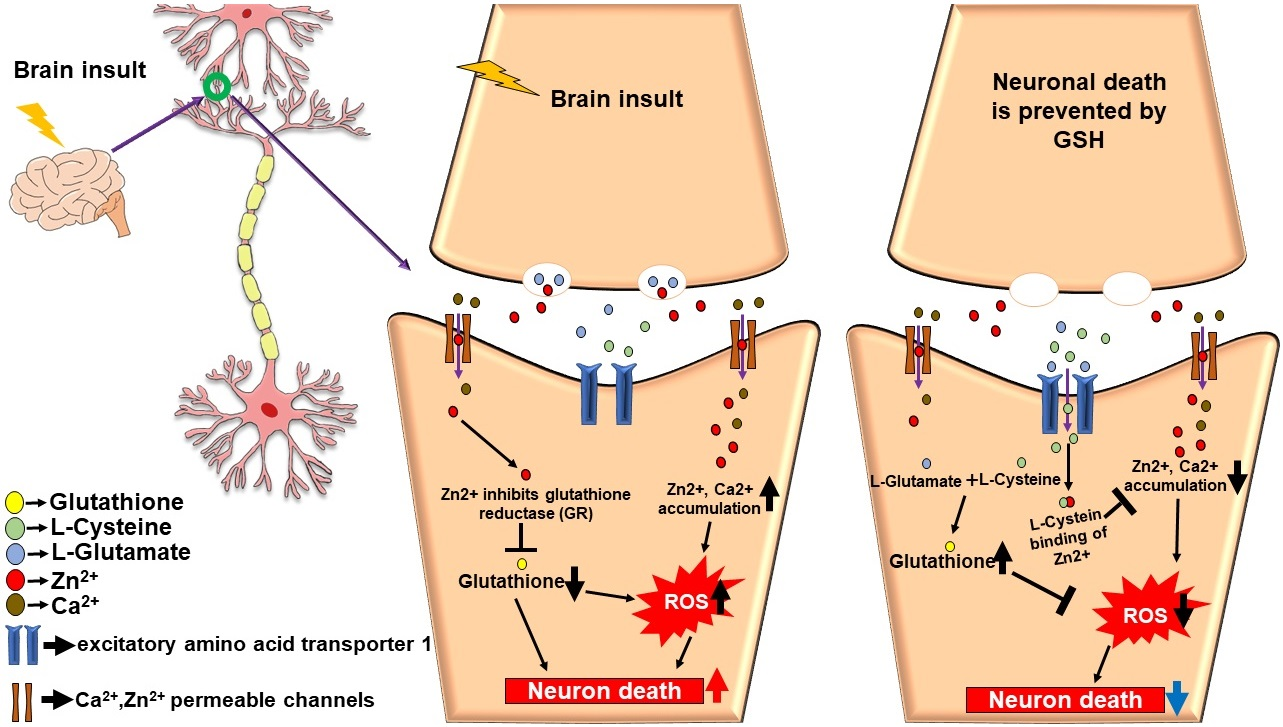
1. Introduction
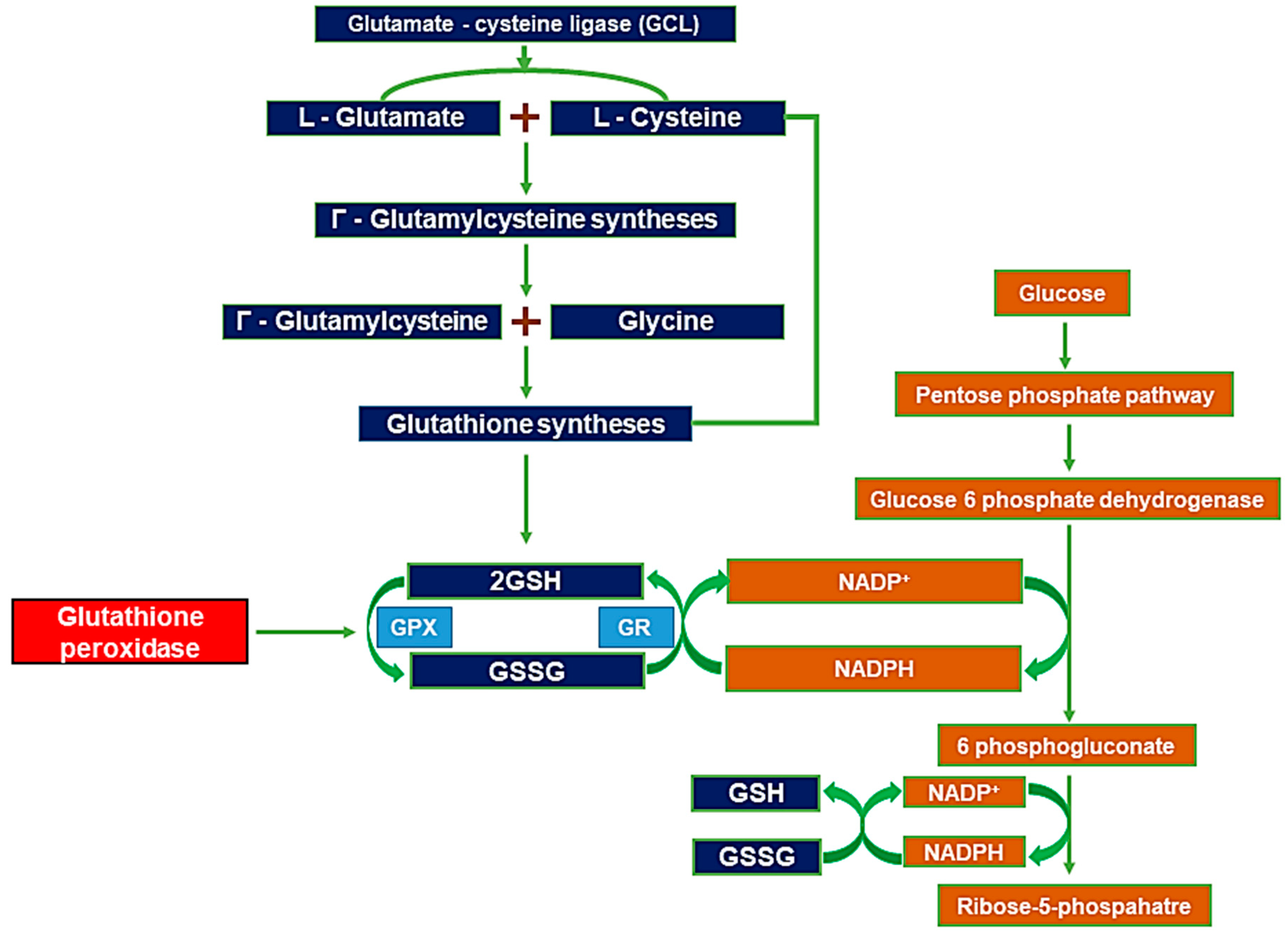
2. Glutathione Synthesis
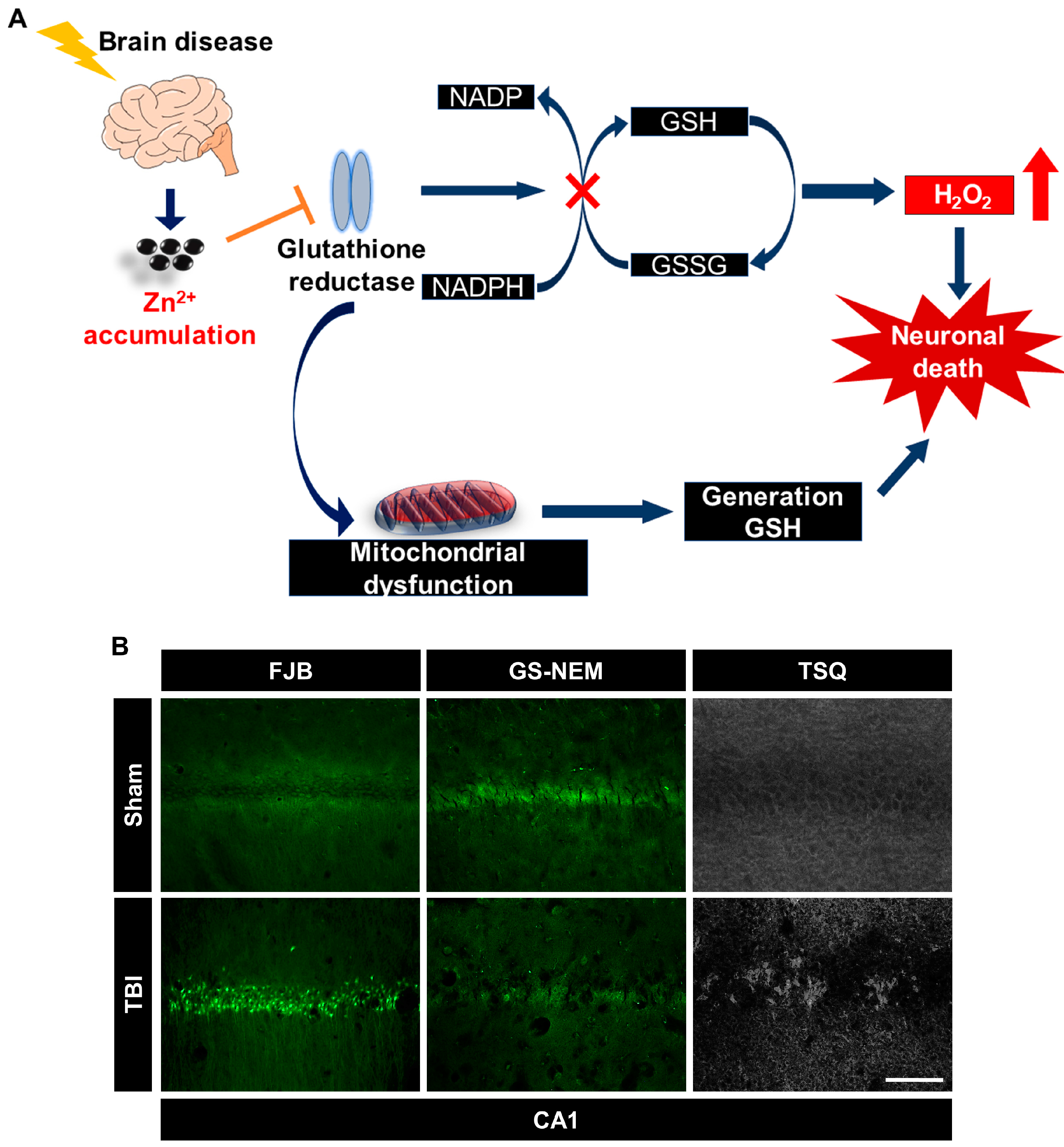
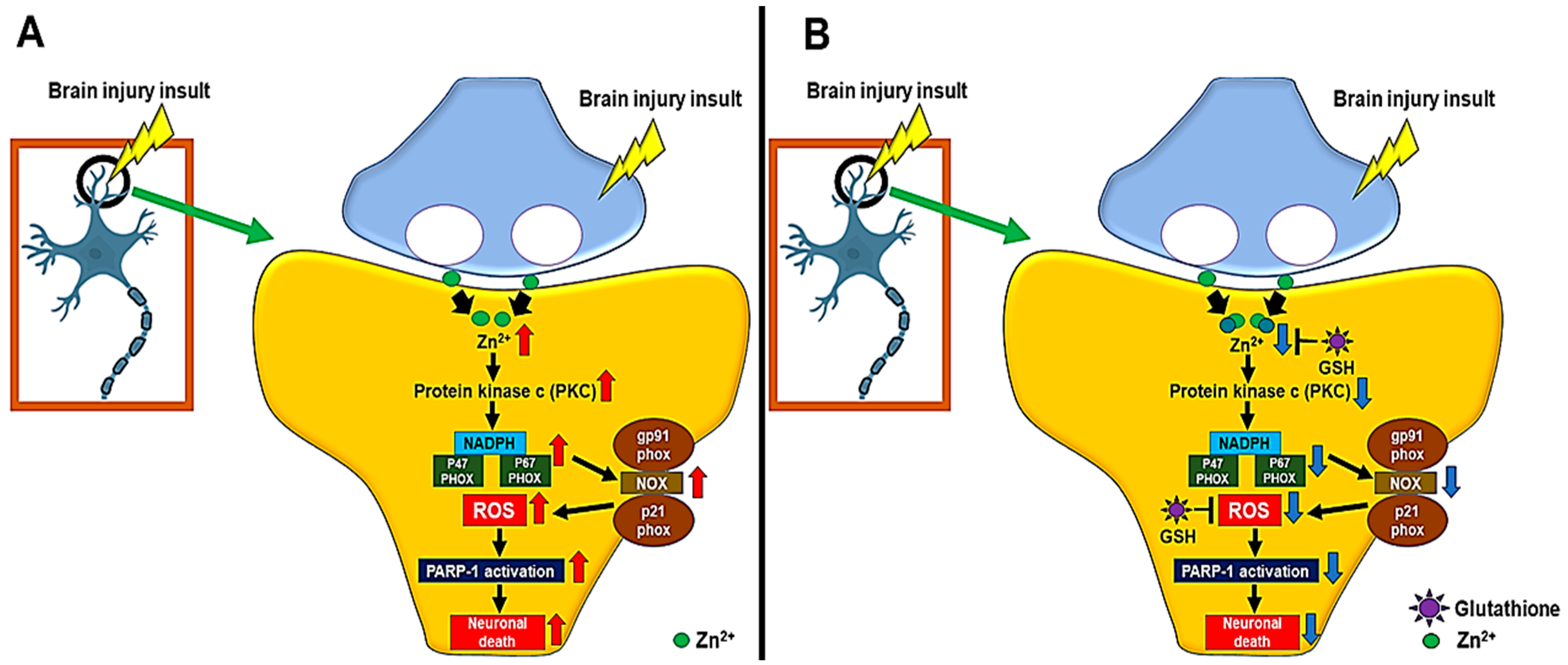
3. Zinc-Induced Glutathione Deficiency
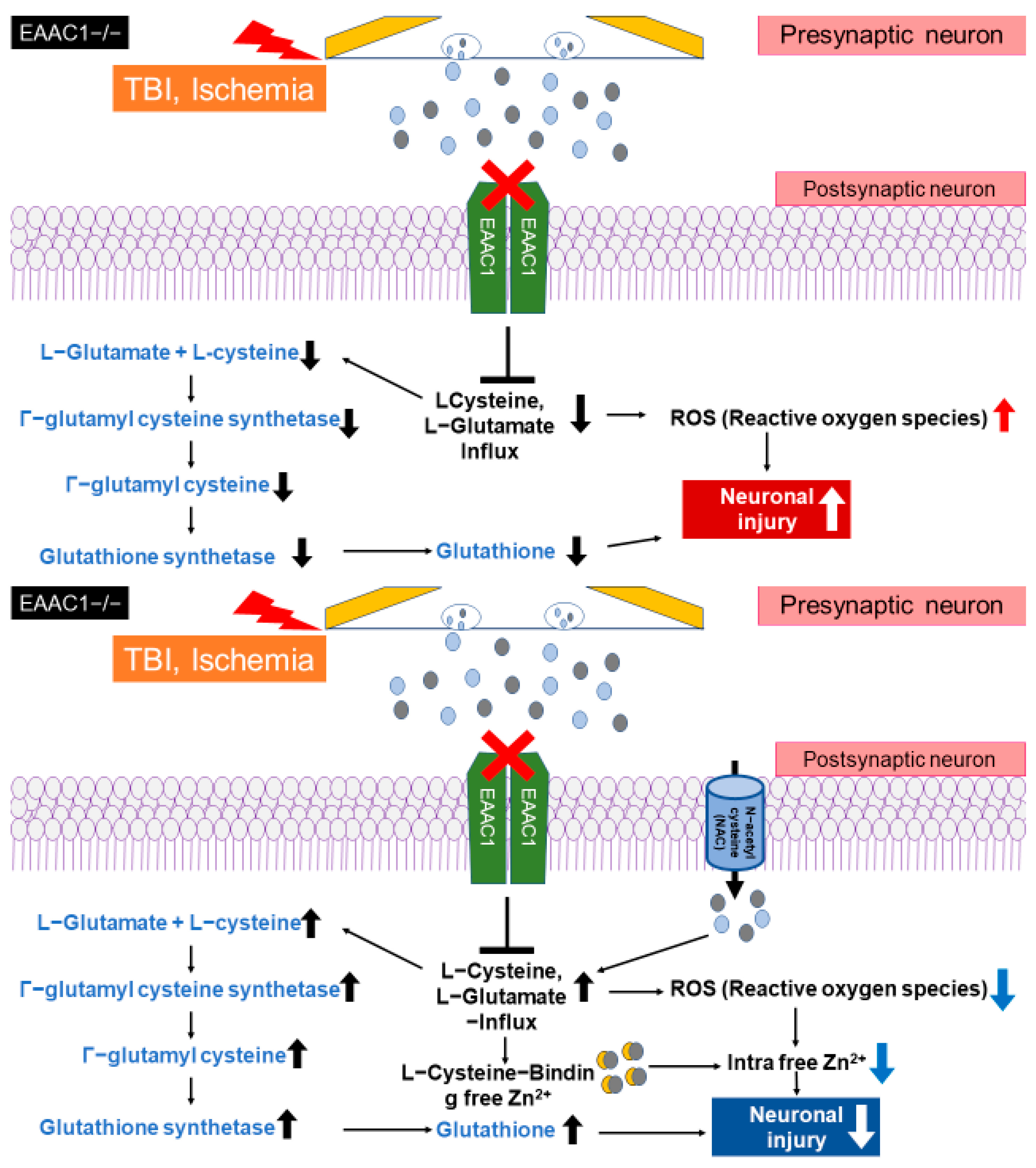
4. EAAC1 and Glutathione Formation
5. The Role of Glutathione in Zinc-Induced Neuron Death after Brain Injury
5.1. Zinc and Glutathione in Stroke
5.2. Zinc and Glutathione in Traumatic Brain Injury
5.3. Zinc and Glutathione in Hypoglycemia
5.4. Zinc and Glutathione in Epilepsy
5.5. Zinc and Glutathione in Brain Injuries
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, K.H.; Cha, M.; Lee, B.H. Neuroprotective Effect of Antioxidants in the Brain. Int. J. Mol. Sci. 2020, 21, 7152. [Google Scholar] [CrossRef] [PubMed]
- Porteous, J.W. Glutamate, glutamine, aspartate, asparagine, glucose and ketone-body metabolism in chick intestinal brush-border cells. Biochem. J. 1980, 188, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sanchez-Perez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef]
- Anjaneyulu, M.; Berent-Spillson, A.; Russell, J.W. Metabotropic glutamate receptors (mGluRs) and diabetic neuropathy. Curr. Drug Targets 2008, 9, 85–93. [Google Scholar] [CrossRef]
- Jiang, L.J.; Maret, W.; Vallee, B.L. The glutathione redox couple modulates zinc transfer from metallothionein to zinc-depleted sorbitol dehydrogenase. Proc. Natl. Acad. Sci. USA 1998, 95, 3483–3488. [Google Scholar] [CrossRef]
- Choi, S.; Hong, D.K.; Choi, B.Y.; Suh, S.W. Zinc in the Brain: Friend or Foe? Int. J. Mol. Sci. 2020, 21, 8941. [Google Scholar] [CrossRef]
- MacDonald, R.S. The role of zinc in growth and cell proliferation. J. Nutr. 2000, 130 (Suppl. S5), 1500S–1508S. [Google Scholar] [CrossRef]
- Beyersmann, D.; Haase, H. Functions of zinc in signaling, proliferation and differentiation of mammalian cells. Biometals 2001, 14, 331–341. [Google Scholar] [CrossRef]
- Besser, L.; Chorin, E.; Sekler, I.; Silverman, W.F.; Atkin, S.; Russell, J.T.; Hershfinkel, M. Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J. Neurosci. 2009, 29, 2890–2901. [Google Scholar] [CrossRef]
- Morris, D.R.; Levenson, C.W. Ion channels and zinc: Mechanisms of neurotoxicity and neurodegeneration. J. Toxicol. 2012, 2012, 785647. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Hong, D.K.; Jeong, J.H.; Lee, B.E.; Koh, J.Y.; Suh, S.W. Zinc transporter 3 modulates cell proliferation and neuronal differentiation in the adult hippocampus. Stem Cells 2020, 38, 994–1006. [Google Scholar] [CrossRef] [PubMed]
- Jang, B.G.; Won, S.J.; Kim, J.H.; Choi, B.Y.; Lee, M.W.; Sohn, M.; Song, H.K.; Suh, S.W. EAAC1 gene deletion alters zinc homeostasis and enhances cortical neuronal injury after transient cerebral ischemia in mice. J. Trace Elem. Med. Biol. 2012, 26, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Suh, S.W. Antimicrotubule Agent-Induced Zinc Neurotoxicity. Biol. Pharm. Bull. 2018, 41, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Hong, D.K.; Suh, S.W. ZnT3 Gene Deletion Reduces Colchicine-Induced Dentate Granule Cell Degeneration. Int. J. Mol. Sci. 2017, 18, 2189. [Google Scholar] [CrossRef]
- Magi, S.; Piccirillo, S.; Amoroso, S.; Lariccia, V. Excitatory Amino Acid Transporters (EAATs): Glutamate Transport and Beyond. Int. J. Mol. Sci. 2019, 20, 5674. [Google Scholar] [CrossRef]
- Malik, A.R.; Willnow, T.E. Excitatory Amino Acid Transporters in Physiology and Disorders of the Central Nervous System. Int. J. Mol. Sci. 2019, 20, 5671. [Google Scholar] [CrossRef]
- Kim, K.; Lee, S.G.; Kegelman, T.P.; Su, Z.Z.; Das, S.K.; Dash, R.; Dasgupta, S.; Barral, P.M.; Hedvat, M.; Diaz, P.; et al. Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: Opportunities for developing novel therapeutics. J. Cell. Physiol. 2011, 226, 2484–2493. [Google Scholar] [CrossRef]
- Parkin, G.M.; Udawela, M.; Gibbons, A.; Dean, B. Glutamate transporters, EAAT1 and EAAT2, are potentially important in the pathophysiology and treatment of schizophrenia and affective disorders. World J. Psychiatry 2018, 8, 51–63. [Google Scholar] [CrossRef]
- Furuta, A.; Martin, L.J.; Lin, C.L.; Dykes-Hoberg, M.; Rothstein, J.D. Cellular and synaptic localization of the neuronal glutamate transporters excitatory amino acid transporter 3 and 4. Neuroscience 1997, 81, 1031–1042. [Google Scholar] [CrossRef]
- Lukasiewcz, P.D.; Bligard, G.W.; DeBrecht, J.D. EAAT5 Glutamate Transporter-Mediated Inhibition in the Vertebrate Retina. Front. Cell. Neurosci. 2021, 15, 662859. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Ko, D.G.; Hong, D.K.; Lim, M.S.; Choi, B.Y.; Suh, S.W. Role of Excitatory Amino Acid Carrier 1 (EAAC1) in Neuronal Death and Neurogenesis After Ischemic Stroke. Int. J. Mol. Sci. 2020, 21, 5676. [Google Scholar] [CrossRef]
- Kim, H.B.; Yoo, J.Y.; Yoo, S.Y.; Lee, J.H.; Chang, W.; Kim, H.S.; Baik, T.K.; Woo, R.S. Neuregulin-1 inhibits CoCl2-induced upregulation of excitatory amino acid carrier 1 expression and oxidative stress in SH-SY5Y cells and the hippocampus of mice. Mol. Brain 2020, 13, 153. [Google Scholar] [CrossRef]
- Choi, B.Y.; Kim, I.Y.; Kim, J.H.; Lee, B.E.; Lee, S.H.; Kho, A.R.; Jung, H.J.; Sohn, M.; Song, H.K.; Suh, S.W. Decreased cysteine uptake by EAAC1 gene deletion exacerbates neuronal oxidative stress and neuronal death after traumatic brain injury. Amino Acids 2016, 48, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Nakaki, T. Inhibition of GTRAP3-18 may increase neuroprotective glutathione (GSH) synthesis. Int. J. Mol. Sci. 2012, 13, 12017–12035. [Google Scholar] [CrossRef] [PubMed]
- Kim, K. Glutathione in the Nervous System as a Potential Therapeutic Target to Control the Development and Progression of Amyotrophic Lateral Sclerosis. Antioxidants 2021, 10, 1011. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K. Glutathione in the Brain. Int. J. Mol. Sci. 2021, 22, 5010. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.; Sandhu, J.K.; Harper, M.E.; Cuperlovic-Culf, M. Role of Glutathione in Cancer: From Mechanisms to Therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef]
- Toroser, D.; Yarian, C.S.; Orr, W.C.; Sohal, R.S. Mechanisms of gamma-glutamylcysteine ligase regulation. Biochim. Biophys. Acta 2006, 1760, 233–244. [Google Scholar] [CrossRef]
- Krejsa, C.M.; Franklin, C.C.; White, C.C.; Ledbetter, J.A.; Schieven, G.L.; Kavanagh, T.J. Rapid activation of glutamate cysteine ligase following oxidative stress. J. Biol. Chem. 2010, 285, 16116–16124. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hasanuzzaman, M.; Nahar, K.; Anee, T.I.; Fujita, M. Glutathione in plants: Biosynthesis and physiological role in environmental stress tolerance. Physiol. Mol. Biol. Plants 2017, 23, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S. Neuroprotective Function of High Glycolytic Activity in Astrocytes: Common Roles in Stroke and Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 6568. [Google Scholar] [CrossRef] [PubMed]
- Tu, D.; Gao, Y.; Yang, R.; Guan, T.; Hong, J.S.; Gao, H.M. The pentose phosphate pathway regulates chronic neuroinflammation and dopaminergic neurodegeneration. J. Neuroinflamm. 2019, 16, 255. [Google Scholar] [CrossRef]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef]
- Takahashi, S.; Mashima, K. Neuroprotection and Disease Modification by Astrocytes and Microglia in Parkinson Disease. Antioxidants 2022, 11, 170. [Google Scholar] [CrossRef]
- Ookhtens, M.; Kaplowitz, N. Role of the liver in interorgan homeostasis of glutathione and cyst(e)ine. Semin. Liver Dis. 1998, 18, 313–329. [Google Scholar] [CrossRef]
- Lauterburg, B.H.; Adams, J.D.; Mitchell, J.R. Hepatic glutathione homeostasis in the rat: Efflux accounts for glutathione turnover. Hepatology 1984, 4, 586–590. [Google Scholar] [CrossRef]
- Bannai, S.; Tateishi, N. Role of membrane transport in metabolism and function of glutathione in mammals. J. Membr. Biol. 1986, 89, 1–8. [Google Scholar] [CrossRef]
- Rahantaniaina, M.S.; Li, S.; Chatel-Innocenti, G.; Tuzet, A.; Mhamdi, A.; Vanacker, H.; Noctor, G. Glutathione oxidation in response to intracellular H2O2: Key but overlapping roles for dehydroascorbate reductases. Plant Signal. Behav. 2017, 12, e1356531. [Google Scholar] [CrossRef] [PubMed]
- Roxas, V.P.; Smith, R.K., Jr.; Allen, E.R.; Allen, R.D. Overexpression of glutathione S-transferase/glutathione peroxidase enhances the growth of transgenic tobacco seedlings during stress. Nat. Biotechnol. 1997, 15, 988–991. [Google Scholar] [CrossRef] [PubMed]
- Rouhier, N. Plant glutaredoxins: Pivotal players in redox biology and iron-sulphur centre assembly. New Phytol. 2010, 186, 365–372. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Z.; Hoshino, A.; Zheng, H.D.; Morley, M.; Arany, Z.; Rabinowitz, J.D. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat. Metab. 2019, 1, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, Y.; Funakoshi, M.; Ishii, K. Determination of reduced nicotinamide adenine dinucleotide phosphate concentration using high-performance liquid chromatography with fluorescence detection: Ratio of the reduced form as a biomarker of oxidative stress. Biol. Pharm. Bull. 2009, 32, 1819–1823. [Google Scholar] [CrossRef]
- Dore, M.P.; Parodi, G.; Portoghese, M.; Pes, G.M. The Controversial Role of Glucose-6-Phosphate Dehydrogenase Deficiency on Cardiovascular Disease: A Narrative Review. Oxidative Med. Cell. Longev. 2021, 2021, 5529256. [Google Scholar] [CrossRef]
- Hao, S.; Liang, B.; Huang, Q.; Dong, S.; Wu, Z.; He, W.; Shi, M. Metabolic networks in ferroptosis. Oncol. Lett. 2018, 15, 5405–5411. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Canli, O.; Alankus, Y.B.; Grootjans, S.; Vegi, N.; Hultner, L.; Hoppe, P.S.; Schroeder, T.; Vandenabeele, P.; Bornkamm, G.W.; Greten, F.R. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood 2016, 127, 139–148. [Google Scholar] [CrossRef]
- Ribas, V.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- Roohani, N.; Hurrell, R.; Kelishadi, R.; Schulin, R. Zinc and its importance for human health: An integrative review. J. Res. Med. Sci. 2013, 18, 144–157. [Google Scholar] [PubMed]
- Plum, L.M.; Rink, L.; Haase, H. The essential toxin: Impact of zinc on human health. Int. J. Environ. Res. Public Health 2010, 7, 1342–1365. [Google Scholar] [CrossRef] [PubMed]
- Li, M.S.; Adesina, S.E.; Ellis, C.L.; Gooch, J.L.; Hoover, R.S.; Williams, C.R. NADPH oxidase-2 mediates zinc deficiency-induced oxidative stress and kidney damage. Am. J. Physiol. Cell Physiol. 2017, 312, C47–C55. [Google Scholar] [CrossRef]
- Noh, K.M.; Koh, J.Y. Induction and activation by zinc of NADPH oxidase in cultured cortical neurons and astrocytes. J. Neurosci. 2000, 20, RC111. [Google Scholar] [CrossRef]
- Trevisan, R.; Flesch, S.; Mattos, J.J.; Milani, M.R.; Bainy, A.C.; Dafre, A.L. Zinc causes acute impairment of glutathione metabolism followed by coordinated antioxidant defenses amplification in gills of brown mussels Perna perna. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2014, 159, 22–30. [Google Scholar] [CrossRef]
- Slepchenko, K.G.; Lu, Q.; Li, Y.V. Cross talk between increased intracellular zinc (Zn(2+)) and accumulation of reactive oxygen species in chemical ischemia. Am. J. Physiol. Cell Physiol. 2017, 313, C448–C459. [Google Scholar] [CrossRef]
- Bishop, G.M.; Dringen, R.; Robinson, S.R. Zinc stimulates the production of toxic reactive oxygen species (ROS) and inhibits glutathione reductase in astrocytes. Free Radic. Biol. Med. 2007, 42, 1222–1230. [Google Scholar] [CrossRef]
- Ryu, R.; Shin, Y.; Choi, J.W.; Min, W.; Ryu, H.; Choi, C.R.; Ko, H. Depletion of intracellular glutathione mediates zinc-induced cell death in rat primary astrocytes. Exp. Brain Res. 2002, 143, 257–263. [Google Scholar] [CrossRef]
- Mize, C.E.; Langdon, R.G. Hepatic glutathione reductase. I. Purification and general kinetic properties. J. Biol. Chem. 1962, 237, 1589–1595. [Google Scholar] [CrossRef]
- Chen, C.J.; Liao, S.L. Zinc toxicity on neonatal cortical neurons: Involvement of glutathione chelation. J. Neurochem. 2003, 85, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Aratake, T.; Shimizu, T.; Shimizu, S.; Saito, M. Protective Role of Glutathione in the Hippocampus after Brain Ischemia. Int. J. Mol. Sci. 2021, 22, 7765. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.K.; Kho, A.R.; Lee, S.H.; Jeong, J.H.; Kang, B.S.; Kang, D.H.; Park, M.K.; Park, K.H.; Lim, M.S.; Choi, B.Y.; et al. Transient Receptor Potential Melastatin 2 (TRPM2) Inhibition by Antioxidant, N-Acetyl-l-Cysteine, Reduces Global Cerebral Ischemia-Induced Neuronal Death. Int. J. Mol. Sci. 2020, 21, 6026. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Nakaki, T. Glutathione in Cellular Redox Homeostasis: Association with the Excitatory Amino Acid Carrier 1 (EAAC1). Molecules 2015, 20, 8742–8758. [Google Scholar] [CrossRef] [PubMed]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Won, S.J.; Yoo, B.H.; Brennan, A.M.; Shin, B.S.; Kauppinen, T.M.; Berman, A.E.; Swanson, R.A.; Suh, S.W. EAAC1 gene deletion alters zinc homeostasis and exacerbates neuronal injury after transient cerebral ischemia. J. Neurosci. 2010, 30, 15409–15418. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126. [Google Scholar] [CrossRef]
- Aoyama, K.; Wang, F.; Matsumura, N.; Kiyonari, H.; Shioi, G.; Tanaka, K.; Kinoshita, C.; Kikuchi-Utsumi, K.; Watabe, M.; Nakaki, T. Increased neuronal glutathione and neuroprotection in GTRAP3-18-deficient mice. Neurobiol. Dis. 2012, 45, 973–982. [Google Scholar] [CrossRef]
- Watabe, M.; Aoyama, K.; Nakaki, T. Regulation of glutathione synthesis via interaction between glutamate transport-associated protein 3-18 (GTRAP3-18) and excitatory amino acid carrier-1 (EAAC1) at plasma membrane. Mol. Pharmacol. 2007, 72, 1103–1110. [Google Scholar] [CrossRef]
- Watabe, M.; Aoyama, K.; Nakaki, T. A dominant role of GTRAP3-18 in neuronal glutathione synthesis. J. Neurosci. 2008, 28, 9404–9413. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Won, S.J.; Kim, J.H.; Sohn, M.; Song, H.K.; Chung, T.N.; Kim, T.Y.; Suh, S.W. EAAC1 gene deletion reduces adult hippocampal neurogenesis after transient cerebral ischemia. Sci. Rep. 2018, 8, 6903. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Kim, J.H.; Kim, H.J.; Lee, B.E.; Kim, I.Y.; Sohn, M.; Suh, S.W. EAAC1 gene deletion increases neuronal death and blood brain barrier disruption after transient cerebral ischemia in female mice. Int. J. Mol. Sci. 2014, 15, 19444–19457. [Google Scholar] [CrossRef] [PubMed]
- Bavarsad Shahripour, R.; Harrigan, M.R.; Alexandrov, A.V. N-acetylcysteine (NAC) in neurological disorders: Mechanisms of action and therapeutic opportunities. Brain Behav. 2014, 4, 108–122. [Google Scholar] [CrossRef]
- Kho, A.R.; Choi, B.Y.; Lee, S.H.; Hong, D.K.; Lee, S.H.; Jeong, J.H.; Park, K.H.; Song, H.K.; Choi, H.C.; Suh, S.W. Effects of Protocatechuic Acid (PCA) on Global Cerebral Ischemia-Induced Hippocampal Neuronal Death. Int. J. Mol. Sci. 2018, 19, 1420. [Google Scholar] [CrossRef]
- Lee, S.H.; Choi, B.Y.; Lee, S.H.; Kho, A.R.; Jeong, J.H.; Hong, D.K.; Suh, S.W. Administration of Protocatechuic Acid Reduces Traumatic Brain Injury-Induced Neuronal Death. Int. J. Mol. Sci. 2017, 18, 2510. [Google Scholar] [CrossRef]
- Lee, S.H.; Choi, B.Y.; Kho, A.R.; Jeong, J.H.; Hong, D.K.; Lee, S.H.; Lee, S.Y.; Lee, M.W.; Song, H.K.; Choi, H.C.; et al. Protective Effects of Protocatechuic Acid on Seizure-Induced Neuronal Death. Int. J. Mol. Sci. 2018, 19, 187. [Google Scholar] [CrossRef]
- Kho, A.R.; Choi, B.Y.; Kim, J.H.; Lee, S.H.; Hong, D.K.; Lee, S.H.; Jeong, J.H.; Sohn, M.; Suh, S.W. Prevention of hypoglycemia-induced hippocampal neuronal death by N-acetyl-L-cysteine (NAC). Amino Acids 2017, 49, 367–378. [Google Scholar] [CrossRef]
- Choi, B.Y.; Kim, J.H.; Kim, H.J.; Yoo, J.H.; Song, H.K.; Sohn, M.; Won, S.J.; Suh, S.W. Pyruvate administration reduces recurrent/moderate hypoglycemia-induced cortical neuron death in diabetic rats. PLoS ONE 2013, 8, e81523. [Google Scholar] [CrossRef]
- Choi, B.Y.; Lee, S.H.; Choi, H.C.; Lee, S.K.; Yoon, H.S.; Park, J.B.; Chung, W.S.; Suh, S.W. Alcohol dependence treating agent, acamprosate, prevents traumatic brain injury-induced neuron death through vesicular zinc depletion. Transl. Res. 2019, 207, 1–18. [Google Scholar] [CrossRef]
- Park, M.K.; Choi, B.Y.; Kho, A.R.; Lee, S.H.; Hong, D.K.; Jeong, J.H.; Kang, D.H.; Kang, B.S.; Suh, S.W. Effects of Transient Receptor Potential Cation 5 (TRPC5) Inhibitor, NU6027, on Hippocampal Neuronal Death after Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 8256. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Jang, B.G.; Kim, J.H.; Lee, B.E.; Sohn, M.; Song, H.K.; Suh, S.W. Prevention of traumatic brain injury-induced neuronal death by inhibition of NADPH oxidase activation. Brain Res. 2012, 1481, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Choi, B.Y.; Kho, A.R.; Jeong, J.H.; Hong, D.K.; Kang, D.H.; Kang, B.S.; Song, H.K.; Choi, H.C.; Suh, S.W. Inhibition of NADPH Oxidase Activation by Apocynin Rescues Seizure-Induced Reduction of Adult Hippocampal Neurogenesis. Int. J. Mol. Sci. 2018, 19, 3087. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jang, B.G.; Choi, B.Y.; Kim, H.S.; Sohn, M.; Chung, T.N.; Choi, H.C.; Song, H.K.; Suh, S.W. Post-treatment of an NADPH oxidase inhibitor prevents seizure-induced neuronal death. Brain Res. 2013, 1499, 163–172. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disorder | Animal Model | Genetic Manipulation | Treatment | Result | References |
|---|---|---|---|---|---|
| Excitatory amino-acid carrier 1 (EAAC1) | Protocatechuic acid (PCA) | ROS ↓ | |||
| Stroke | C57BL/6J SD rat | Glutamate transporter-associated protein 3–18 (GTRAP3-18) | N-acetylcysteine (NAC) | activity of microglia and astrocytes ↓ neuronal cell death ↓ | [13,22,73,75] |
| zinc accumulation ↓ GSH levels ↑ | |||||
| - | ischemic brain injury ↓ | ||||
| Traumatic brain injury (TBI) | C57BL/6J SD rat | Excitatory amino-acid carrier 1 (EAAC1) Zinc transporter 3 (ZnT3) | Protocatechuic acid (PCA) N-acetylcysteine (NAC) | ROS ↓ activity of microglia and astrocytes ↓ neuronal cell death ↓ zinc accumulation ↓ traumatic brain injury ↓ | [15,24,76] |
| Epilepsy | SD rat | Protocatechuic acid (PCA) | activity of microglia and astrocytes ↓ neuronal cell death ↓ GSH levels ↑ ROS ↓ | [77] | |
| Hypoglycemia | SD rat | Pyruvic acid N-acetylcysteine (NAC) | ROS ↓ activity of microglia and astrocytes ↓ neuronal cell death ↓ zinc accumulation ↓ GSH levels ↑ disruption of the blood–brain barrier (BBB) ↓ | [78,79] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, M.K.; Choi, B.Y.; Kho, A.R.; Lee, S.H.; Hong, D.K.; Kang, B.S.; Lee, S.H.; Suh, S.W. The Protective Role of Glutathione on Zinc-Induced Neuron Death after Brain Injuries. Int. J. Mol. Sci. 2023, 24, 2950. https://doi.org/10.3390/ijms24032950
Park MK, Choi BY, Kho AR, Lee SH, Hong DK, Kang BS, Lee SH, Suh SW. The Protective Role of Glutathione on Zinc-Induced Neuron Death after Brain Injuries. International Journal of Molecular Sciences. 2023; 24(3):2950. https://doi.org/10.3390/ijms24032950
Chicago/Turabian StylePark, Min Kyu, Bo Young Choi, A Ra Kho, Song Hee Lee, Dae Ki Hong, Beom Seok Kang, Si Hyun Lee, and Sang Won Suh. 2023. "The Protective Role of Glutathione on Zinc-Induced Neuron Death after Brain Injuries" International Journal of Molecular Sciences 24, no. 3: 2950. https://doi.org/10.3390/ijms24032950
APA StylePark, M. K., Choi, B. Y., Kho, A. R., Lee, S. H., Hong, D. K., Kang, B. S., Lee, S. H., & Suh, S. W. (2023). The Protective Role of Glutathione on Zinc-Induced Neuron Death after Brain Injuries. International Journal of Molecular Sciences, 24(3), 2950. https://doi.org/10.3390/ijms24032950