
Toll-like Receptor 2 Mediates VEGF Overexpression and Mesothelial Hyperpermeability in Tuberculous Pleural Effusion

Abstract
1. Introduction
2. Results
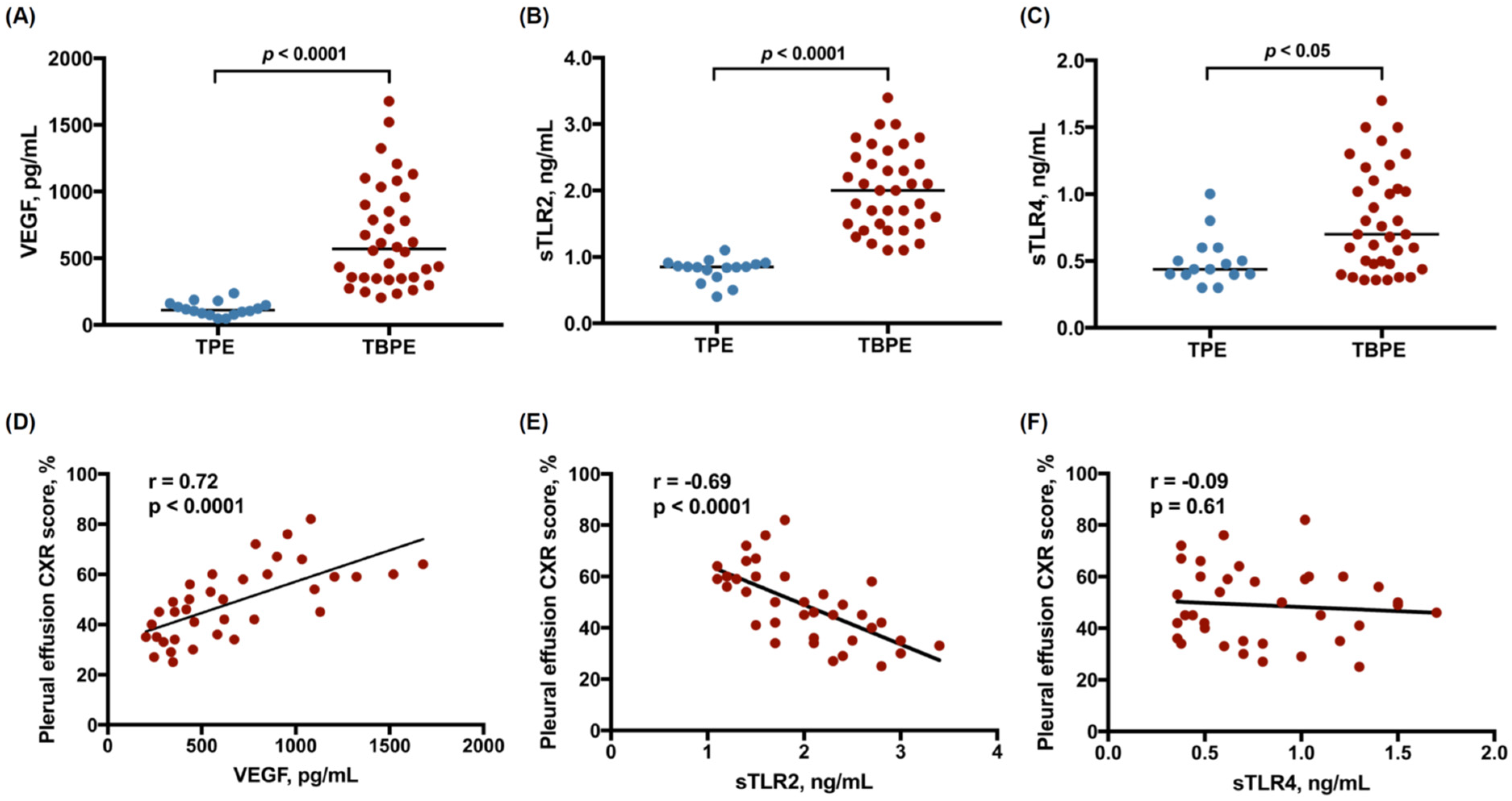
2.1. Levels of VEGF, Soluble TLR2, and Soluble TLR4 between Transudative and Tuberculous Pleural Effusions
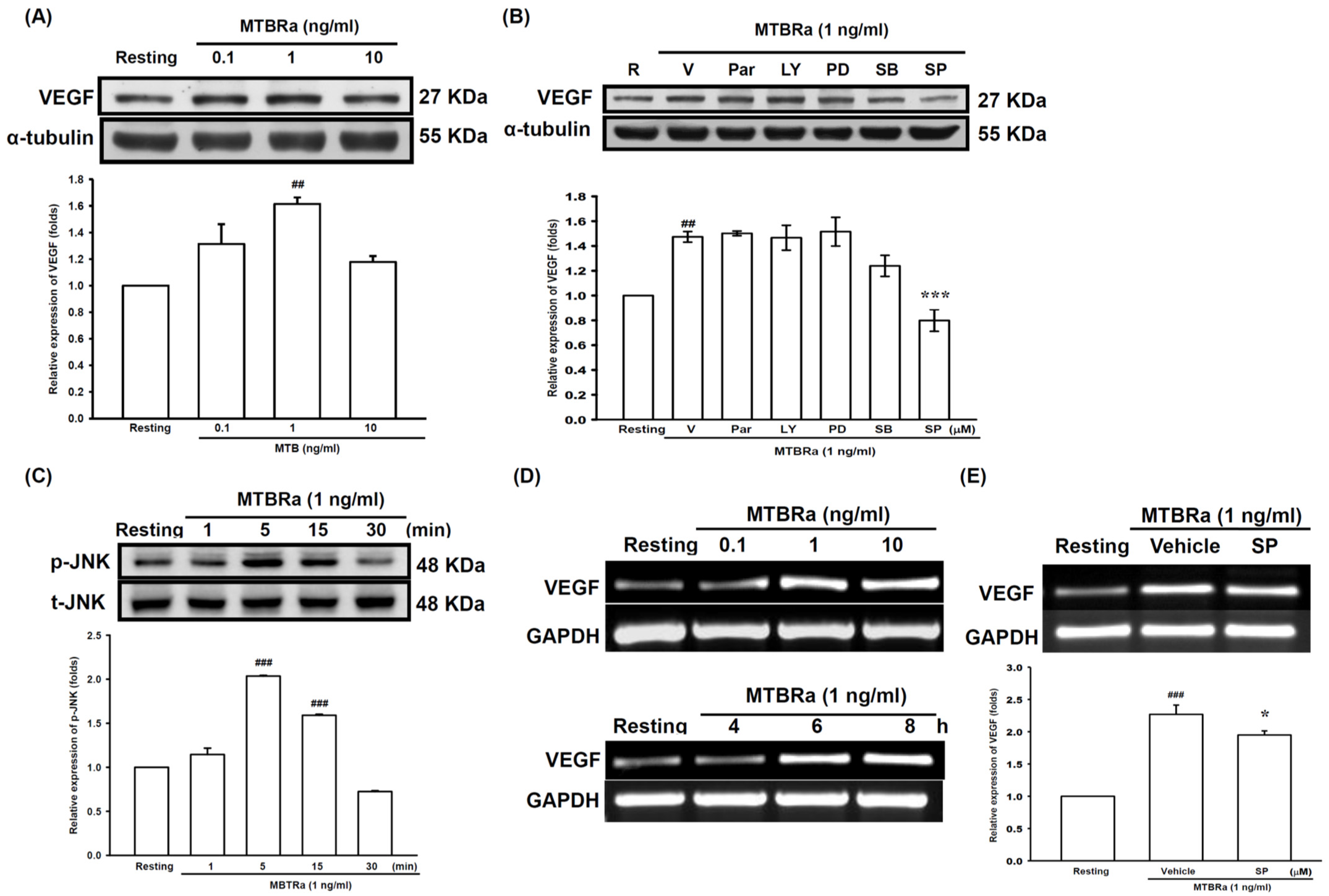
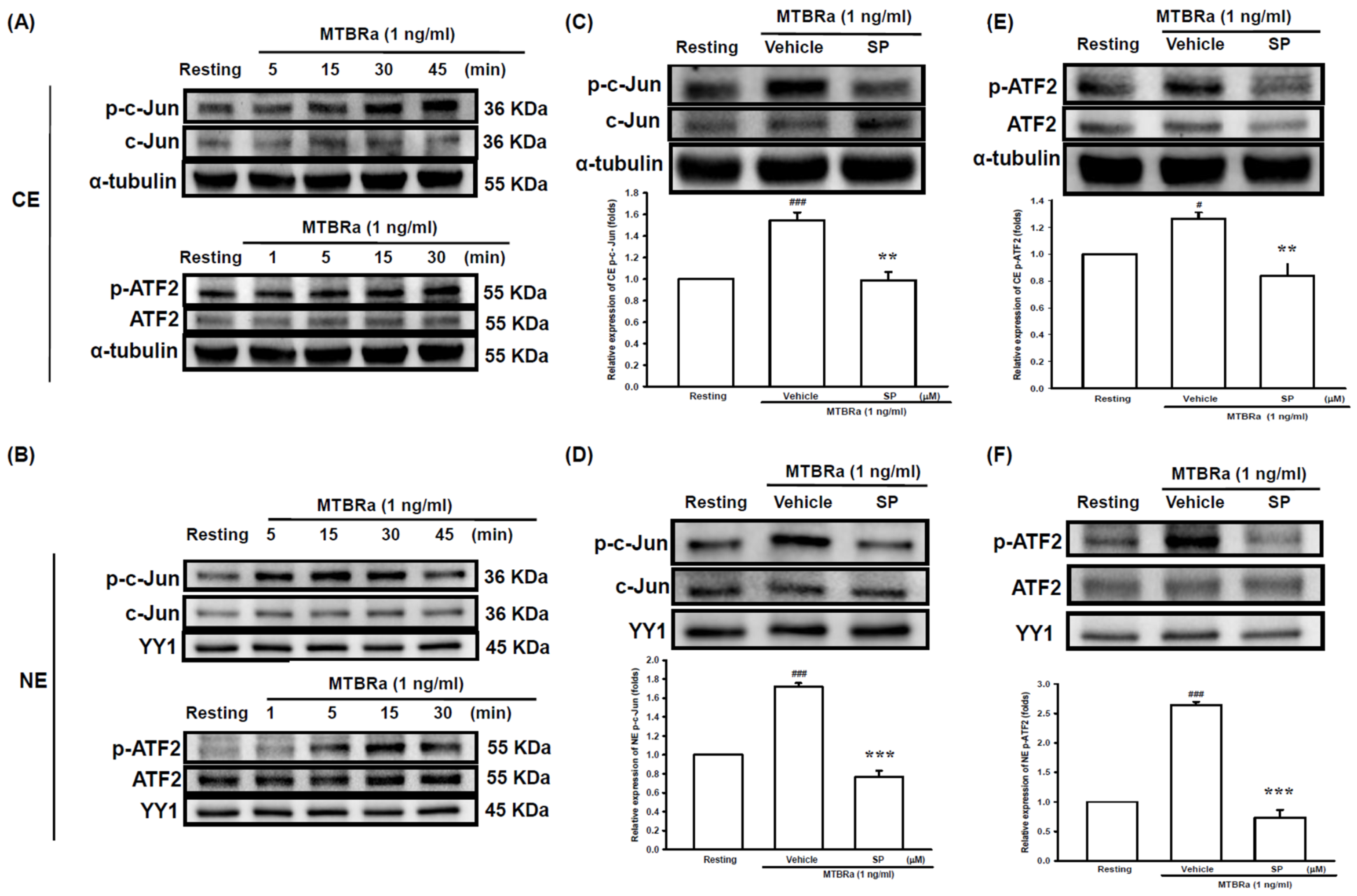
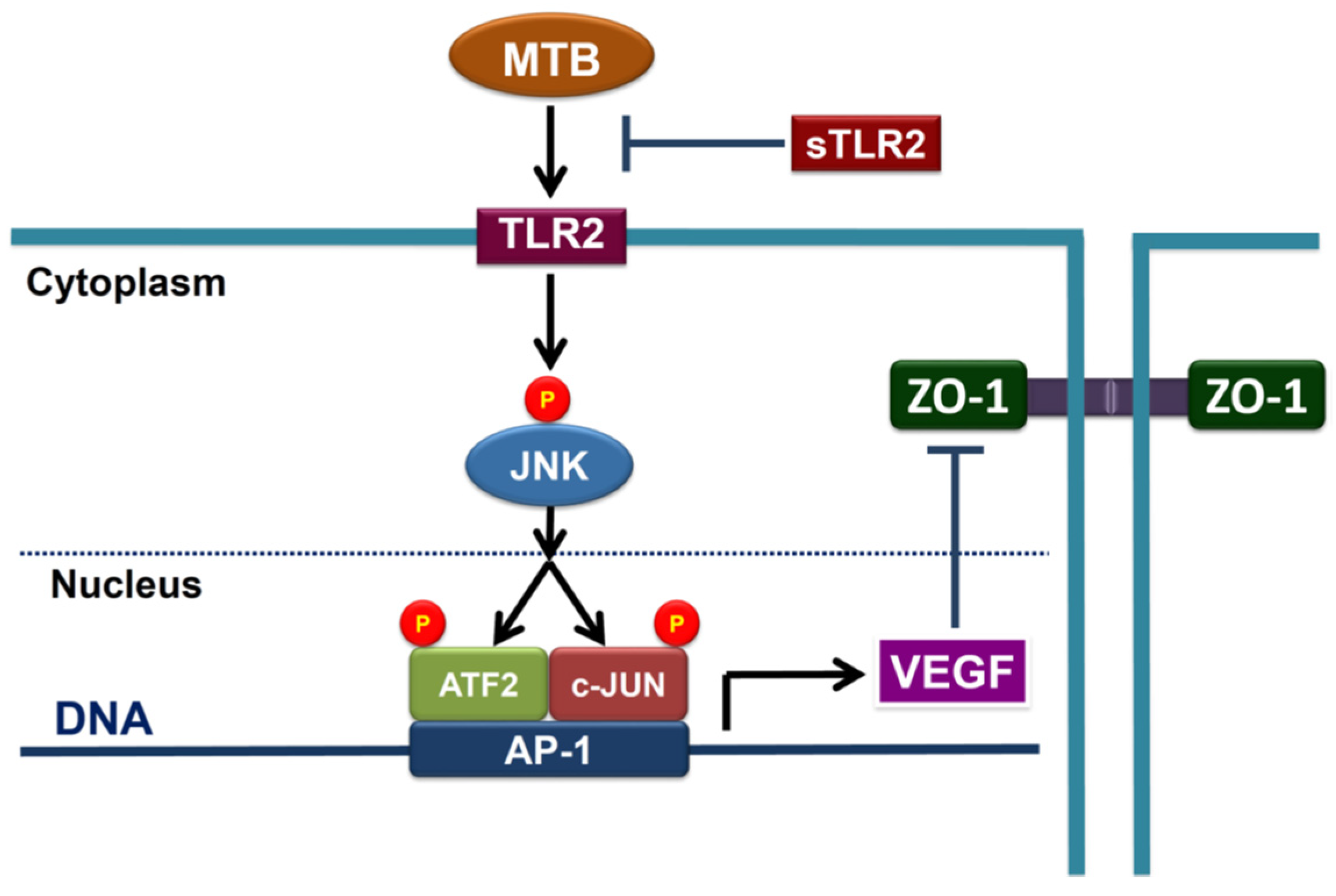
2.2. Heat-Killed Mycobacterium Tuberculosis H37Ra Increased VEGF Expression in Human PMCs through JNK/AP-1 Signaling
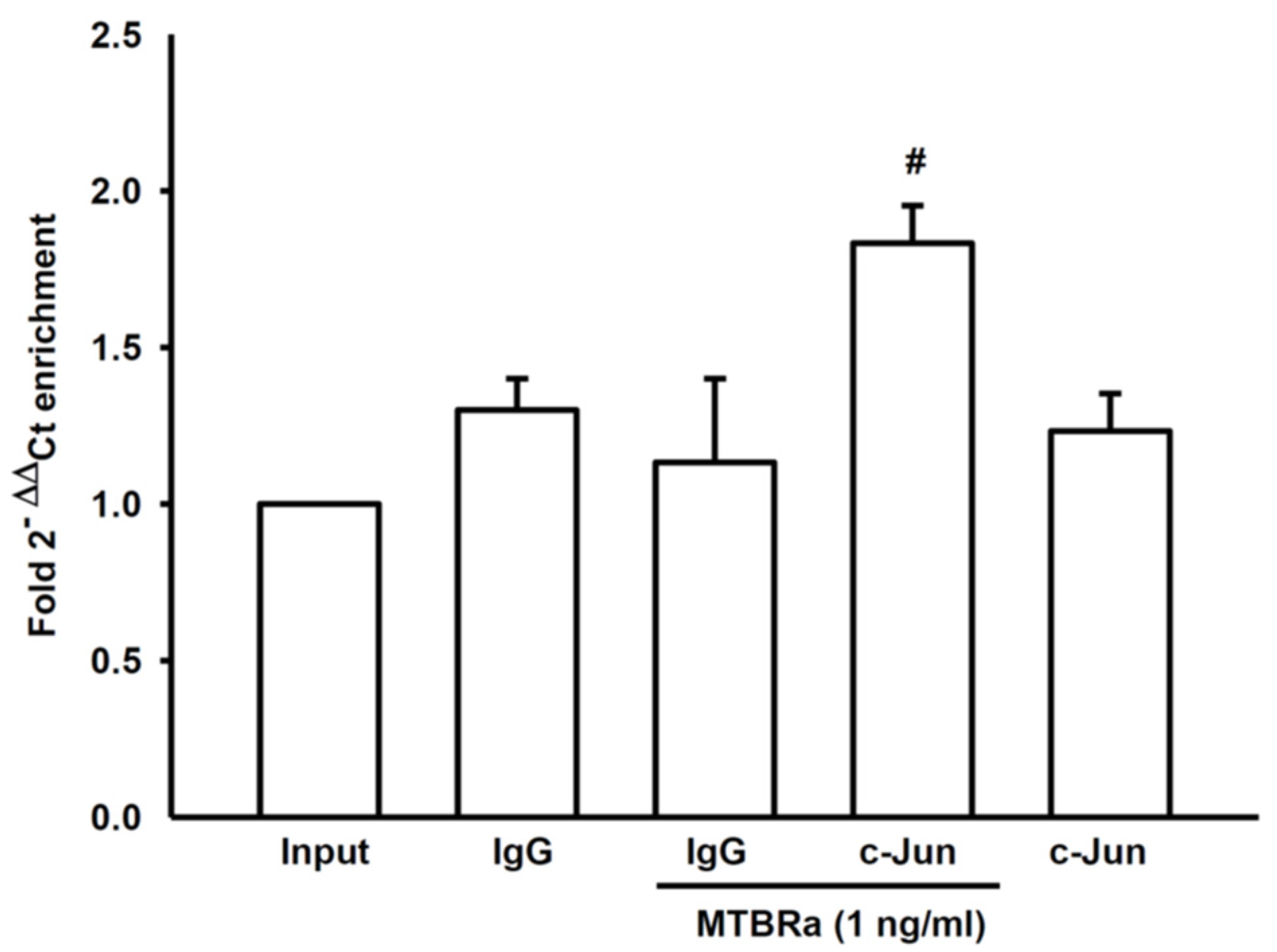
2.3. MTBRa Stimulats Binding of AP-1 to VEGF Promotor in Human PMCs
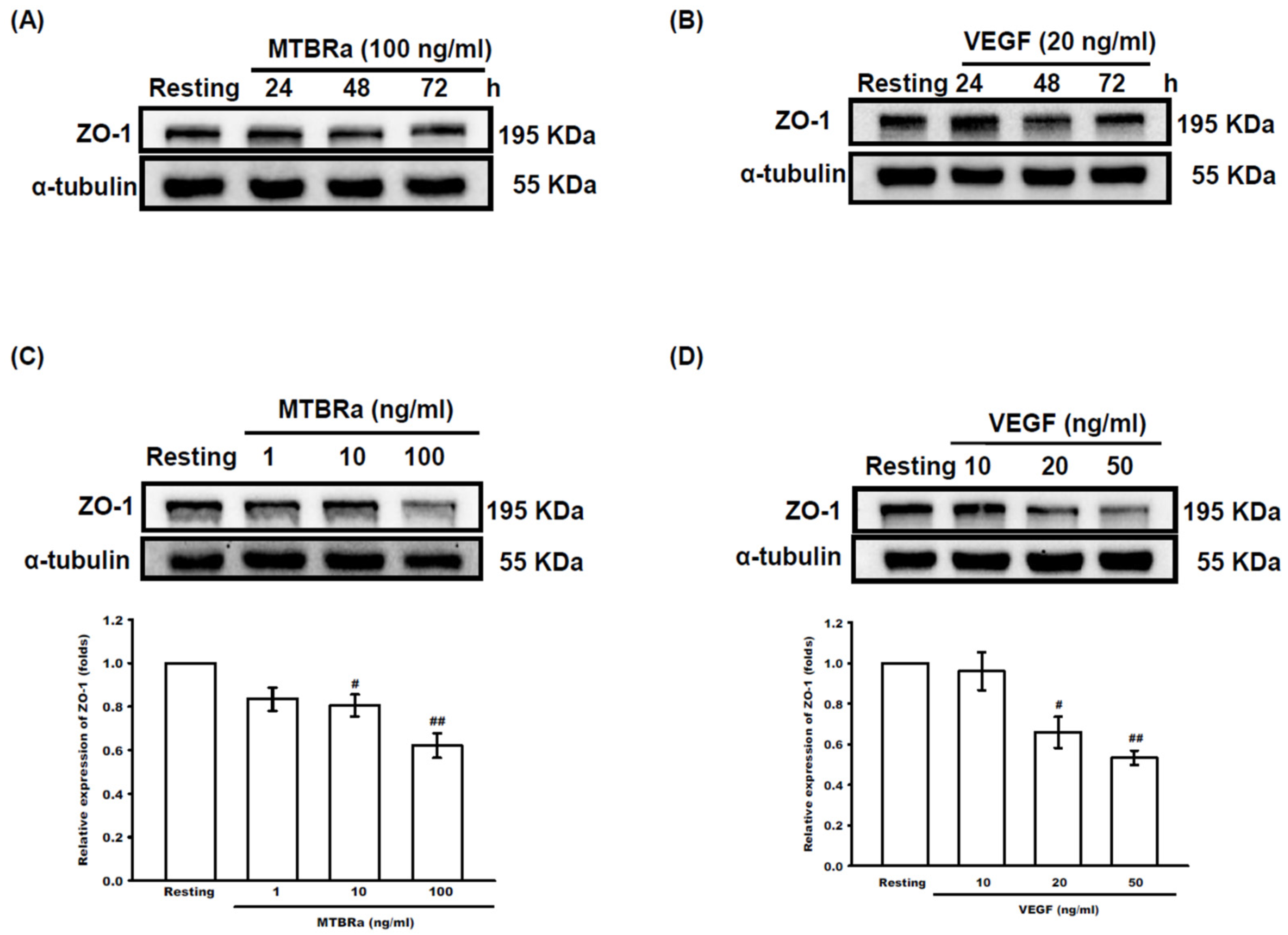
2.4. MTBRa and VEGF Downregulate ZO-1 Expression in Human PMCs
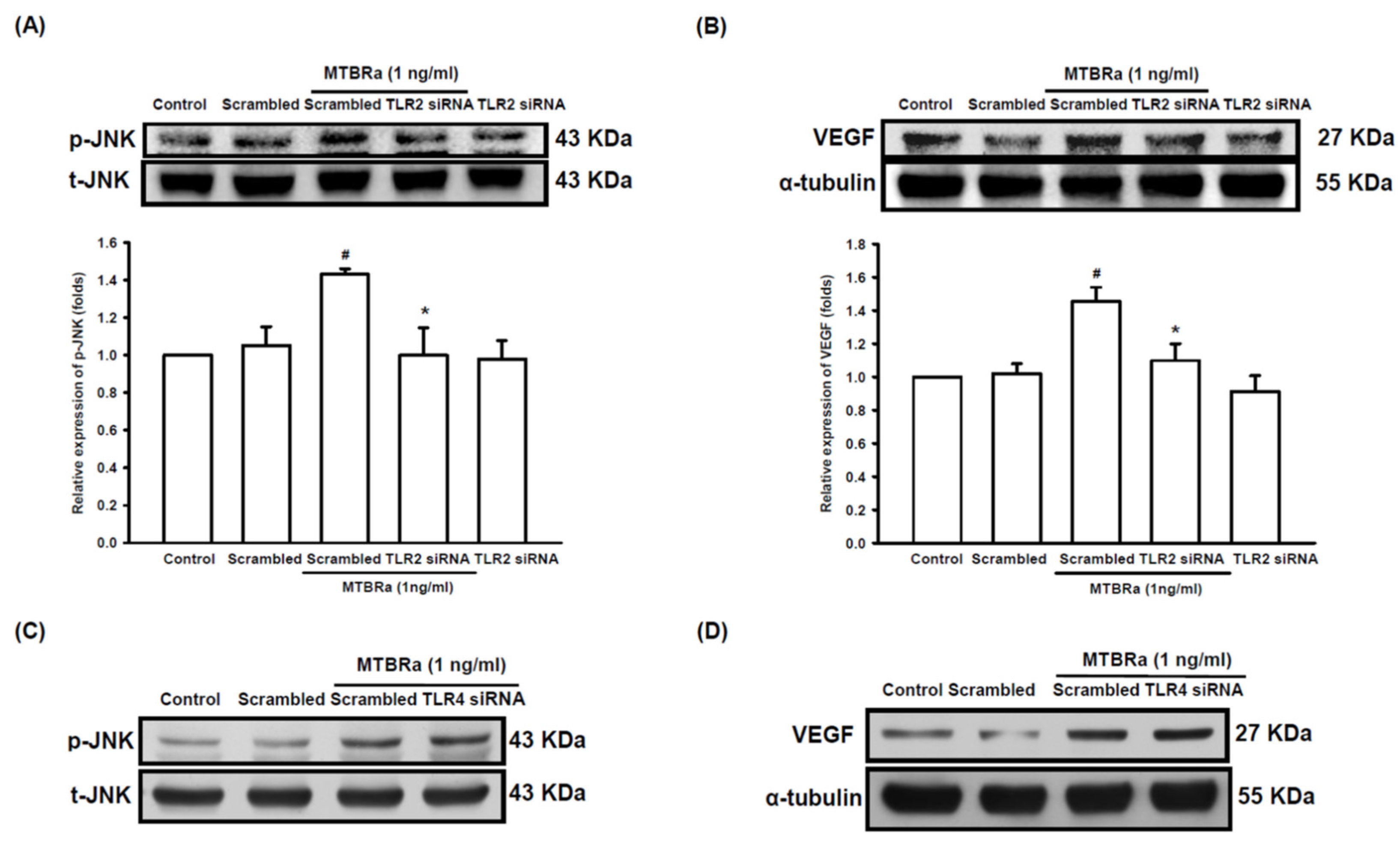
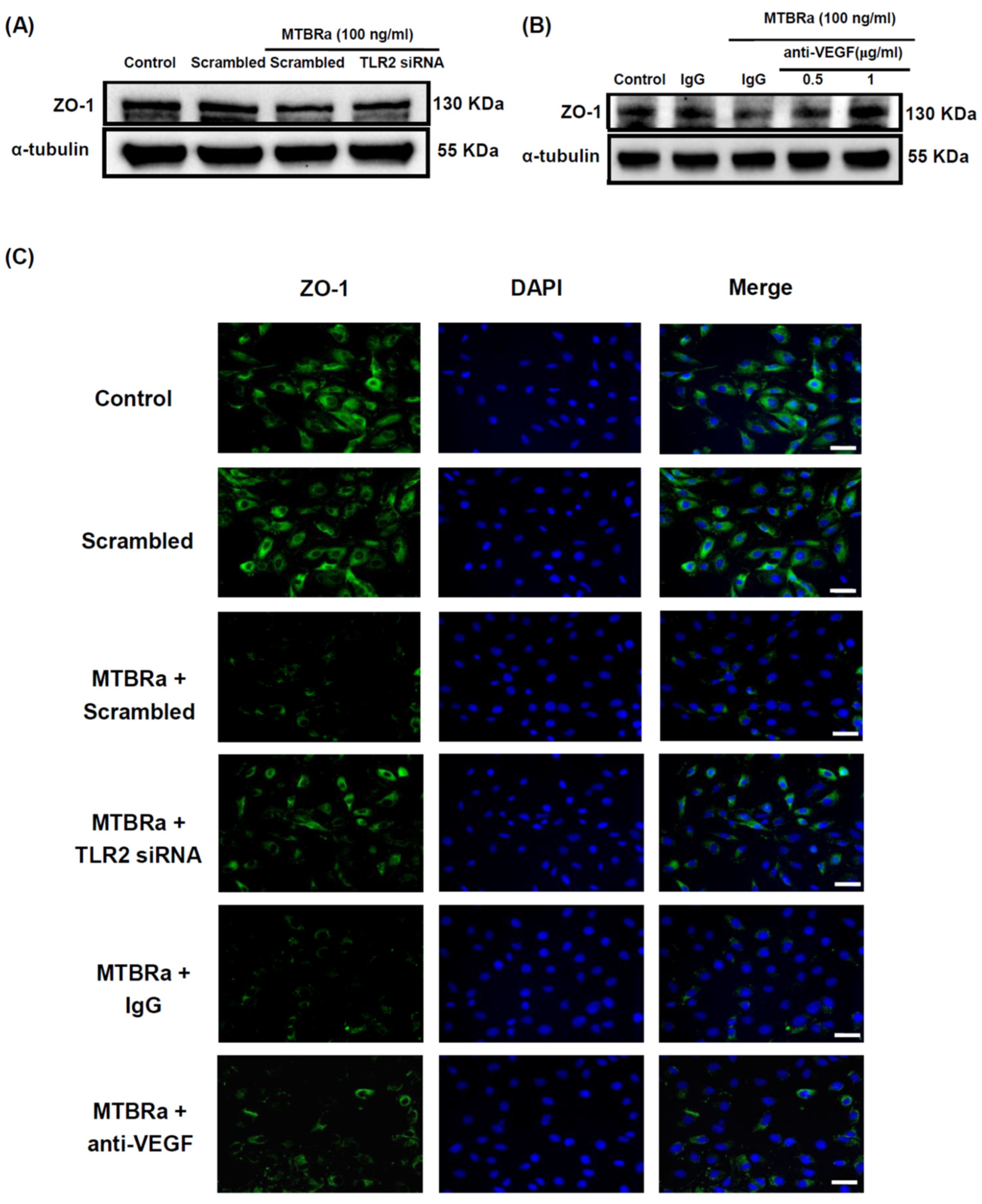
2.5. TLR2 Mediates Upregulation of VEGF and Downregulation of ZO-1 in MTBRa-Stimulated Human PMCs
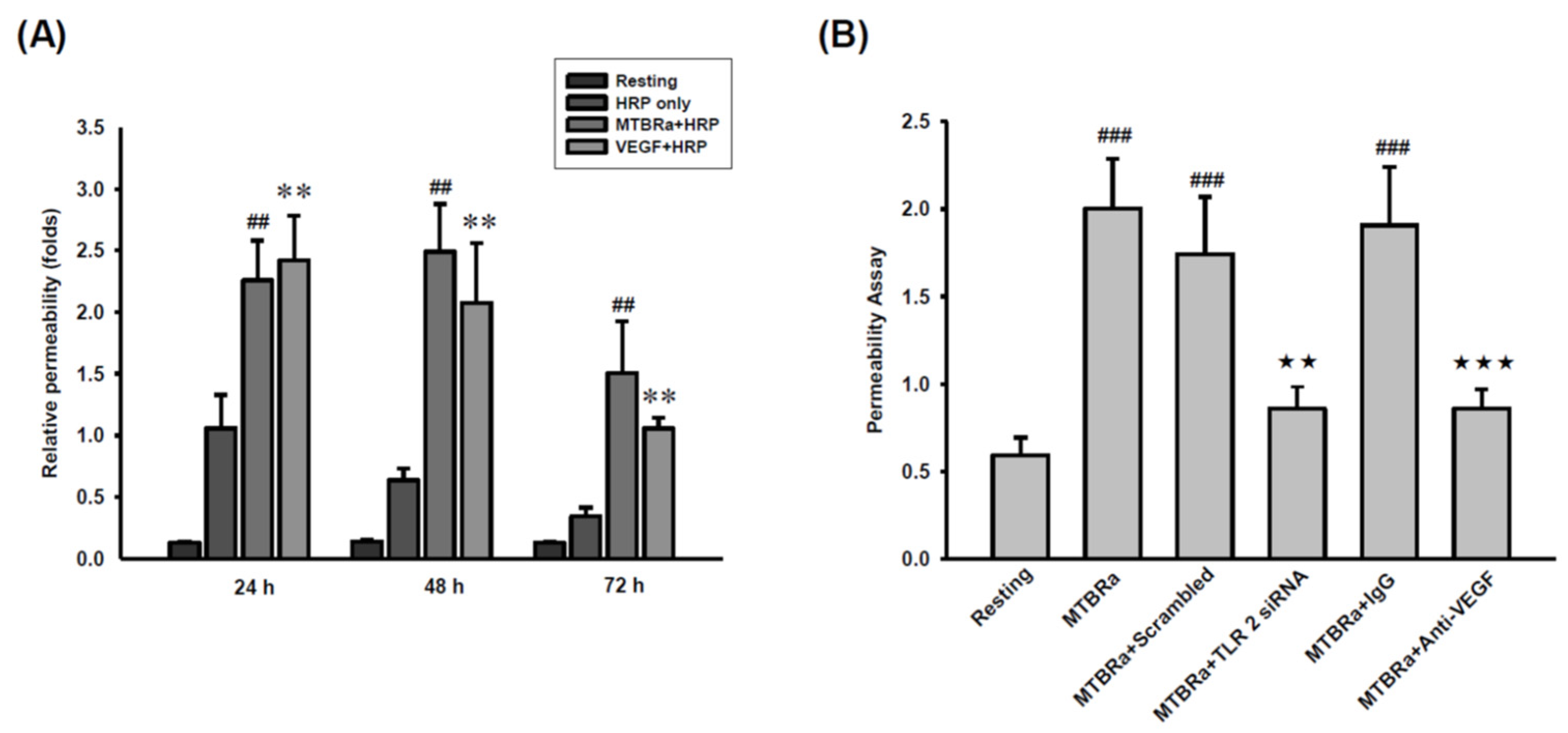
2.6. MTBRa Induces Pleural Mesothelial Hyperpermeability through TLR2/VEGF Pathway
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Patient Enrolment
4.3. Thoracentesis and Pleural Fluid Analysis
4.4. Pleural Effusion Size Chest Radiograph Score
4.5. Measurement of VEGF and Soluble TLRs in Pleural Fluids
4.6. Human Pleural Mesothelial Cell (PMC)
4.7. Western Blot
4.8. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
4.9. Nuclear and Cytoplasmic Proteins Extraction
4.10. Chromatin Immunoprecipitation-Quantitative PCR (ChIP-qPCR)
4.11. RNA Interference
4.12. Mesothelial Permeability
4.13. Immunofluorescence Staining
4.14. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dye, C.; Williams, B.G. The population dynamics and control of tuberculosis. Science 2010, 328, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Jeon, D. Tuberculous pleurisy: An update. Tuberc. Respir. Dis. 2014, 76, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Batra, H.; Antony, V.B. Pleural mesothelial cells in pleural and lung diseases. J. Thorac. Dis. 2015, 7, 964980. [Google Scholar]
- Chen, W.L.; Sheu, J.R.; Chen, R.J.; Hsiao, S.H.; Hsiao, C.J.; Chou, Y.C.; Chung, C.L.; Hsiao, G. Mycobacterium tuberculosis upregulates TNF-α expression via TLR2/ERK signaling and induces MMP-1 and MMP-9 production in human pleural mesothelial cells. PLoS ONE 2015, 10, e0137979. [Google Scholar] [CrossRef]
- Hwanga, E.H.; Kim, T.H.; Park, J.Y.; Hong, J.J.; Kim, D.H.; Ha, S.J.; Yang, S.J.; Shin, S.J.; Park, J.H. TLR2 contributes to trigger immune response of pleural mesothelial cells against Mycobacterium bovis BCG and M. tuberculosis infection. Cytokine 2017, 95, 80–87. [Google Scholar] [CrossRef]
- Henrick, B.M.; Yao, X.D.; Taha, A.Y.; German, J.B.; Rosenthal, K.L. Insights into soluble Toll-like receptor 2 as a downregulator of virally induced inflammation. Front. Immunol. 2016, 7, 291. [Google Scholar] [CrossRef] [PubMed]
- Markov, A.G.; Amasheh, S. Tight junction physiology of pleural mesothelium. Front. Physiol. 2014, 5, 221. [Google Scholar] [CrossRef]
- Mohammed, K.A.; Nasreen, N.; Hardwick, J.; Logie, C.S.; Patterson, C.E.; Antony, V.B. Bacterial induction of pleural mesothelial monolayer barrier dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L119–L125. [Google Scholar] [CrossRef]
- Gary Lee, Y.C.; Melkerneker, D.; Thompson, P.J.; Light, R.W.; Lane, K.B. Transforming growth factor-β induces vascular endothelial growth factor elaboration from pleural mesothelial cells in vivo and in vitro. Am. J. Respir. Crit. Care Med. 2002, 165, 88–94. [Google Scholar]
- Acencio, M.M.; Vargas, F.S.; Marchi, E.; Carnevale, G.G.; Teixeira, L.R.; Antonangelo, L.; Broaddus, V.C. Pleural mesothelial cells mediate inflammatory and profibrotic responses in talc-induced pleurodesis. Lung 2007, 185, 343–348. [Google Scholar] [CrossRef]
- Thickett, D.R.; Armstrong, L.; Millar, A.B. Vascular endothelial growth factor (VEGF) in inflammatory and malignant pleural effusions. Thorax 1999, 54, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Esser, S.; Lampugnani, M.G.; Corada, M.; Dejana, E.; Risau, W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J. Cell Sci. 1998, 111, 1853–1865. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Dentler, W.L.; Borchardt, R.T. VEGF increases BMEC monolayer permeability by affecting occludin expression and tight junction assembly. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H434–H440. [Google Scholar] [CrossRef]
- Leung, J.C.; Chan, L.Y.; Li, F.F.; Tang, S.C.; Chan, K.W.; Chan, T.M.; Lam, M.F.; Wieslander, A.; Lai, K.N. Glucose degradation products downregulate ZO-1 expression in human peritoneal mesothelial cells: The role of VEGF. Nephrol. Dial. Transplant. 2005, 20, 1336–1349. [Google Scholar] [CrossRef]
- Bien, M.Y.; Wu, M.P.; Chen, W.L.; Chung, C.L. VEGF correlates with inflammation and fibrosis in tuberculous pleural effusion. Sci. World J. 2015, 2015, 417124. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, K.A.; Nasreen, N.; Hardwick, J.; Van Horn, R.D.; Sanders, K.L.; Antony, V.B. Mycobacteria induces pleural mesothelial permeability by down-regulating beta-catenin expression. Lung 2003, 181, 57–66. [Google Scholar] [CrossRef]
- Iwaki, D.; Mitsuzawa, H.; Murakami, S.; Sano, H.; Konishi, M.; Akino, T.; Kuroki, Y. The extracellular toll-like receptor 2 domain directly binds peptidoglycan derived from Staphylococcus aureus. J. Biol. Chem. 2002, 277, 24315–24320. [Google Scholar] [CrossRef]
- Raby, A.C.; Le Bouder, E.; Colmont, C.; Davies, J.; Richards, P.; Coles, B.; George, C.H.; Jones, S.A.; Brennan, P.; Topley, N.; et al. Soluble TLR2 reduces inflammation without compromising bacterial clearance by disrupting TLR2 triggering. J. Immunol. 2009, 183, 506–517. [Google Scholar] [CrossRef]
- Ten Oever, J.; Kox, M.; van de Veerdonk, F.L.; Mothapo, K.M.; Slavcovici, A.; Jansen, T.L.; Tweehuysen, L.; Giamarellos-Bourboulis, E.J.; Schneeberger, P.M.; Wever, P.C.; et al. The discriminative capacity of soluble Toll-like receptor (sTLR)2 and sTLR4 in inflammatory diseases. BMC Immunol. 2014, 15, 55. [Google Scholar] [CrossRef]
- Lin, Y.; Feng, T.; Lan, J.; Chen, C.; Qin, Z.; Wu, Y.; Shi, H.; Ye, J.; Wei, C.; Wang, W.; et al. Expression of Toll-like receptor 2 and Toll-like receptor 4 in tuberculous pleural effusion. Med. Chem. 2017, 13, 569–576. [Google Scholar] [CrossRef]
- Lima, C.X.; Souza, D.G.; Amaral, F.A.; Fagundes, C.T.; Rodrigues, I.P.; Alves-Filho, J.C.; Kosco-Vilbois, M.; Ferlin, W.; Shang, L.; Elson, G.; et al. Therapeutic effects of treatment with anti-TLR2 and anti-TLR4 monoclonal antibodies in polymicrobial sepsis. PLoS ONE 2015, 10, e0132336. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.; Laird, M.H.; Schwarz, R.S.; Greene, S.; Dyson, T.; Snyder, G.A.; Xiao, T.S.; Chauhan, J.; Fletcher, S.; Toshchakov, V.Y.; et al. Inhibition of TLR2 signaling by small molecule inhibitors targeting a pocket within the TLR2 TIR domain. Proc. Natl. Acad. Sci. USA 2015, 112, 5455–5460. [Google Scholar] [CrossRef] [PubMed]
- Ebner, S.; Trieb, M.; Schönfeld, M.; Wietzorrek, G.; Santos-Sierra, S. Decoy peptides derived from the extracellular domain of toll-like receptor 2 (TLR2) show anti-inflammatory properties. Bioorg. Med. Chem. 2018, 26, 4615–4623. [Google Scholar] [CrossRef] [PubMed]
- Raby, A.C.; Colmont, C.S.; Kift-Morgan, A.; Köhl, J.; Eberl, M.; Fraser, D.; Topley, N.; Labéta, M.O. Toll-like receptors 2 and 4 are potential therapeutic targets in peritoneal dialysis-associated fibrosis. J. Am. Soc. Nephrol. 2017, 28, 461–478. [Google Scholar] [CrossRef]
- Simpson, M.E.; Petri, W.A., Jr. TLR2 as a therapeutic target in bacterial infection. Trends Mol. Med. 2020, 26, 715–717. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, S.C.; Vargas, F.S.; Antonangelo, L.; Marchi, E.; Genofre, E.H.; Acencio, M.M.; Teixeira, L.R. Monoclonal anti-vascular endothelial growth factor antibody reduces fluid volume in an experimental model of inflammatory pleural effusion. Respirology 2009, 14, 1188–1193. [Google Scholar] [CrossRef]
- Noro, R.; Kobayashi, K.; Usuki, J.; Yomota, M.; Nishitsuji, M.; Shimokawa, T.; Ando, M.; Hino, M.; Hagiwara, K.; Miyanaga, A.; et al. Bevacizumab plus chemotherapy in nonsquamous non-small cell lung cancer patients with malignant pleural effusion uncontrolled by tube drainage or pleurodesis: A phase II study North East Japan Study group trial NEJ013B. Thorac. Cancer 2020, 11, 1876–1884. [Google Scholar] [CrossRef]
- Pichelmayer, O.; Zielinski, C.; Raderer, M. Response of a nonmalignant pleural effusion to bevacizumab. N. Engl. J. Med. 2005, 353, 740–741. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TPE | TBPE | p-Value | |
|---|---|---|---|
| Subjects, n | 16 | 36 | |
| Age, years | 83 (54–95) | 78 (34–90) | 0.058 |
| Male, n (%) | 9 (57) | 21 (58) | 0.888 |
| Pleural effusion CXR score, % | 53 (46–65) | 46 (19–87) | 0.107 |
| Pleural fluid | |||
| pH value | 7.38 (7.32–7.42) | 7.36 (6.90–7.51) | 0.084 |
| Glucose, mg/dL | 120 (73–152) | 122 (16–188) | 0.691 |
| LDH, IU/dL | 118 (57–135) | 293 (64–1999) | <0.0001 |
| Leukocyte count, cells/uL | 354 (115–490) | 1044 (81–15,840) | 0.001 |
| ADA, IU/L | 25 (6–39) | 124 (48–262) | <0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.-L.; Lee, K.-L.; Lai, K.S.; Tsai, J.-H.; Hsiao, S.-H.; Chung, C.-L. Toll-like Receptor 2 Mediates VEGF Overexpression and Mesothelial Hyperpermeability in Tuberculous Pleural Effusion. Int. J. Mol. Sci. 2023, 24, 2846. https://doi.org/10.3390/ijms24032846
Chen W-L, Lee K-L, Lai KS, Tsai J-H, Hsiao S-H, Chung C-L. Toll-like Receptor 2 Mediates VEGF Overexpression and Mesothelial Hyperpermeability in Tuberculous Pleural Effusion. International Journal of Molecular Sciences. 2023; 24(3):2846. https://doi.org/10.3390/ijms24032846
Chicago/Turabian StyleChen, Wei-Lin, Kai-Ling Lee, Kevin S. Lai, Jie-Heng Tsai, Shih-Hsin Hsiao, and Chi-Li Chung. 2023. "Toll-like Receptor 2 Mediates VEGF Overexpression and Mesothelial Hyperpermeability in Tuberculous Pleural Effusion" International Journal of Molecular Sciences 24, no. 3: 2846. https://doi.org/10.3390/ijms24032846
APA StyleChen, W.-L., Lee, K.-L., Lai, K. S., Tsai, J.-H., Hsiao, S.-H., & Chung, C.-L. (2023). Toll-like Receptor 2 Mediates VEGF Overexpression and Mesothelial Hyperpermeability in Tuberculous Pleural Effusion. International Journal of Molecular Sciences, 24(3), 2846. https://doi.org/10.3390/ijms24032846