
Functional Characterization of a Familial ALS-Associated Missense TBK1 (p-Arg573Gly) Mutation in Patient-Derived Lymphoblasts

 , ,
, ,  and
and 
Abstract
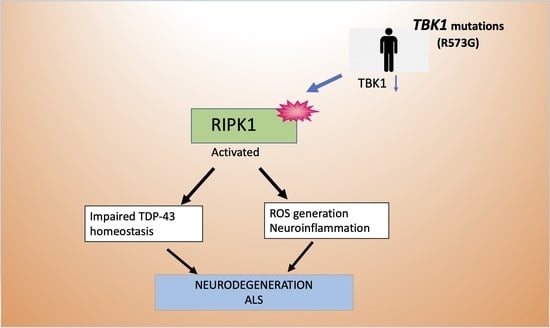
1. Introduction
2. Results
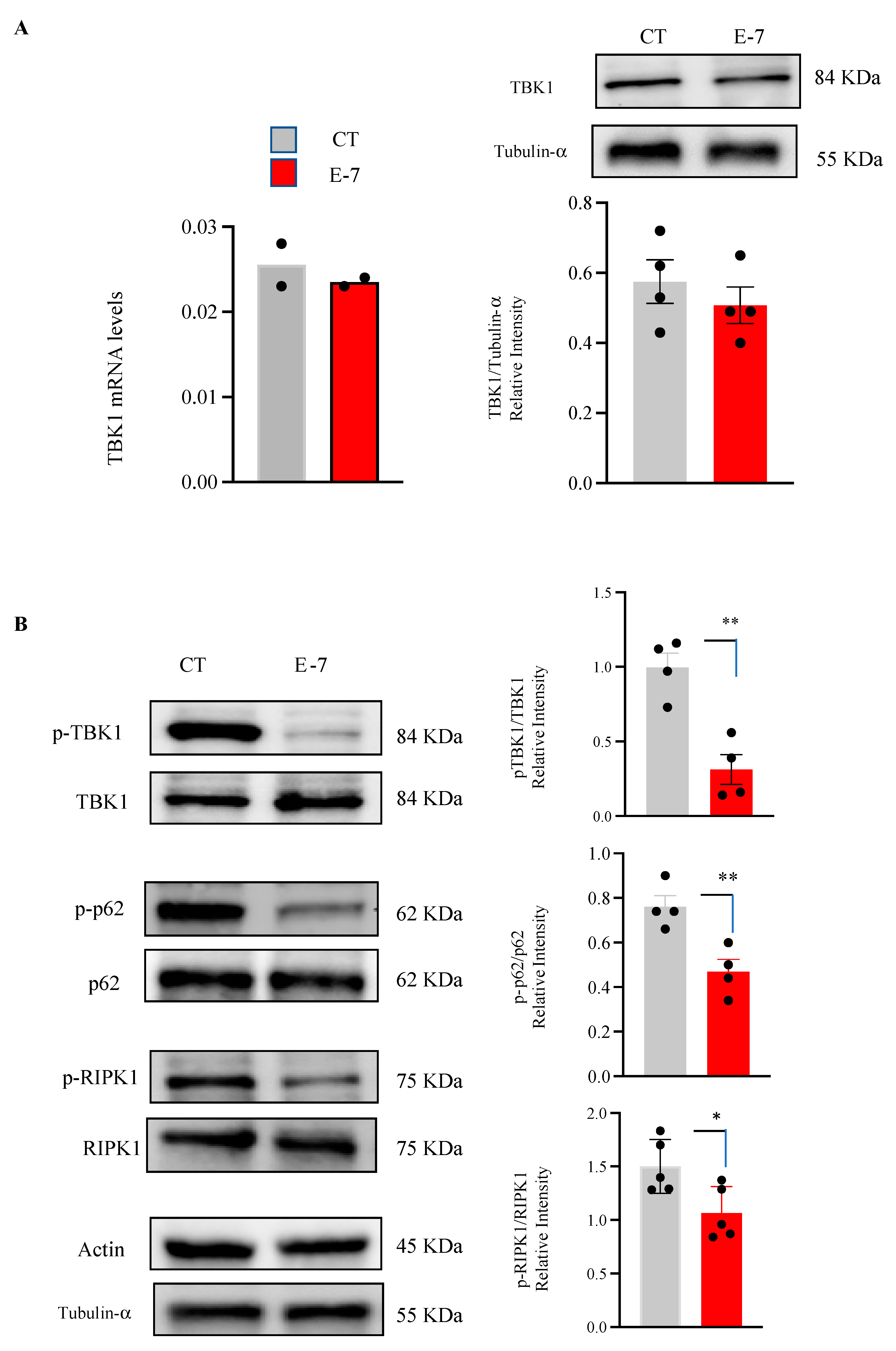
2.1. Influence of R573G TBK1 Mutation on Expression Levels and Activity of TBK1
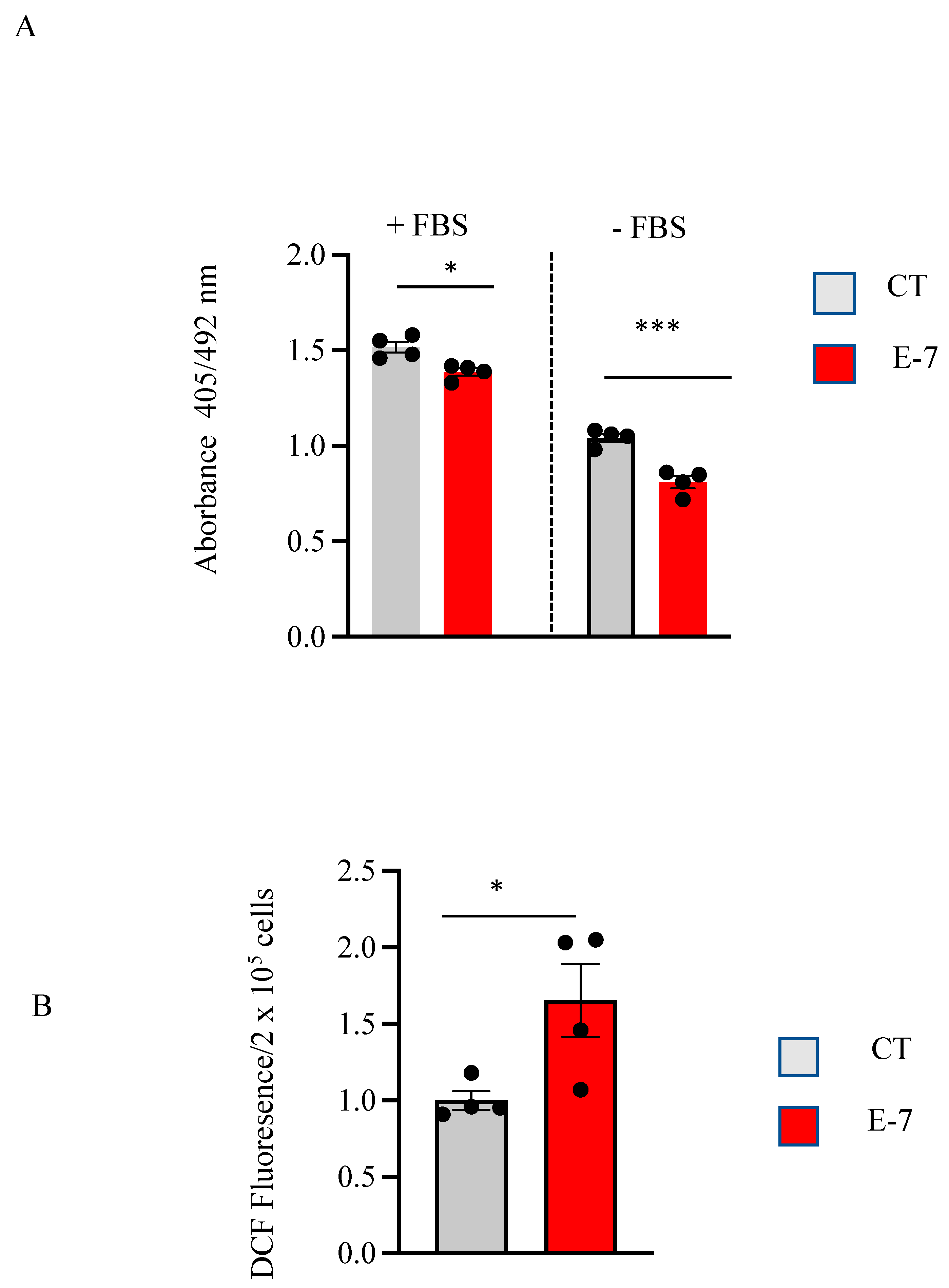
2.2. Cell Viability and ROS Generation in R573G Mutant Lymphoblasts
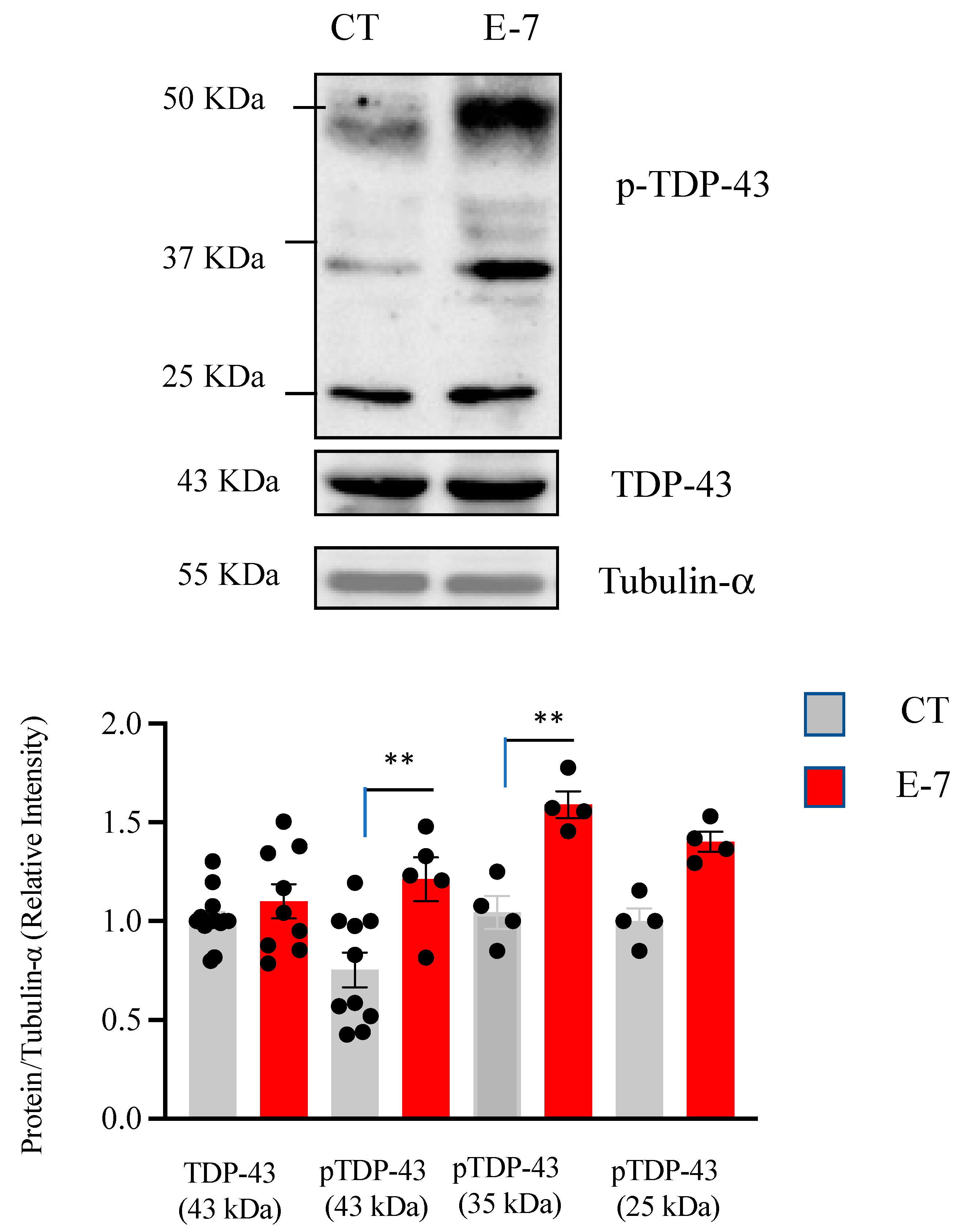
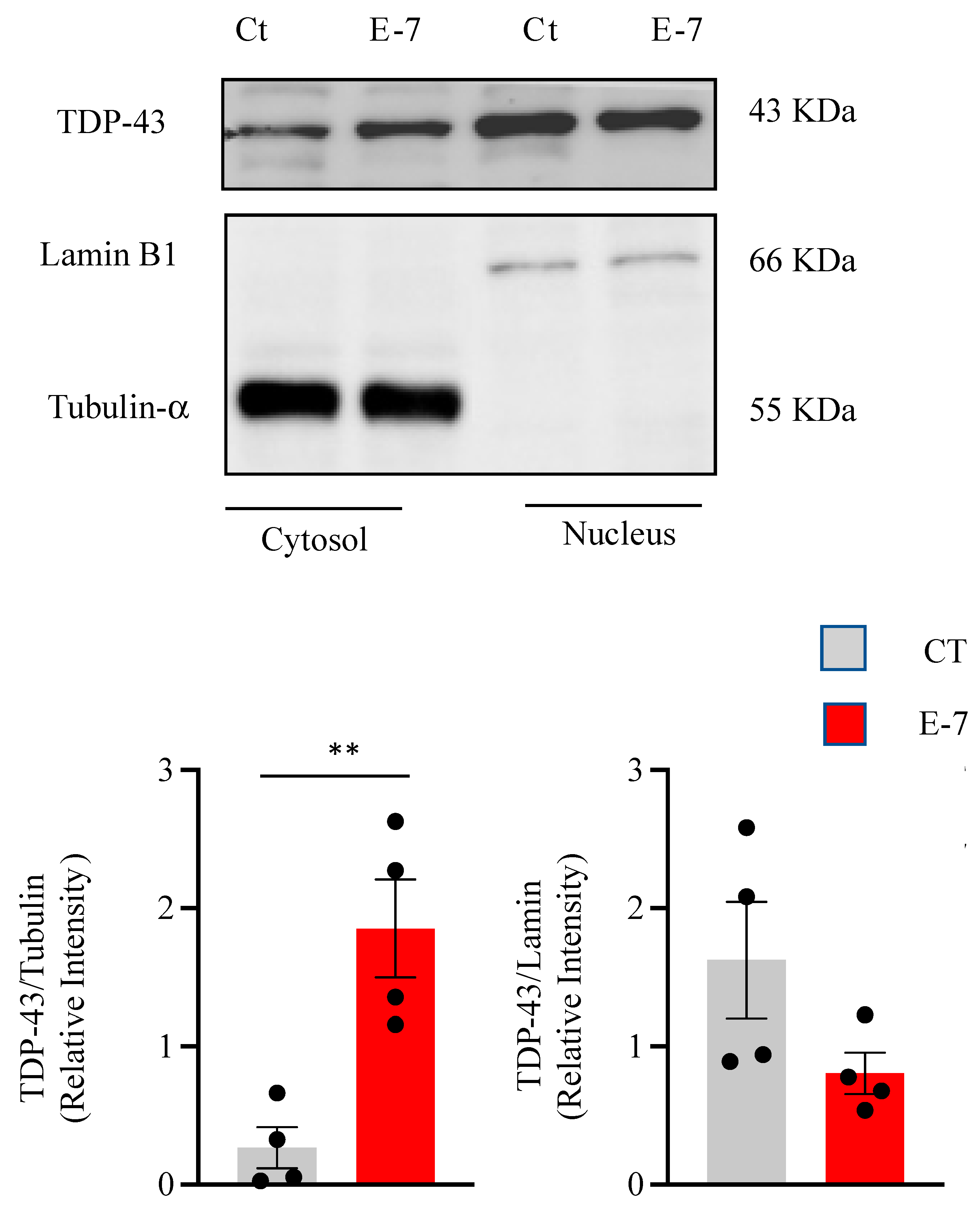
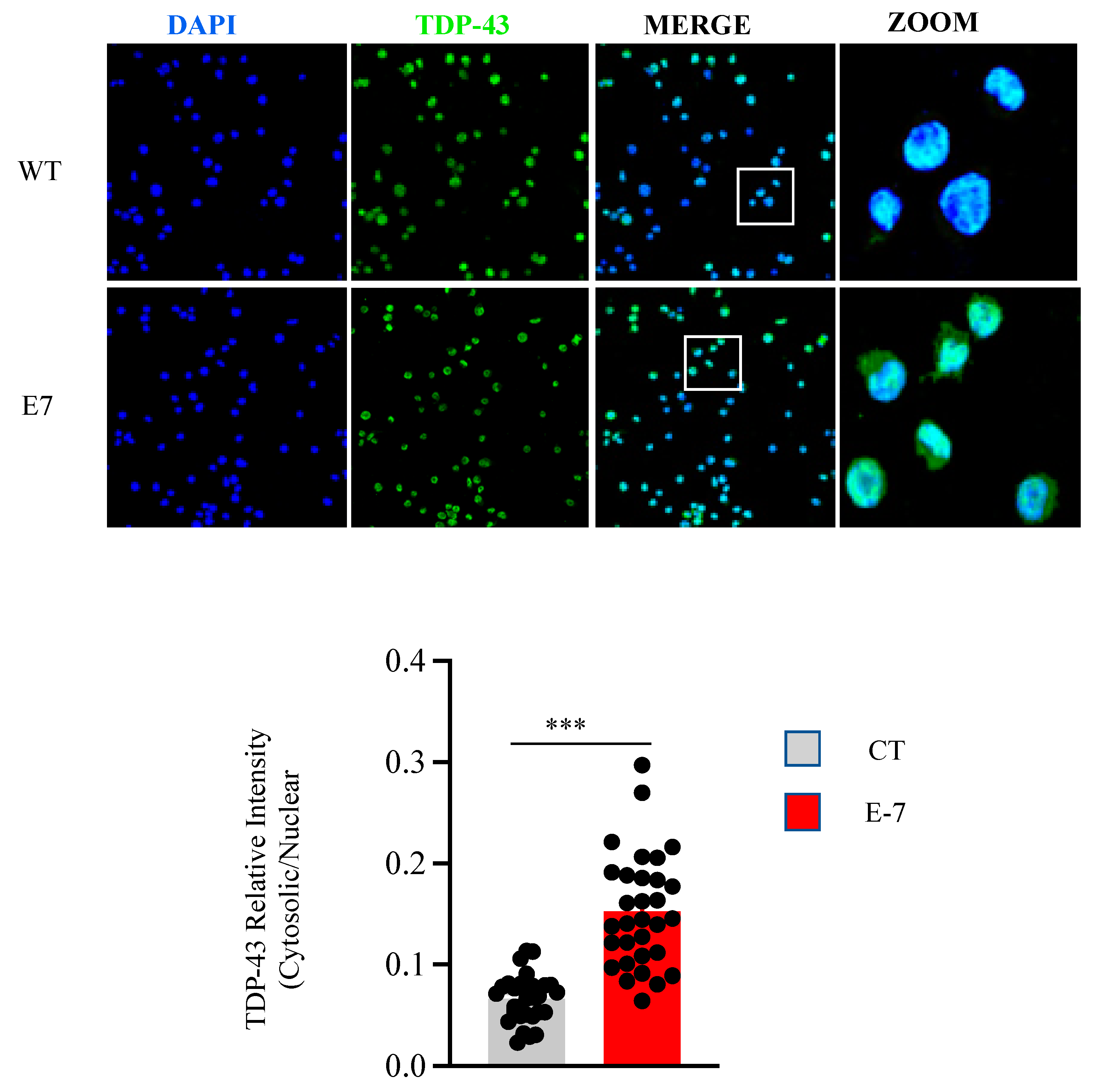
2.3. R573G TBK1 Mutation Alters TDP-43 Homeostasis
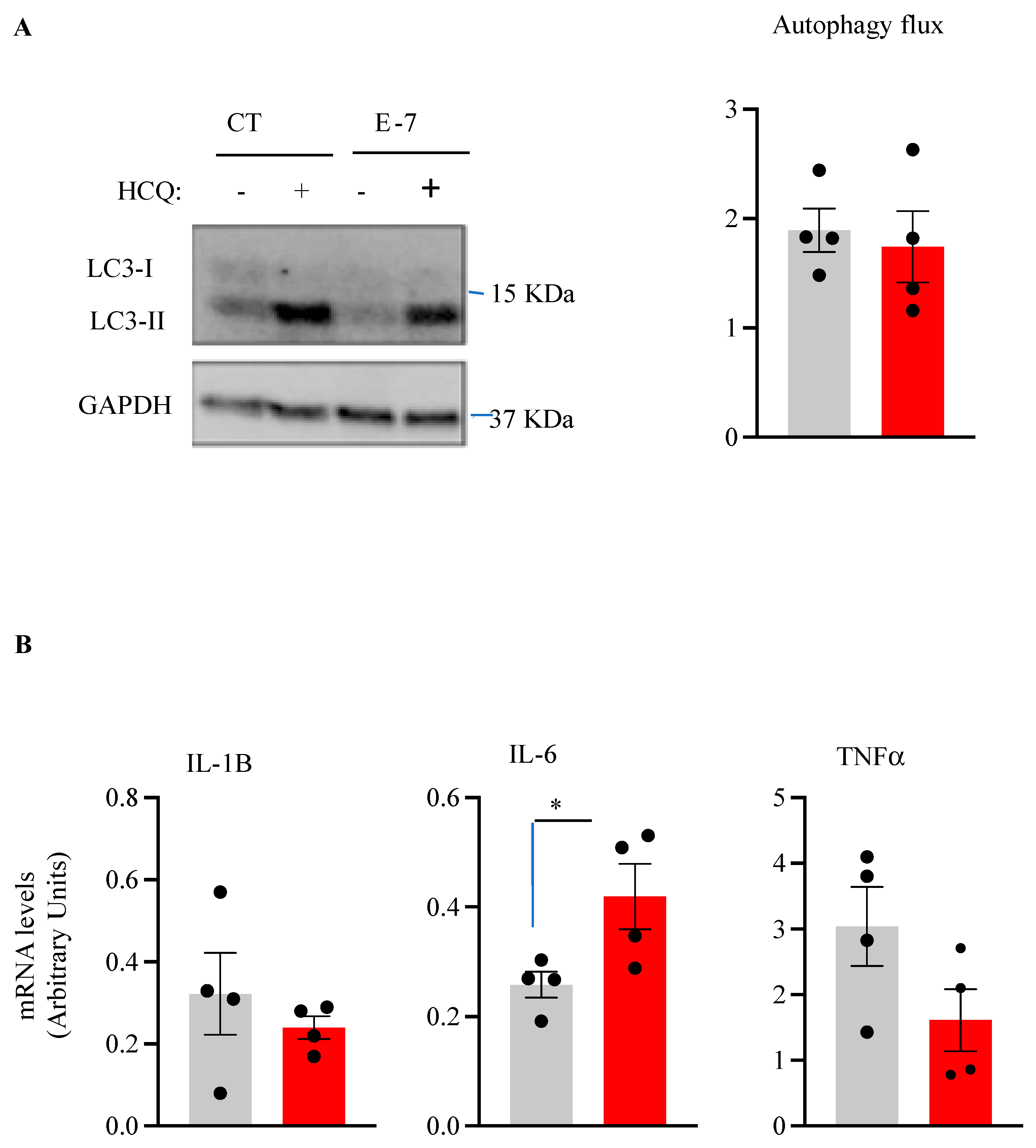
2.4. Influence of R573G TBK1 Mutation on Autophagic Flux and Pro-Inflammatory Cytokine Levels
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Culture of Human Lymphoblasts
4.3. Determination of Cell Viability
4.4. Analysis of mRNA Levels by Quantitative Real-Time PCR (q-RT-PCR)
4.5. Immunoblotting Analysis
4.6. Immunofluorescence
4.7. Determination of Reactive Oxygen Species (ROS)
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.F.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A Comprehensive Review of Amyotrophic Lateral Sclerosis. Surg. Neurol. Int. 2015, 6, 171. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef]
- Tiwari, A.; Xu, Z.; Hayward, L.J. Aberrantly Increased Hydrophobicity Shared by Mutants of Cu,Zn-Superoxide Dismutase in Familial Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2005, 280, 29771–29779. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.-F.; Wang, Q.; Krueger, B.J.; et al. Exome Sequencing in Amyotrophic Lateral Sclerosis Identifies Risk Genes and Pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef]
- le Ber, I.; de Septenville, A.; Millecamps, S.; Camuzat, A.; Caroppo, P.; Couratier, P.; Blanc, F.; Lacomblez, L.; Sellal, F.; Fleury, M.-C.; et al. TBK1 Mutation Frequencies in French Frontotemporal Dementia and Amyotrophic Lateral Sclerosis Cohorts. Neurobiol. Aging 2015, 36, 3116.e5–3116.e8. [Google Scholar] [CrossRef] [PubMed]
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A New Player in ALS Linking Autophagy and Neuroinflammation. Mol. Brain 2017, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Cheung, J.; Gerbino, V.; Ahlsén, G.; Zimanyi, C.; Hirsh, D.; Maniatis, T. Effects of ALS-Associated TANK Binding Kinase 1 Mutations on Protein–Protein Interactions and Kinase Activity. Proc. Natl. Acad. Sci. USA 2019, 116, 24517–24526. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Helgason, E.; Phung, Q.T.; Quan, C.L.; Iyer, R.S.; Lee, M.W.; Bowman, K.K.; Starovasnik, M.A.; Dueber, E.C. Molecular Basis of Tank-Binding Kinase 1 Activation by Transautophosphorylation. Proc. Natl. Acad. Sci. USA 2012, 109, 9378–9383. [Google Scholar] [CrossRef]
- Tu, D.; Zhu, Z.; Zhou, A.Y.; Yun, C.; Lee, K.-E.; Toms, A.V.; Li, Y.; Dunn, G.P.; Chan, E.; Thai, T.; et al. Structure and Ubiquitination-Dependent Activation of TANK-Binding Kinase 1. Cell Rep. 2013, 3, 747–758. [Google Scholar] [CrossRef]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 Causes Familial ALS and Fronto-Temporal Dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- de Majo, M.; Topp, S.D.; Smith, B.N.; Nishimura, A.L.; Chen, H.-J.; Gkazi, A.S.; Miller, J.; Wong, C.H.; Vance, C.; Baas, F.; et al. ALS-Associated Missense and Nonsense TBK1 Mutations Can Both Cause Loss of Kinase Function. Neurobiol. Aging 2018, 71, 266.e1–266.e10. [Google Scholar] [CrossRef]
- Gómez-Tortosa, E.; van der Zee, J.; Ruggiero, M.; Gijselinck, I.; Esteban-Pérez, J.; García-Redondo, A.; Borrego-Hernández, D.; Navarro, E.; Sainz, M.J.; Pérez-Pérez, J.; et al. Familial Primary Lateral Sclerosis or Dementia Associated with Arg573Gly TBK1 Mutation. J. Neurol. Neurosurg. Psychiatry 2017, 88, 996–997. [Google Scholar] [CrossRef] [PubMed]
- Posa, D.; Martínez-González, L.; Bartolomé, F.; Nagaraj, S.; Porras, G.; Martínez, A.; Martín-Requero, Á. Recapitulation of Pathological TDP-43 Features in Immortalized Lymphocytes from Sporadic ALS Patients. Mol. Neurobiol. 2018, 56, 2424–2432. [Google Scholar] [CrossRef]
- Martínez-González, L.; Rodríguez-Cueto, C.; Cabezudo, D.; Bartolomé, F.; Andrés-Benito, P.; Ferrer, I.; Gil, C.; Martín-Requero, Á.; Fernández-Ruiz, J.; Martínez, A.; et al. Motor Neuron Preservation and Decrease of in Vivo TDP-43 Phosphorylation by Protein CK-1δ Kinase Inhibitor Treatment. Sci. Rep. 2020, 10, 4449. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Cheng, X.; Zhong, S.; Zhang, X.; Liu, C.; Liu, F.; Zhao, C. Peripheral and Central Nervous System Immune Response Crosstalk in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2020, 14, 575. [Google Scholar] [CrossRef] [PubMed]
- Robberecht, W.; Philips, T. The Changing Scene of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef]
- Matsumoto, G.; Shimogori, T.; Hattori, N.; Nukina, N. TBK1 Controls Autophagosomal Engulfment of Polyubiquitinated Mitochondria through P62/SQSTM1 Phosphorylation. Hum. Mol. Genet. 2015, 24, 4429–4442. [Google Scholar] [CrossRef]
- Geng, J.; Ito, Y.; Shi, L.; Amin, P.; Chu, J.; Ouchida, A.T.; Mookhtiar, A.K.; Zhao, H.; Xu, D.; Shan, B.; et al. Regulation of RIPK1 Activation by TAK1-Mediated Phosphorylation Dictates Apoptosis and Necroptosis. Nat. Commun. 2017, 8, 359. [Google Scholar] [CrossRef]
- Scotter, E.L.; Chen, H.J.; Shaw, C.E. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics 2015, 12, 352–363. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine Inhibits Autophagic Flux by Decreasing Autophagosome-Lysosome Fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Cleveland, D.W. Tuning Apoptosis and Neuroinflammation: TBK1 Restrains RIPK1. Cell 2018, 174, 1339–1341. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Shaw, P.J. Oxidative Stress in ALS: Key Role in Motor Neuron Injury and Therapeutic Target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Porras, G.; Arribas-Blázquez, M.; Maestro, I.; Borrego-Hernández, D.; Boya, P.; Cerdán, S.; García-Redondo, A.; Martínez, A.; Martin-Requero, Á. Molecular Alterations in Sporadic and Sod1-Als Immortalized Lymphocytes: Towards a Personalized Therapy. Int. J. Mol. Sci. 2021, 22, 3007. [Google Scholar] [CrossRef] [PubMed]
- Said Ahmed, M.; Hung, W.-Y.; Zu, J.S.; Hockberger, P.; Siddique, T. Increased reactive oxygen species in familial amyotrophic lateral sclerosis with mutations in SOD1. J. Neurol. Sci. 2000, 176, 88–94. [Google Scholar] [CrossRef]
- Onesto, E.; Colombrita, C.; Gumina, V.; Borghi, M.O.; Dusi, S.; Doretti, A.; Fagiolari, G.; Invernizzi, F.; Moggio, M.; Tiranti, V.; et al. Gene-Specific Mitochondria Dysfunctions in Human TARDBP and C9ORF72 Fibroblasts. Acta Neuropathol. Commun. 2016, 4, 47. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.A.; Rademakers, R. The Role of Transactive Response DNA-Binding Protein-43 in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Curr. Opin. Neurol. 2008, 21, 693–700. [Google Scholar] [CrossRef]
- Vaca, G.; Martinez-Gonzalez, L.; Fernandez, A.; Rojas-Prats, E.; Porras, G.; Cuevas, E.P.; Gil, C.; Martinez, A.; Martin-Requero, Á. Therapeutic Potential of Novel Cell Division Cycle Kinase 7 Inhibitors on TDP-43-Related Pathogenesis Such as Frontotemporal Lobar Degeneration (FTLD) and Amyotrophic Lateral Sclerosis (ALS). J. Neurochem. 2021, 156, 379–390. [Google Scholar] [CrossRef]
- de Marco, G.; Lupino, E.; Calvo, A.; Moglia, C.; Buccinnà, B.; Grifoni, S.; Ramondetti, C.; Lomartire, A.; Rinaudo, M.T.; Piccinini, M.; et al. Cytoplasmic Accumulation of TDP-43 in Circulating Lymphomonocytes of ALS Patients with and without TARDBP Mutations. Acta Neuropathol. 2011, 121, 611–622. [Google Scholar] [CrossRef]
- Duan, W.; Guo, M.; Yi, L.; Zhang, J.; Bi, Y.; Liu, Y.; Li, Y.; Li, Z.; Ma, Y.; Zhang, G.; et al. Deletion of Tbk1 disrupts autophagy and reproduces behavioral and locomotor symptoms of FTD-ALS in mice. Aging 2019, 11, 2457–2476. [Google Scholar] [CrossRef]
- Ahmad, L.; Zhang, S.Y.; Casanova, J.L.; Sancho-Shimizu, V. Human TBK1: A Gatekeeper of Neuroinflammation. Trends Mol. Med. 2016, 22, 511–527. [Google Scholar] [CrossRef]
- Lafont, E.; Draber, P.; Rieser, E.; Reichert, M.; Kupka, S.; de Miguel, D.; Draberova, H.; von Mässenhausen, A.; Bhamra, A.; Henderson, S.; et al. TBK1 and IKKε Prevent TNF-Induced Cell Death by RIPK1 Phosphorylation. Nat. Cell Biol. 2018, 20, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Jin, T.; Zhu, H.; Chen, H.; Ofengeim, D.; Zou, C.; Mifflin, L.; Pan, L.; Amin, P.; Li, W.; et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell 2018, 174, 1477–1491.e19. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Sammut, B.; Wang, F.M.; Kurihara, N.; Windle, J.J.; Roodman, G.D.; Galson, D.L. TBK1 Mediates Critical Effects of Measles Virus Nucleocapsid Protein (MVNP) on Pagetic Osteoclast Formation. J. Bone Miner. Res. 2014, 29, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Tortelli, R.; Zecca, C.; Piccininni, M.; Benmahamed, S.; Dell’Abate, M.T.; Barulli, M.R.; Capozzo, R.; Battista, P.; Logroscino, G. Plasma Inflammatory Cytokines Are Elevated in ALS. Front. Neurol. 2020, 11, 552295. [Google Scholar] [CrossRef]
- Pronto-Laborinho, A.; Pinto, S.; Gromicho, M.; Pereira, M.; Swash, M.; de Carvalho, M. Interleukin-6 and Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 2019, 398, 50–53. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial Revisited: Revised Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Denizot, F.; Lang, R. Rapid Colorimetric Assay for Cell Growth and Survival: Modifications to the Tetrazolium Dye Procedure Giving Improved Sensitivity and Reliability. J. Immunol. Mrthods 1986, 89, 271–277. [Google Scholar] [CrossRef]
- Alquezar, C.; Salado, I.G.; de La Encarnación, A.; Pérez, D.I.; Moreno, F.; Gil, C.; de Munain, A.L.; Martínez, A.; Martín-Requero, Á. Targeting TDP-43 Phosphorylation by Casein Kinase-1δ Inhibitors: A Novel Strategy for the Treatment of Frontotemporal Dementia. Mol. Neurodegener. 2016, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Gender | Age | Clinical Presentation | Motor Neuron Affected |
|---|---|---|---|---|
| C105 | Female | 54 | NA 1 | NA |
| C106 | Female | 57 | NA | NA |
| C112 | Male | 71 | NA | NA |
| C126 | Male | 73 | NA | NA |
| E7 | Male | 69 | Spinal | MNS + MSI 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porras, G.; Ruiz, S.; Maestro, I.; Borrego-Hernández, D.; Redondo, A.G.; Martínez, A.; Martín-Requero, Á. Functional Characterization of a Familial ALS-Associated Missense TBK1 (p-Arg573Gly) Mutation in Patient-Derived Lymphoblasts. Int. J. Mol. Sci. 2023, 24, 2847. https://doi.org/10.3390/ijms24032847
Porras G, Ruiz S, Maestro I, Borrego-Hernández D, Redondo AG, Martínez A, Martín-Requero Á. Functional Characterization of a Familial ALS-Associated Missense TBK1 (p-Arg573Gly) Mutation in Patient-Derived Lymphoblasts. International Journal of Molecular Sciences. 2023; 24(3):2847. https://doi.org/10.3390/ijms24032847
Chicago/Turabian StylePorras, Gracia, Silvana Ruiz, Inés Maestro, Daniel Borrego-Hernández, Alberto G. Redondo, Ana Martínez, and Ángeles Martín-Requero. 2023. "Functional Characterization of a Familial ALS-Associated Missense TBK1 (p-Arg573Gly) Mutation in Patient-Derived Lymphoblasts" International Journal of Molecular Sciences 24, no. 3: 2847. https://doi.org/10.3390/ijms24032847
APA StylePorras, G., Ruiz, S., Maestro, I., Borrego-Hernández, D., Redondo, A. G., Martínez, A., & Martín-Requero, Á. (2023). Functional Characterization of a Familial ALS-Associated Missense TBK1 (p-Arg573Gly) Mutation in Patient-Derived Lymphoblasts. International Journal of Molecular Sciences, 24(3), 2847. https://doi.org/10.3390/ijms24032847