



Overnight Corticosterone and Gene Expression in Mouse Hippocampus: Time Course during Resting Period

 ,
,
Abstract
1. Introduction
2. Results
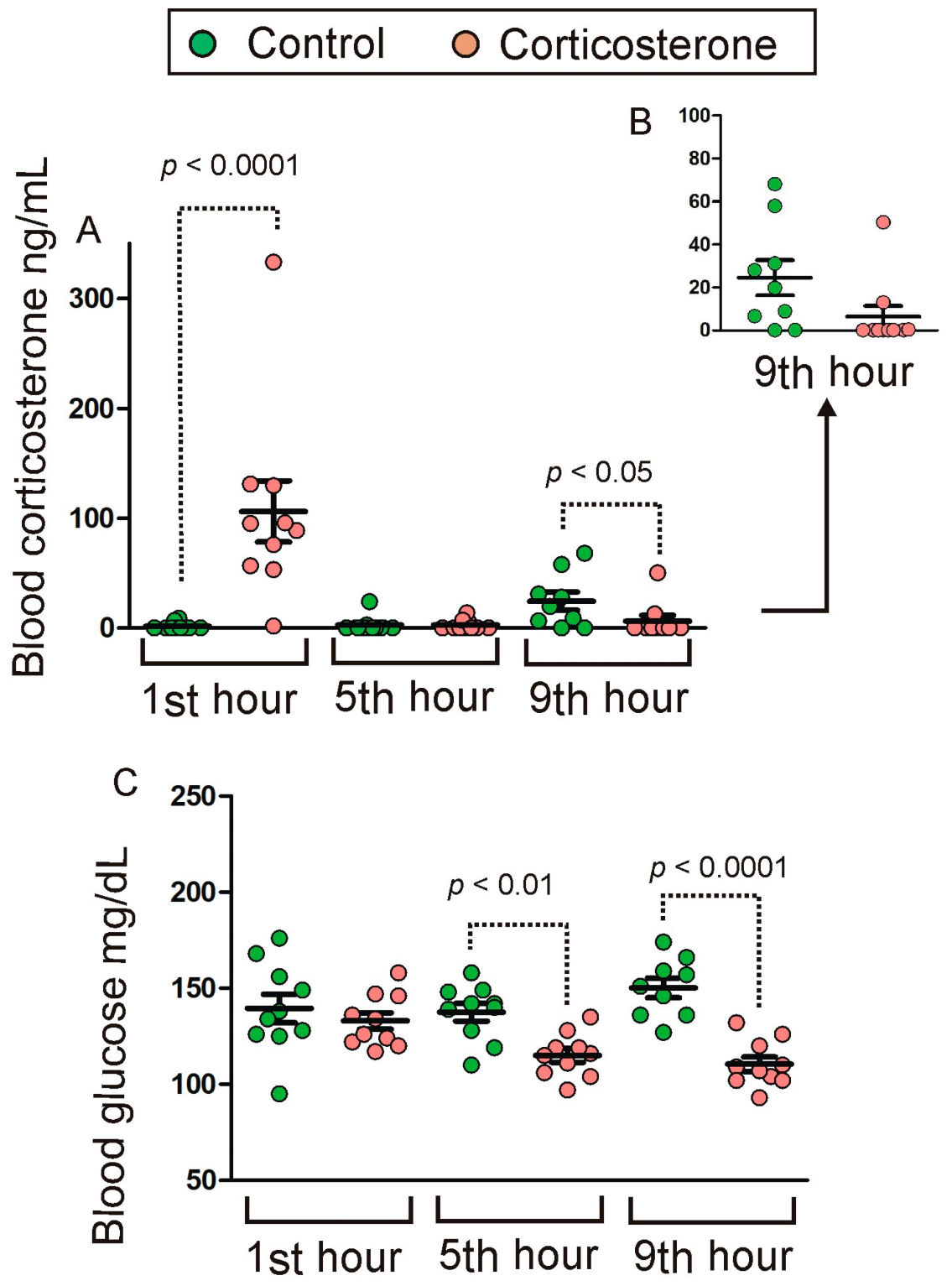
2.1. Blood Corticosterone and Glucose
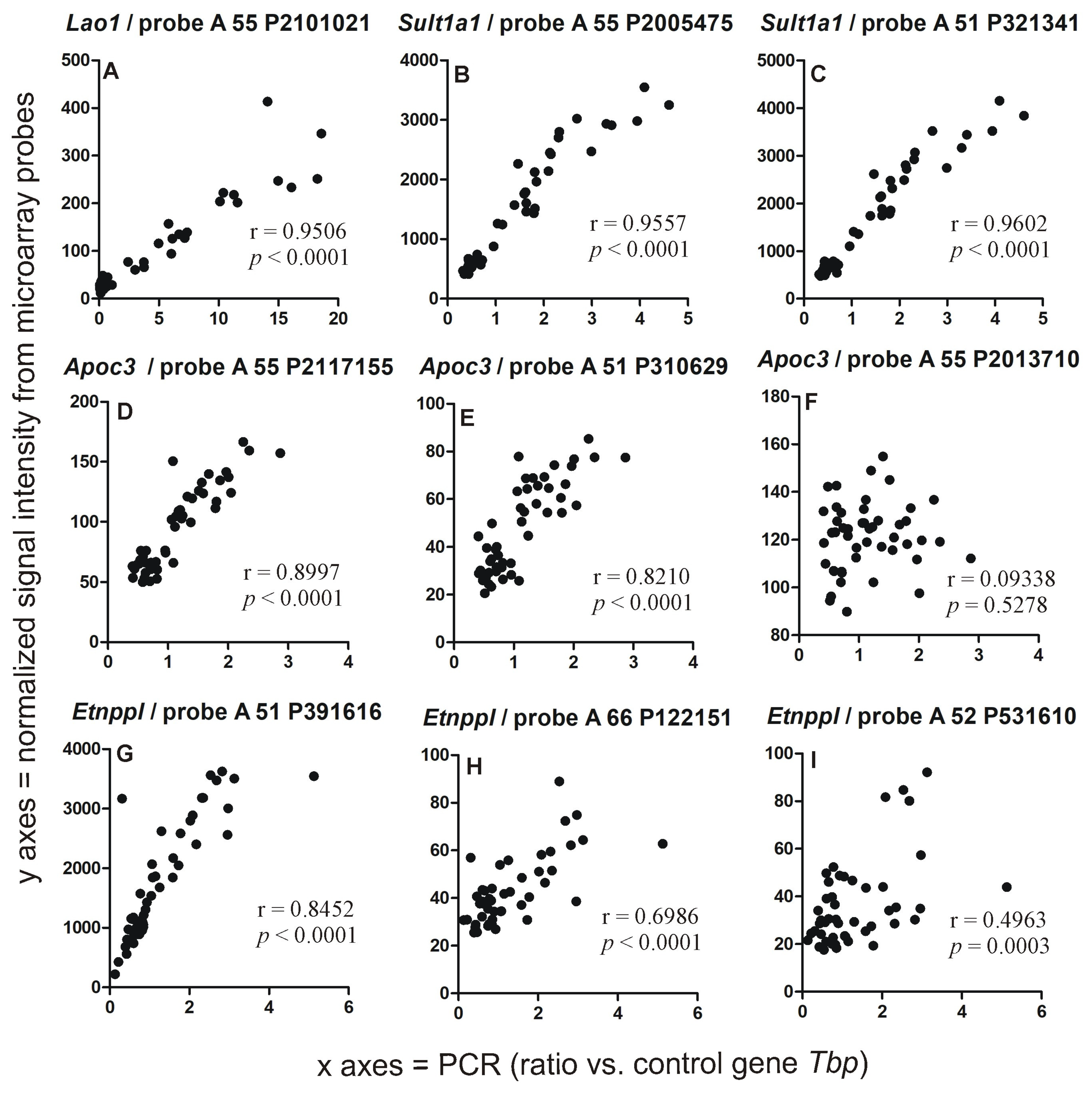
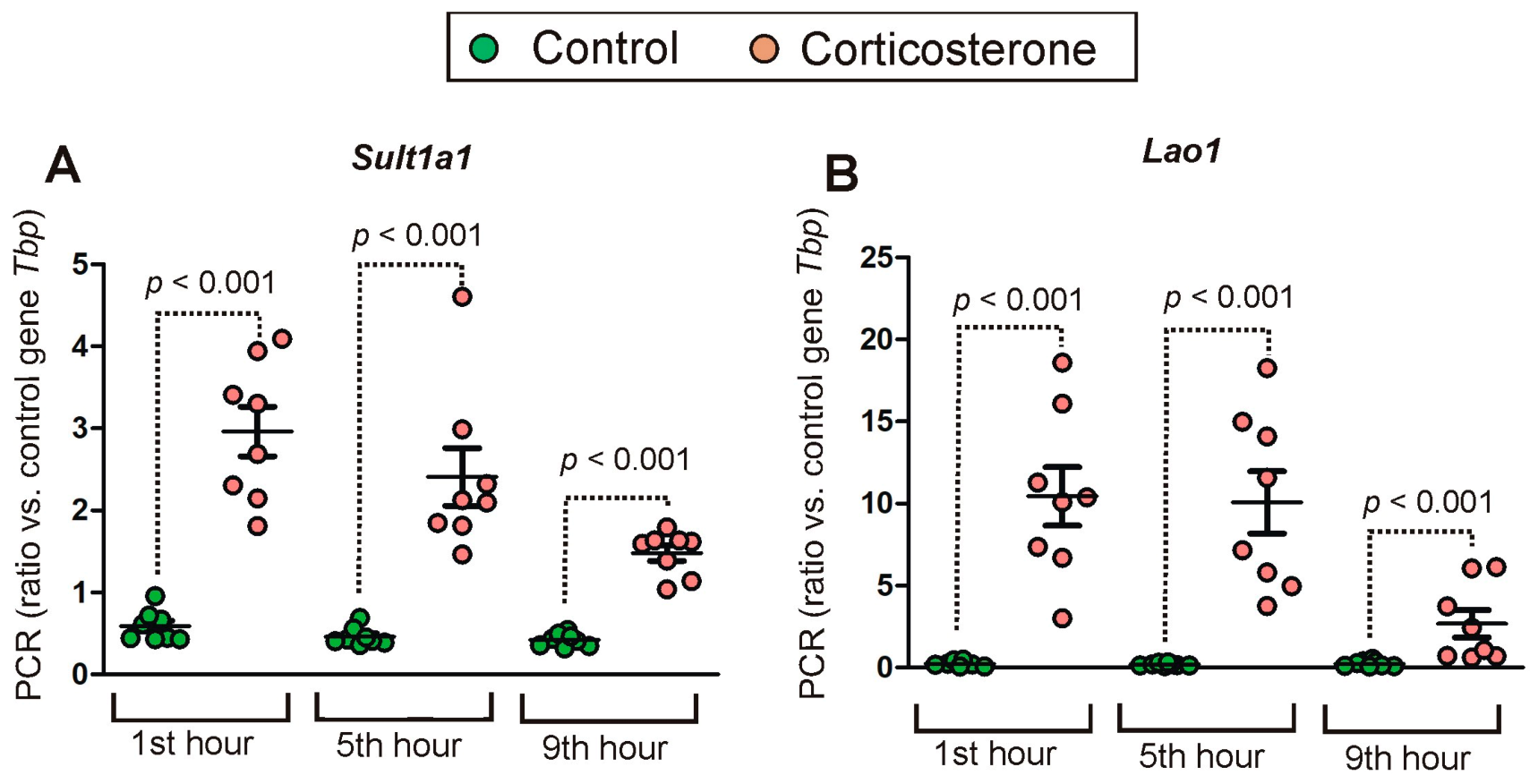
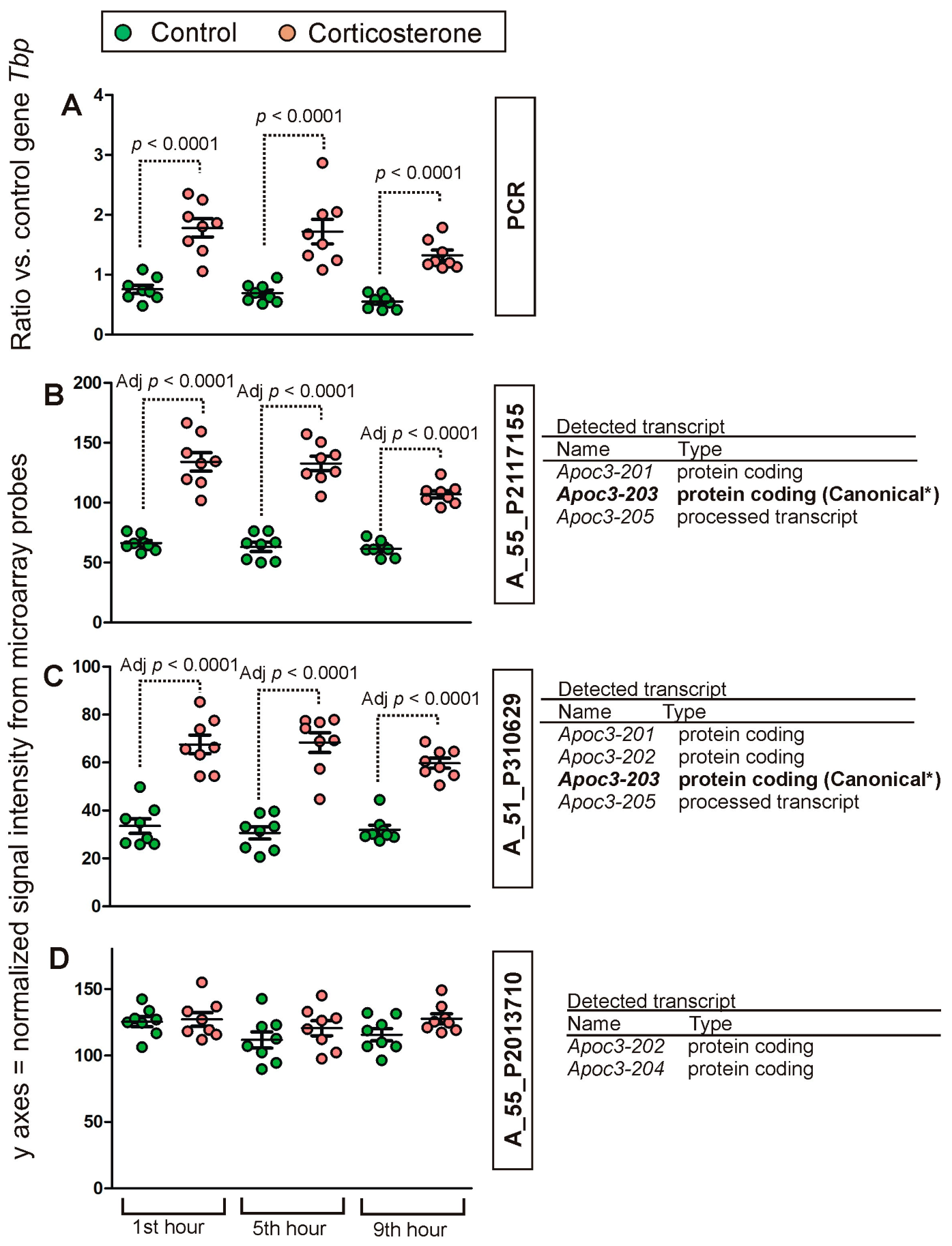
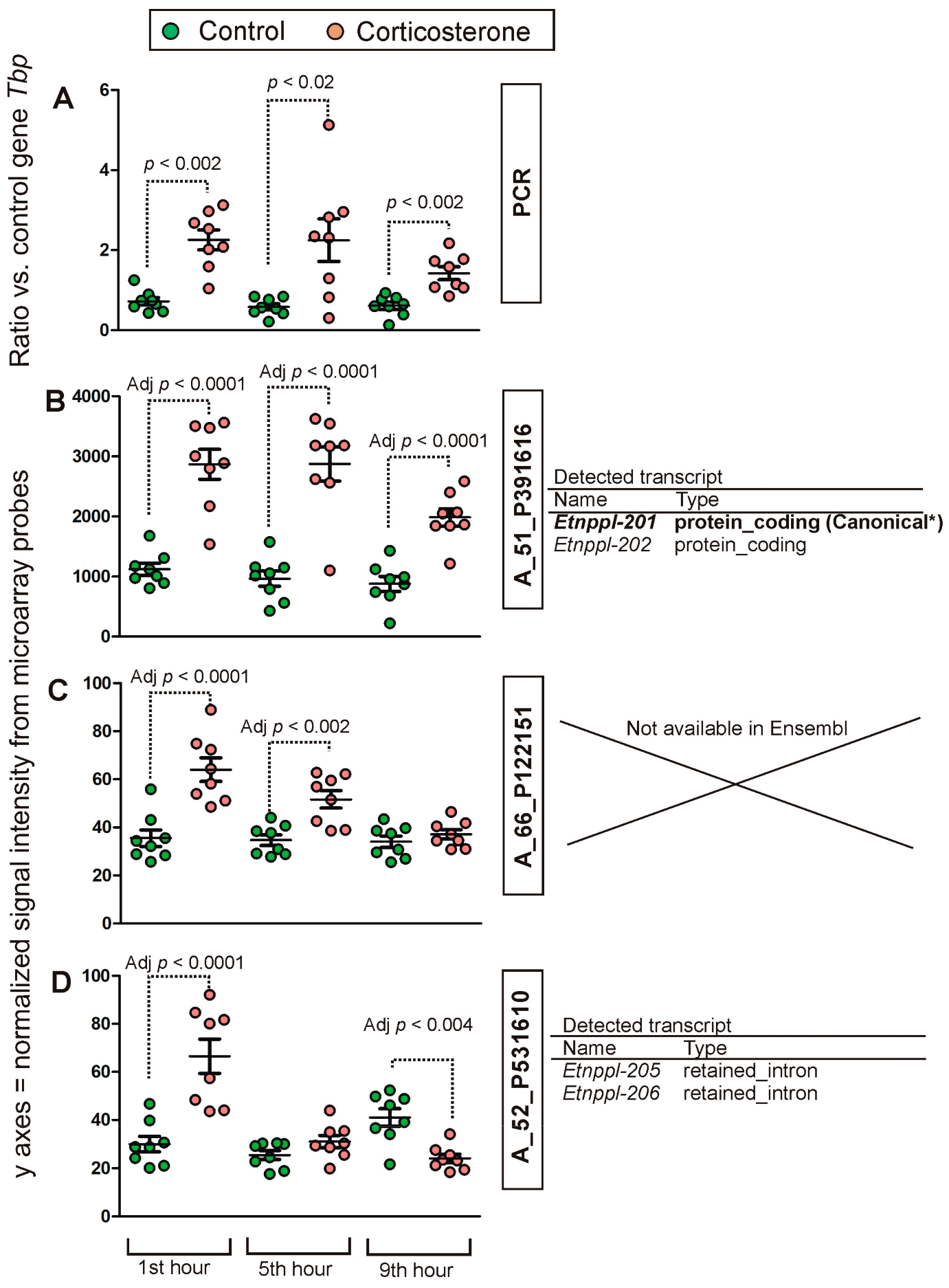
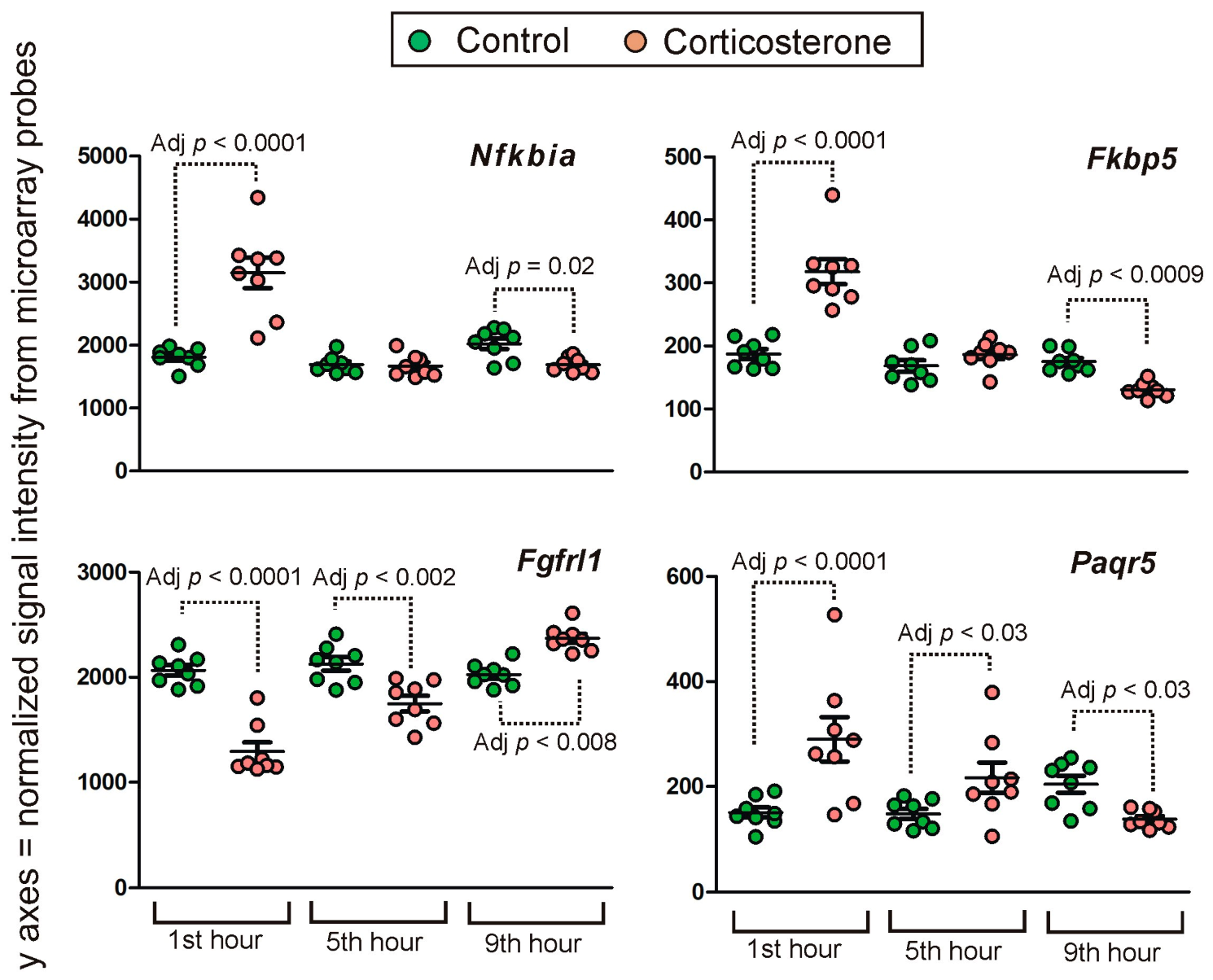
2.2. PCR Validation of Microarray Results
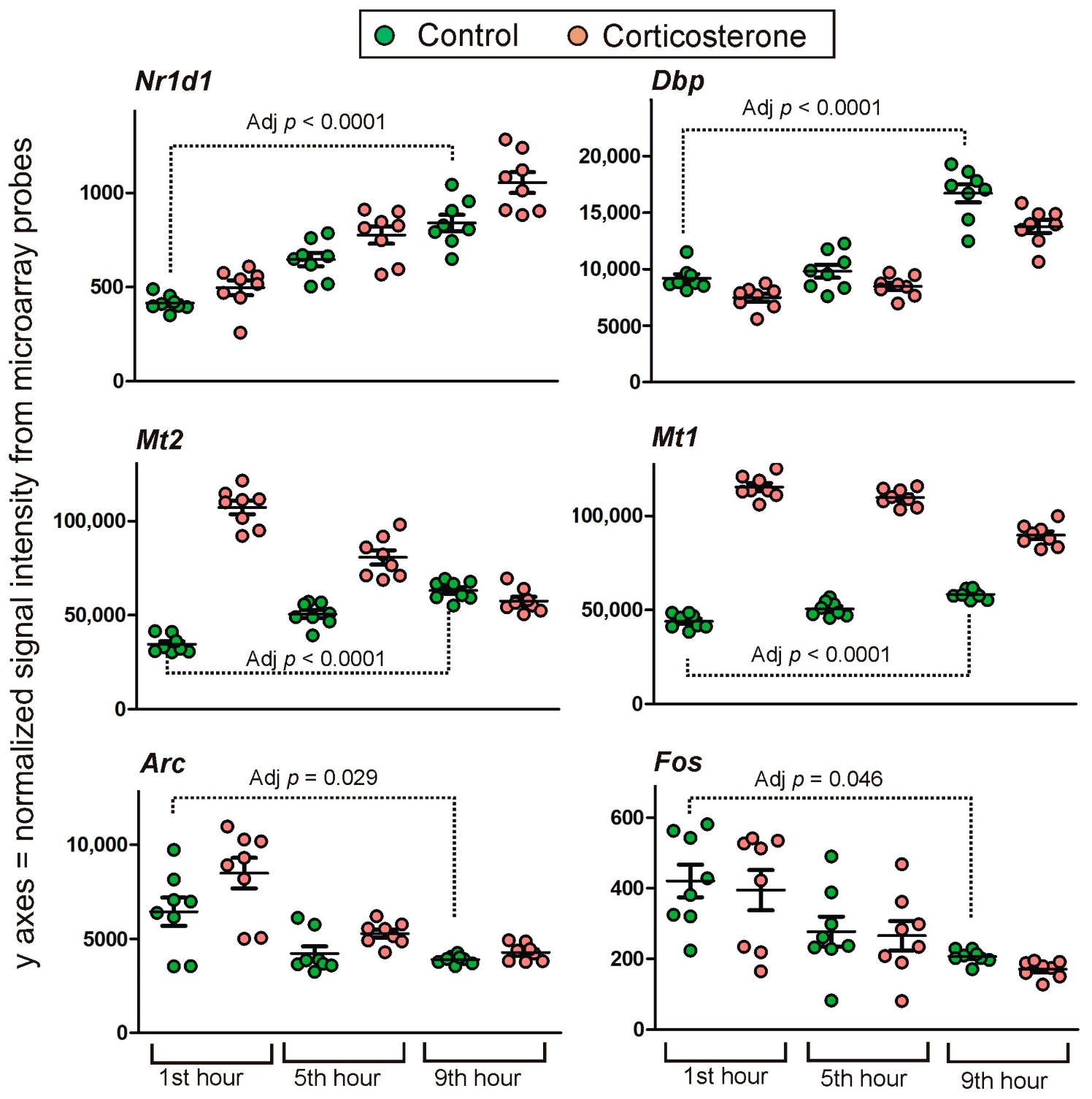
2.3. Transcriptomic Changes in Control Animals during the Resting Period
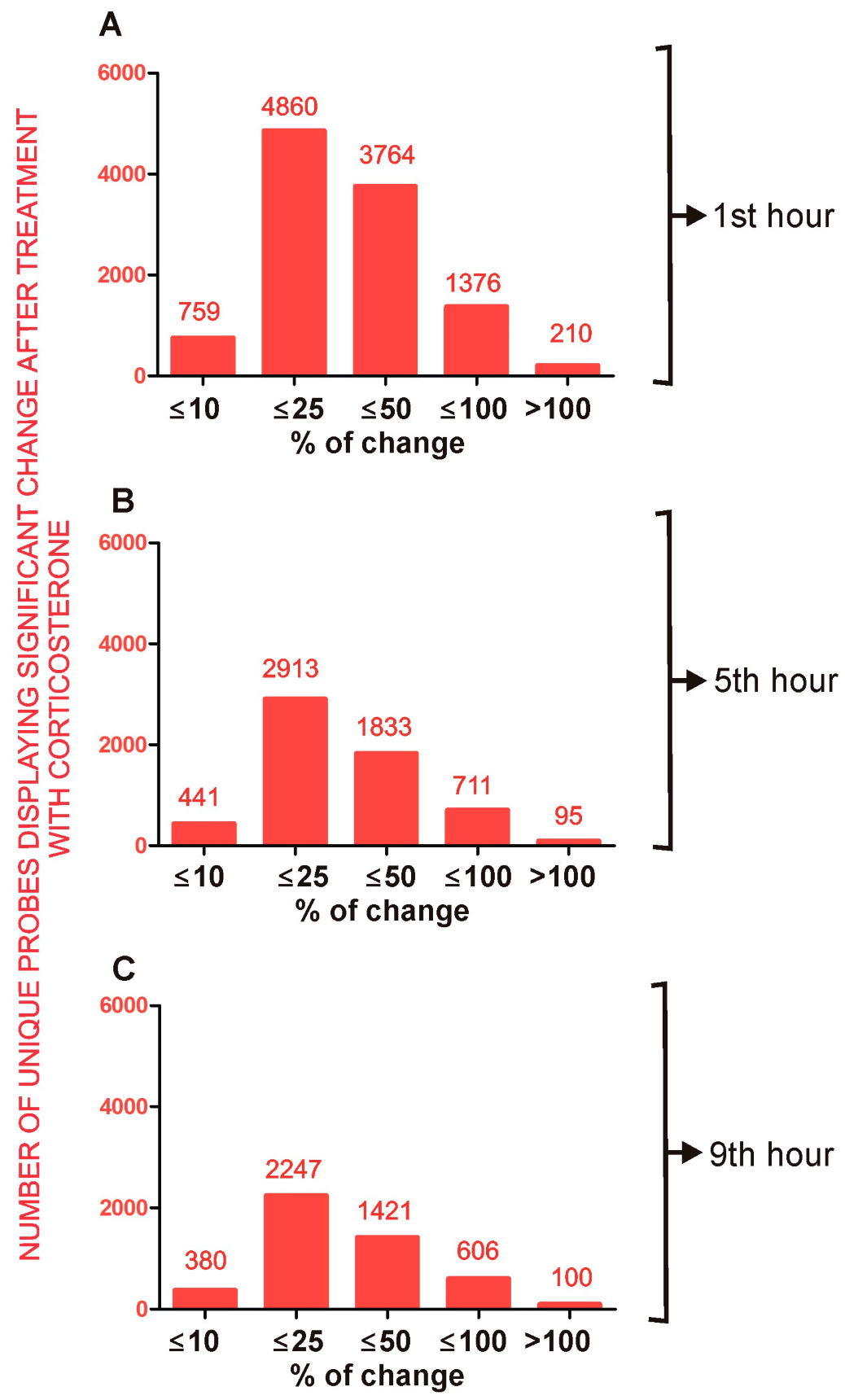
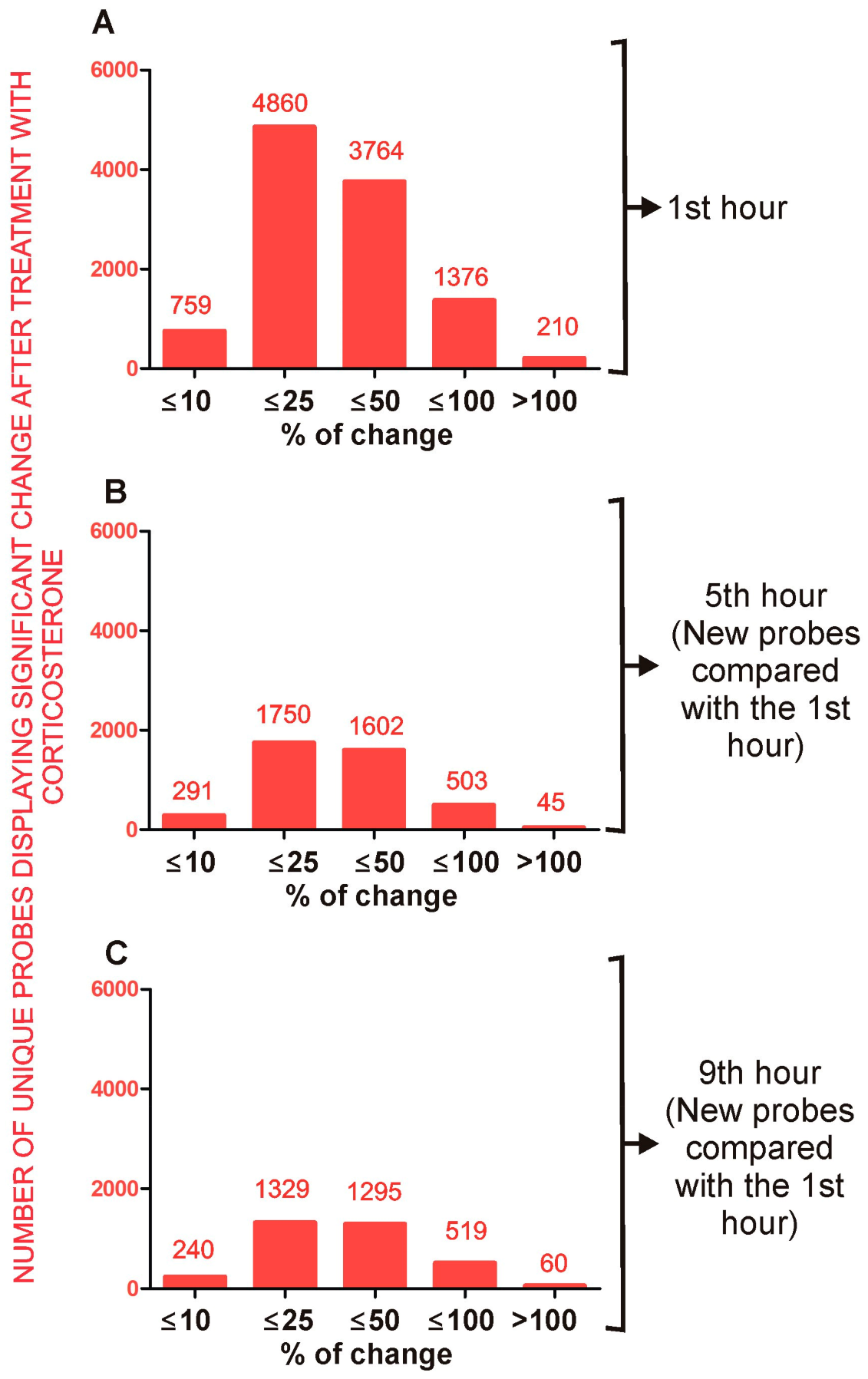
2.4. Effect of Corticosterone—General Characteristics of Microarray Results
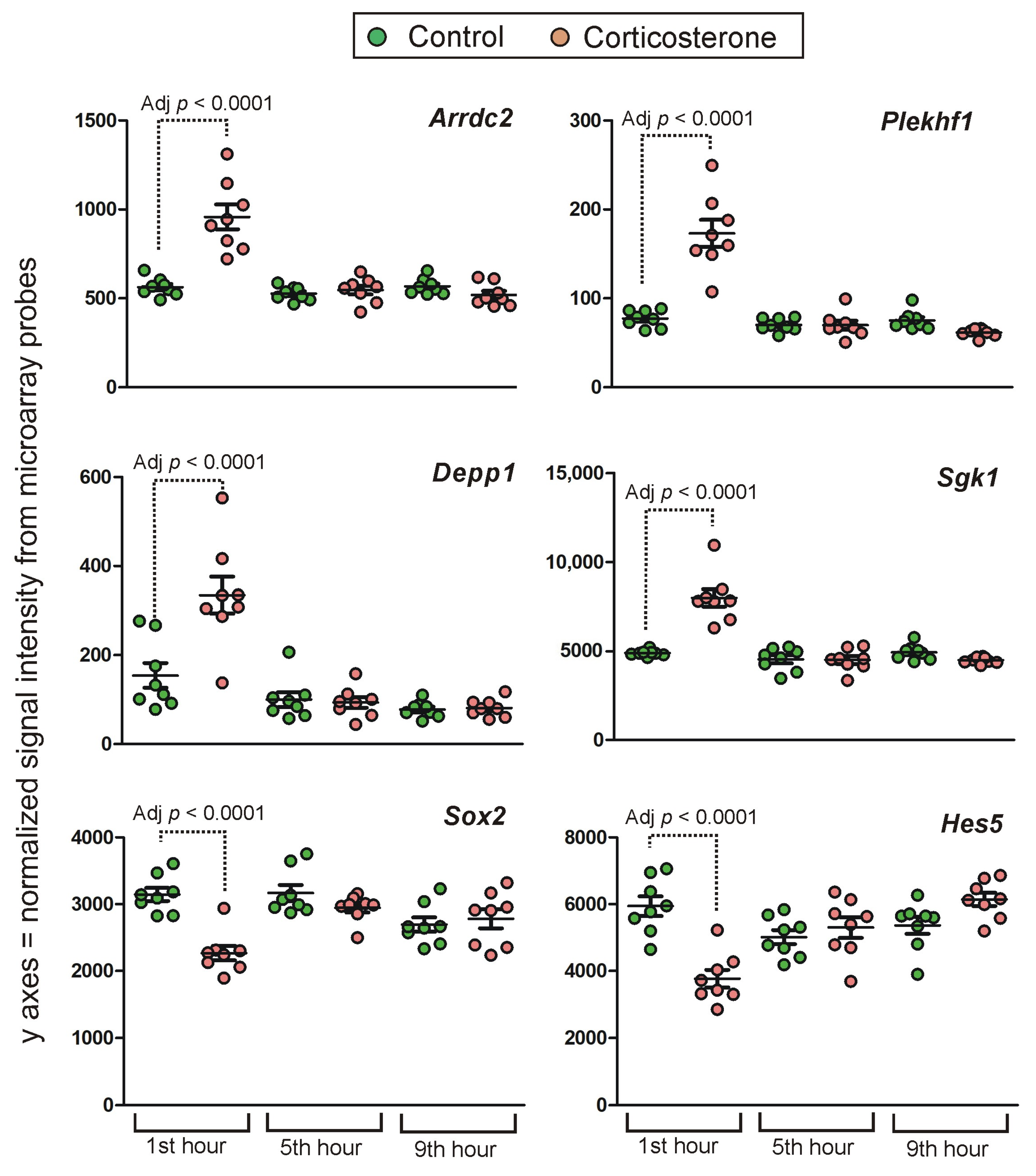
2.4.1. Primary Effects
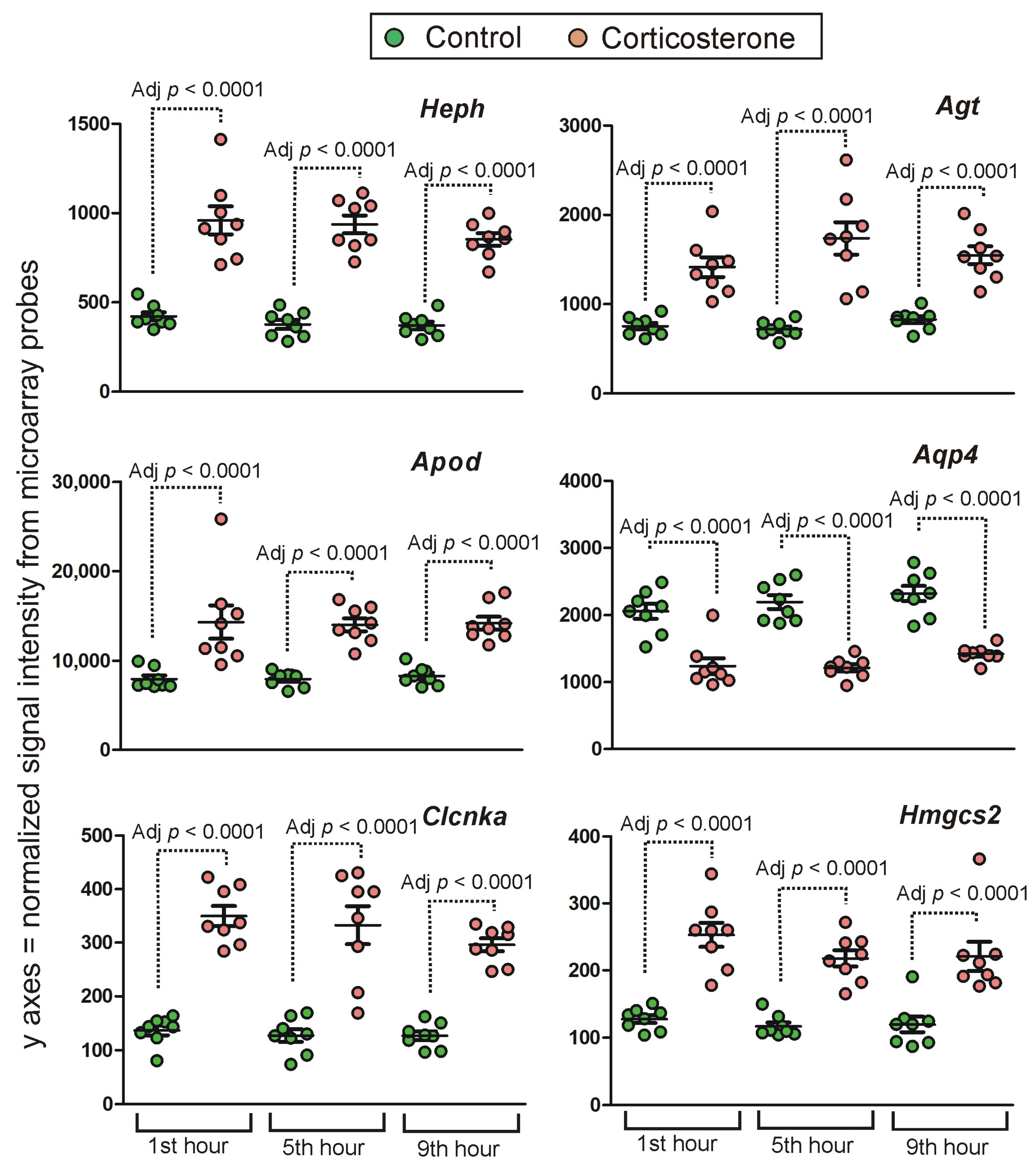
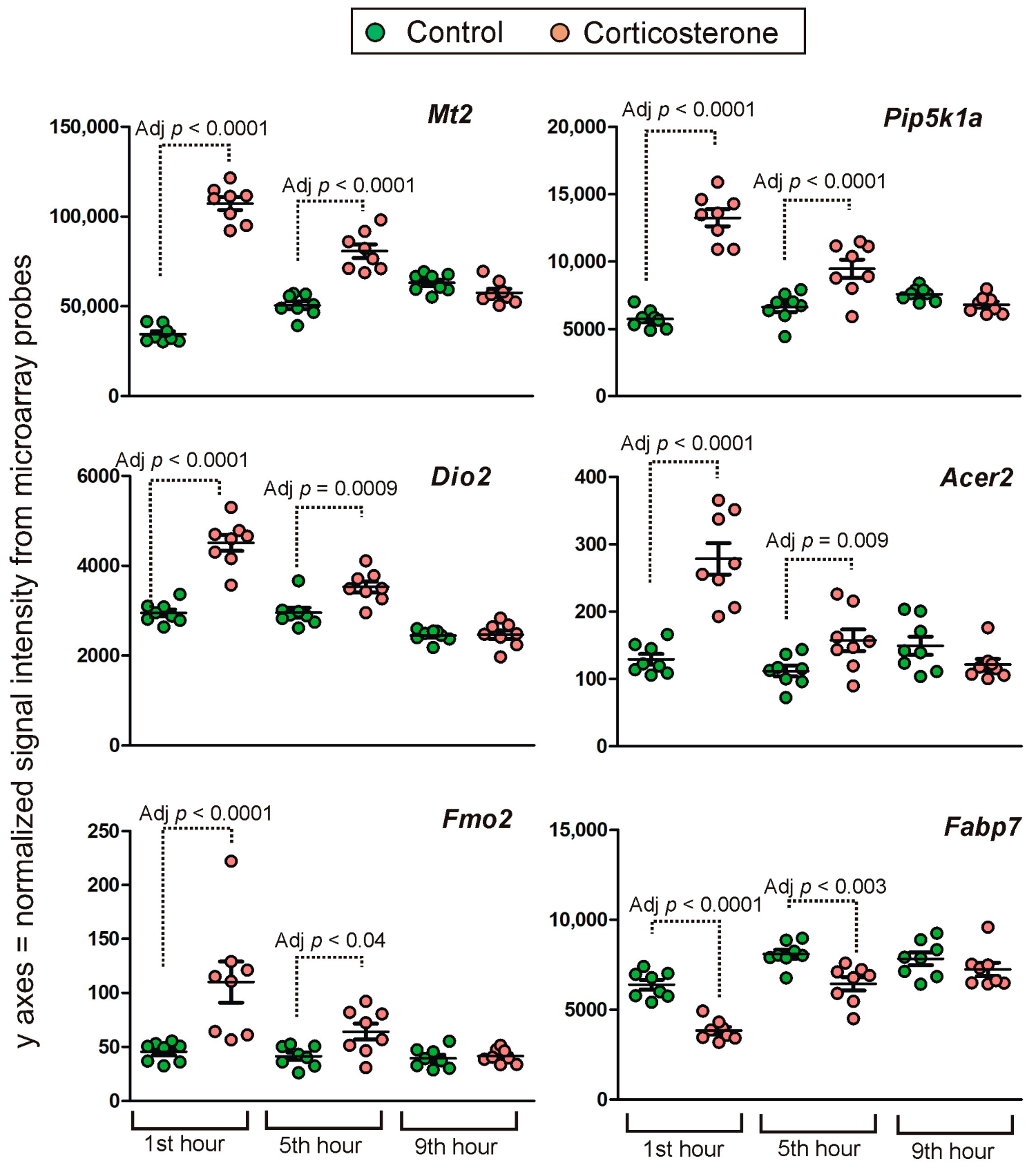
Long-Lasting Primary Effects
Intermediate Primary Effects
Short-Lasting Primary Effects
Time-Dependent Reversal of Primary Effects
2.4.2. Secondary Effects
3. Discussion
3.1. Characteristics of Experimental Model
3.2. General Characteristics of Transcriptomic Response to Corticosterone
3.3. Time-Course of Transcriptomic Responses to Corticosterone during the Resting Period
3.4. Comparison with the List of Established GC-Responsive Genes
3.5. Comparison with Transcriptomic Response to Stress
4. Materials and Methods
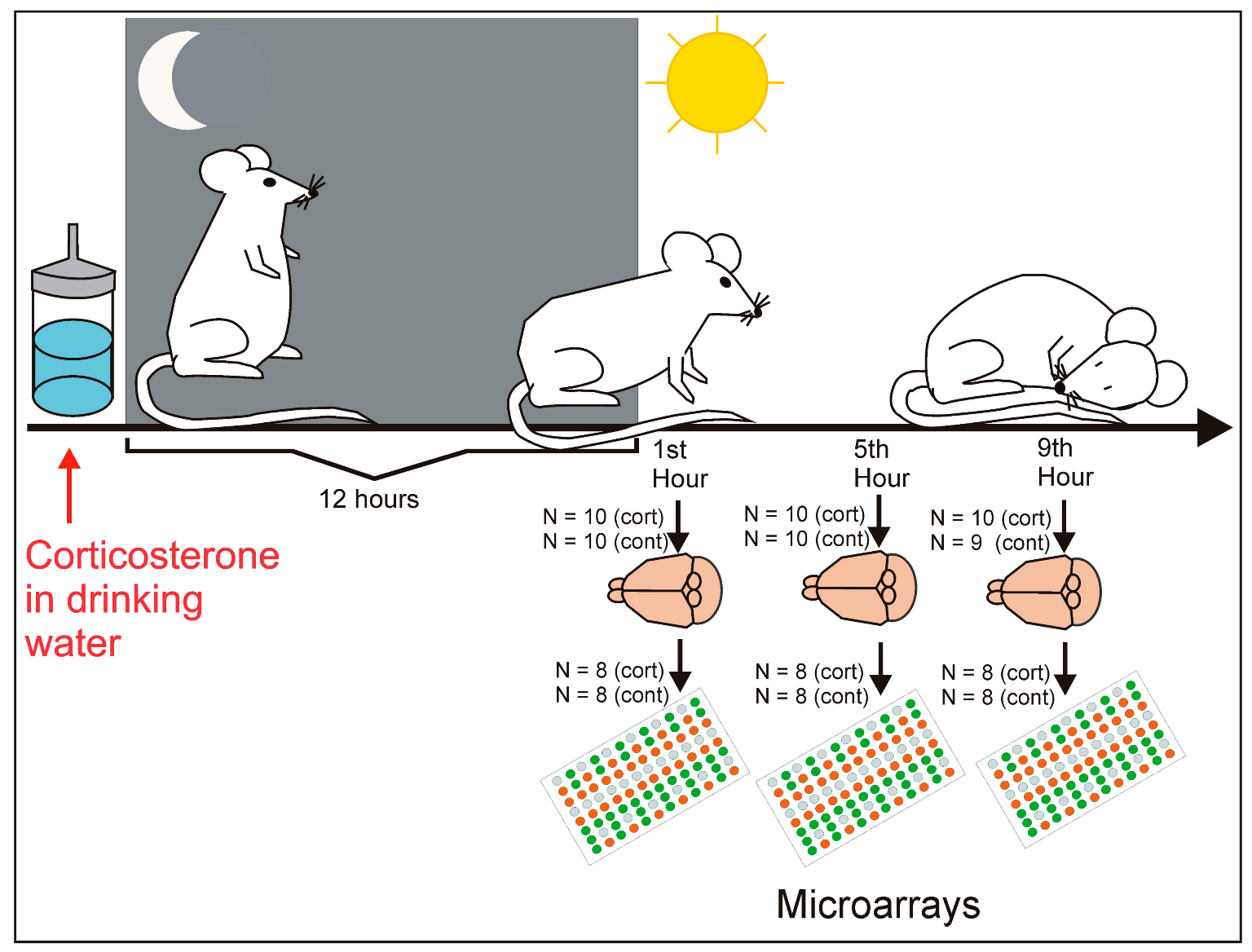
4.1. Animals
4.2. Experimental Procedure
4.3. Analysis of Blood Samples
4.4. RNA Isolation
4.5. Microarrays
4.6. Annotation of Microarray Data
4.7. Microarray Data Analysis
4.8. PCR Validation
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Charmandari, E.; Tsigos, C.; Chrousos, G. Endocrinology of the stress response. Annu. Rev. Physiol. 2005, 67, 259–284. [Google Scholar] [CrossRef]
- Somvanshi, P.R.; Mellon, S.H.; Flory, J.D.; Abu-Amara, D.; Wolkowitz, O.M.; Yehuda, R.; Jett, M.; Hood, L.; Marmar, C.; Doyle, F.J., 3rd. Mechanistic inferences on metabolic dysfunction in posttraumatic stress disorder from an integrated model and multiomic analysis: Role of glucocorticoid receptor sensitivity. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E879–E898. [Google Scholar] [CrossRef] [PubMed]
- Sheerin, C.M.; Lind, M.J.; Bountress, K.E.; Marraccini, M.E.; Amstadter, A.B.; Bacanu, S.A.; Nugent, N.R. Meta-Analysis of Associations Between Hypothalamic-Pituitary-Adrenal Axis Genes and Risk of Posttraumatic Stress Disorder. J. Trauma. Stress 2020, 33, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Szeszko, P.R.; Lehrner, A.; Yehuda, R. Glucocorticoids and Hippocampal Structure and Function in PTSD. Harv. Rev. Psychiatry 2018, 26, 142–157. [Google Scholar] [CrossRef]
- Farrell, C.; Doolin, K.; O’ Leary, N.; Jairaj, C.; Roddy, D.; Tozzi, L.; Morris, D.; Harkin, A.; Frodl, T.; Nemoda, Z.; et al. DNA methylation differences at the glucocorticoid receptor gene in depression are related to functional alterations in hypothalamic-pituitary-adrenal axis activity and to early life emotional abuse. Psychiatry Res. 2018, 265, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Gaffey, A.E.; Walsh, E.C.; Ladd, C.O.; Hoks, R.M.; Abercrombie, H.C. Alterations in Systemic and Cognitive Glucocorticoid Sensitivity in Depression. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2019, 4, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Yan, H.; Wen, Y.; Lai, C.; Shi, L. Association between NR3C1 rs41423247 polymorphism and depression: A PRISMA-compliant meta-analysis. Medicine 2018, 97, e12541. [Google Scholar] [CrossRef]
- Reichardt, S.D.; Amouret, A.; Muzzi, C.; Vettorazzi, S.; Tuckermann, J.P.; Luhder, F.; Reichardt, H.M. The Role of Glucocorticoids in Inflammatory Diseases. Cells 2021, 10, 2921. [Google Scholar] [CrossRef] [PubMed]
- Starkman, M.N. Neuropsychiatric findings in Cushing syndrome and exogenous glucocorticoid administration. Endocrinol. Metab. Clin. N. Am. 2013, 42, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Nieman, L.K. Cushing’s syndrome: Update on signs, symptoms and biochemical screening. Eur. J. Endocrinol. 2015, 173, M33–M38. [Google Scholar] [CrossRef]
- Lonser, R.R.; Nieman, L.; Oldfield, E.H. Cushing’s disease: Pathobiology, diagnosis, and management. J. Neurosurg. 2017, 126, 404–417. [Google Scholar] [CrossRef]
- Jaszczyk, A.; Juszczak, G.R. Glucocorticoids, metabolism and brain activity. Neurosci. Biobehav. Rev. 2021, 126, 113–145. [Google Scholar] [CrossRef]
- Juszczak, G.R.; Stankiewicz, A.M. Glucocorticoids, genes and brain function. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 82, 136–168. [Google Scholar] [CrossRef]
- Carter, B.S.; Meng, F.; Thompson, R.C. Glucocorticoid treatment of astrocytes results in temporally dynamic transcriptome regulation and astrocyte-enriched mRNA changes in vitro. Physiol. Genom. 2012, 44, 1188–1200. [Google Scholar] [CrossRef] [PubMed]
- Kamisoglu, K.; Sukumaran, S.; Nouri-Nigjeh, E.; Tu, C.; Li, J.; Shen, X.; Duan, X.; Qu, J.; Almon, R.R.; DuBois, D.C.; et al. Tandem analysis of transcriptome and proteome changes after a single dose of corticosteroid: A systems approach to liver function in pharmacogenomics. OMICS 2015, 19, 80–91. [Google Scholar] [CrossRef]
- Ayyar, V.S.; Almon, R.R.; DuBois, D.C.; Sukumaran, S.; Qu, J.; Jusko, W.J. Functional proteomic analysis of corticosteroid pharmacodynamics in rat liver: Relationship to hepatic stress, signaling, energy regulation, and drug metabolism. J. Proteom. 2017, 160, 84–105. [Google Scholar] [CrossRef]
- Virgin, C.E., Jr.; Ha, T.P.; Packan, D.R.; Tombaugh, G.C.; Yang, S.H.; Horner, H.C.; Sapolsky, R.M. Glucocorticoids inhibit glucose transport and glutamate uptake in hippocampal astrocytes: Implications for glucocorticoid neurotoxicity. J. Neurochem. 1991, 57, 1422–1428. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, A.M.; Jaszczyk, A.; Goscik, J.; Juszczak, G.R. Stress and the brain transcriptome: Identifying commonalities and clusters in standardized data from published experiments. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2022, 119, 110558. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.; Dumont, F.; Khelfaoui, M.; Foisset, S.R.; Letourneur, F.; Bienvenu, T.; Khwaja, O.; Dorseuil, O.; Billuart, P. Identification of intellectual disability genes showing circadian clock-dependent expression in the mouse hippocampus. Neuroscience 2015, 308, 11–50. [Google Scholar] [CrossRef] [PubMed]
- Annayev, Y.; Adar, S.; Chiou, Y.Y.; Lieb, J.D.; Sancar, A.; Ye, R. Gene model 129 (Gm129) encodes a novel transcriptional repressor that modulates circadian gene expression. J. Biol. Chem. 2014, 289, 5013–5024. [Google Scholar] [CrossRef]
- Grassi-Zucconi, G.; Menegazzi, M.; De Prati, A.C.; Bassetti, A.; Montagnese, P.; Mandile, P.; Cosi, C.; Bentivoglio, M. c-fos mRNA is spontaneously induced in the rat brain during the activity period of the circadian cycle. Eur. J. Neurosci. 1993, 5, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Rath, M.F.; Rohde, K.; Fahrenkrug, J.; Moller, M. Circadian clock components in the rat neocortex: Daily dynamics, localization and regulation. Brain Struct. Funct. 2013, 218, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Gjerstad, J.K.; Lightman, S.L.; Spiga, F. Role of glucocorticoid negative feedback in the regulation of HPA axis pulsatility. Stress 2018, 21, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Dallman, M.F.; Jones, M.T. Corticosteroid feedback control of ACTH secretion: Effect of stress-induced corticosterone ssecretion on subsequent stress responses in the rat. Endocrinology 1973, 92, 1367–1375. [Google Scholar] [CrossRef]
- Qian, X.; Droste, S.K.; Lightman, S.L.; Reul, J.M.; Linthorst, A.C. Circadian and ultradian rhythms of free glucocorticoid hormone are highly synchronized between the blood, the subcutaneous tissue, and the brain. Endocrinology 2012, 153, 4346–4353. [Google Scholar] [CrossRef]
- Watanabe, H.; Passonneau, J.V. Factors affecting the turnover of cerebral glycogen and limit dextrin in vivo. J. Neurochem. 1973, 20, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, A.; Hochgerner, H.; Lonnerberg, P.; Johnsson, A.; Memic, F.; van der Zwan, J.; Haring, M.; Braun, E.; Borm, L.E.; La Manno, G.; et al. Molecular Architecture of the Mouse Nervous System. Cell 2018, 174, 999–1014. [Google Scholar] [CrossRef]
- Guo, Y.; Tian, L.; Liu, X.; He, Y.; Chang, S.; Shen, Y. ERRFI1 Inhibits Proliferation and Inflammation of Nucleus Pulposus and Is Negatively Regulated by miR-2355-5p in Intervertebral Disc Degeneration. Spine 2019, 44, E873–E881. [Google Scholar] [CrossRef] [PubMed]
- Ball, K.L. p21: Structure and functions associated with cyclin-CDK binding. Prog. Cell Cycle Res. 1997, 3, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Dungan, C.M.; Gordon, B.S.; Williamson, D.L. Acute treadmill exercise discriminately improves the skeletal muscle insulin-stimulated growth signaling responses in mice lacking REDD1. Physiol. Rep. 2019, 7, e14011. [Google Scholar] [CrossRef]
- Takeichi, T.; Takarada-Iemata, M.; Hashida, K.; Sudo, H.; Okuda, T.; Kokame, K.; Hatano, T.; Takanashi, M.; Funabe, S.; Hattori, N.; et al. The effect of Ndrg2 expression on astroglial activation. Neurochem. Int. 2011, 59, 21–27. [Google Scholar] [CrossRef]
- Sun, G.; Xue, R.; Yao, F.; Liu, D.; Huang, H.; Chen, C.; Li, Y.; Zeng, J.; Zhang, G.; Dong, Y.; et al. The critical role of Sestrin 1 in regulating the proliferation of cardiac fibroblasts. Arch. Biochem. Biophys 2014, 542, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Sun, G.; Murai, K.; Ye, P.; Li, W.; Asuelime, G.; Cheung, Y.T.; Shi, Y. Wnt7a regulates multiple steps of neurogenesis. Mol. Cell Biol. 2013, 33, 2551–2559. [Google Scholar] [CrossRef]
- Lee, M.; Choi, K.H.; Oh, J.N.; Kim, S.H.; Lee, D.K.; Choe, G.C.; Jeong, J.; Lee, C.K. SOX2 plays a crucial role in cell proliferation and lineage segregation during porcine pre-implantation embryo development. Cell Prolif. 2021, 54, e13097. [Google Scholar] [CrossRef] [PubMed]
- Sarlak, G.; Vincent, B. The Roles of the Stem Cell-Controlling Sox2 Transcription Factor: From Neuroectoderm Development to Alzheimer’s Disease? Mol. Neurobiol. 2016, 53, 1679–1698. [Google Scholar] [CrossRef] [PubMed]
- Bonapersona, V.; Schuler, H.; Damsteegt, R.; Adolfs, Y.; Pasterkamp, R.J.; van den Heuvel, M.P.; Joels, M.; Sarabdjitsingh, R.A. The mouse brain after foot shock in four dimensions: Temporal dynamics at a single-cell resolution. Proc. Natl. Acad. Sci. USA 2022, 119, e2114002119. [Google Scholar] [CrossRef]
- Bremner, J.D. Stress and brain atrophy. CNS Neurol. Disord. Drug Targets 2006, 5, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Singareddy, R.K.; Balon, R. Sleep in posttraumatic stress disorder. Ann. Clin. Psychiatry 2002, 14, 183–190. [Google Scholar] [CrossRef]
- Germain, A. Sleep disturbances as the hallmark of PTSD: Where are we now? Am. J. Psychiatry 2013, 170, 372–382. [Google Scholar] [CrossRef]
- Bajor, L.A.; Balsara, C.; Osser, D.N. An evidence-based approach to psychopharmacology for posttraumatic stress disorder (PTSD)—2022 update. Psychiatry Res. 2022, 317, 114840. [Google Scholar] [CrossRef] [PubMed]
- White, C.J.; Ellis, J.M.; Wolfgang, M.J. The role of ethanolamine phosphate phospholyase in regulation of astrocyte lipid homeostasis. J. Biol. Chem. 2021, 297, 100830. [Google Scholar] [CrossRef]
- Rassart, E.; Desmarais, F.; Najyb, O.; Bergeron, K.F.; Mounier, C. Apolipoprotein D. Gene 2020, 756, 144874. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Kato, R.; Isogai, Y.; Saka, G.; Ohtsuki, M.; Taketomi, Y.; Yamamoto, K.; Tsutsumi, K.; Yamada, J.; Masuda, S.; et al. Analyses of group III secreted phospholipase A2 transgenic mice reveal potential participation of this enzyme in plasma lipoprotein modification, macrophage foam cell formation, and atherosclerosis. J. Biol. Chem. 2008, 283, 33483–33497. [Google Scholar] [CrossRef] [PubMed]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [PubMed]
- Migocka-Patrzalek, M.; Elias, M. Muscle Glycogen Phosphorylase and Its Functional Partners in Health and Disease. Cells 2021, 10, 883. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Jiang, R.; Chen, M.; Zheng, J.; Braidy, N.; Liu, S.; Liu, G.; Maimaitiming, Z.; Shen, T.; Dunaief, J.L.; et al. Multi-copper ferroxidase deficiency leads to iron accumulation and oxidative damage in astrocytes and oligodendrocytes. Sci. Rep. 2019, 9, 9437. [Google Scholar] [CrossRef]
- Vandebroek, A.; Yasui, M. Regulation of AQP4 in the Central Nervous System. Int. J. Mol. Sci. 2020, 21, 1603. [Google Scholar] [CrossRef] [PubMed]
- Rokka, A.; Antonenkov, V.D.; Soininen, R.; Immonen, H.L.; Pirila, P.L.; Bergmann, U.; Sormunen, R.T.; Weckstrom, M.; Benz, R.; Hiltunen, J.K. Pxmp2 is a channel-forming protein in Mammalian peroxisomal membrane. PLoS ONE 2009, 4, e5090. [Google Scholar] [CrossRef]
- Kretzschmar, A.; Schulke, J.P.; Masana, M.; Durre, K.; Muller, M.B.; Bausch, A.R.; Rein, T. The Stress-Inducible Protein DRR1 Exerts Distinct Effects on Actin Dynamics. Int. J. Mol. Sci. 2018, 19, 3993. [Google Scholar] [CrossRef] [PubMed]
- Brew, K.; Dinakarpandian, D.; Nagase, H. Tissue inhibitors of metalloproteinases: Evolution, structure and function. Biochim. Biophys Acta 2000, 1477, 267–283. [Google Scholar] [CrossRef]
- Sheikh, B.N.; Guhathakurta, S.; Tsang, T.H.; Schwabenland, M.; Renschler, G.; Herquel, B.; Bhardwaj, V.; Holz, H.; Stehle, T.; Bondareva, O.; et al. Neural metabolic imbalance induced by MOF dysfunction triggers pericyte activation and breakdown of vasculature. Nat. Cell Biol. 2020, 22, 828–841. [Google Scholar] [CrossRef] [PubMed]
- Rivera, F.B.; Alfonso, P.; Golbin, J.M.; Lo, K.; Lerma, E.; Volgman, A.S.; Kazory, A. The Role of Serum Chloride in Acute and Chronic Heart Failure: A Narrative Review. Cardiorenal Med. 2021, 11, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Riches, Z.; Bloomer, J.C.; Coughtrie, M.W. Comparison of 2-aminophenol and 4-nitrophenol as in vitro probe substrates for the major human hepatic sulfotransferase, SULT1A1, demonstrates improved selectivity with 2-aminophenol. Biochem. Pharmacol. 2007, 74, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Green, W.L.; Huang, W.S.; Hays, M.T.; Chopra, I.J. Alternate pathways of thyroid hormone metabolism. Thyroid 2005, 15, 943–958. [Google Scholar] [CrossRef] [PubMed]
- Vasak, M.; Meloni, G. Chemistry and biology of mammalian metallothioneins. J. Biol. Inorg. Chem. 2011, 16, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Coyle, P.; Philcox, J.C.; Carey, L.C.; Rofe, A.M. Metallothionein: The multipurpose protein. Cell. Mol. Life Sci. 2002, 59, 627–647. [Google Scholar] [CrossRef]
- Ouzzine, M.; Barre, L.; Netter, P.; Magdalou, J.; Fournel-Gigleux, S. The human UDP-glucuronosyltransferases: Structural aspects and drug glucuronidation. Drug Metab. Rev. 2003, 35, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Urade, Y.; Hayaishi, O. Prostaglandin D synthase: Structure and function. Vitam. Horm. 2000, 58, 89–120. [Google Scholar] [CrossRef]
- Cain, D.W.; Cidlowski, J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247. [Google Scholar] [CrossRef]
- Goodwin, J.E. Glucocorticoids and the Cardiovascular System. Adv. Exp. Med. Biol. 2015, 872, 299–314. [Google Scholar] [CrossRef]
- Walker, B.R. Glucocorticoids and cardiovascular disease. Eur. J. Endocrinol. 2007, 157, 545–559. [Google Scholar] [CrossRef]
- Stankiewicz, A.M.; Jaszczyk, A.; Juszczak, G.R. Annotation of illumina microarray probes: Similarities and differences between various bioinformatic tools. Anim. Sci. Pap. Rep. 2019, 37, 323–327. [Google Scholar]
- Kendziorski, C.; Irizarry, R.A.; Chen, K.S.; Haag, J.D.; Gould, M.N. On the utility of pooling biological samples in microarray experiments. Proc. Natl. Acad. Sci. USA 2005, 102, 4252–4257. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.H.; Michalowska, A.M.; Dobbin, K.; Ye, Y.; Qiu, T.H.; Green, J.E. Effects of pooling mRNA in microarray class comparisons. Bioinformatics 2004, 20, 3318–3325. [Google Scholar] [CrossRef] [PubMed]
- Jolly, R.A.; Goldstein, K.M.; Wei, T.; Gao, H.; Chen, P.; Huang, S.; Colet, J.M.; Ryan, T.P.; Thomas, C.E.; Estrem, S.T. Pooling samples within microarray studies: A comparative analysis of rat liver transcription response to prototypical toxicants. Physiol Genom. 2005, 22, 346–355. [Google Scholar] [CrossRef]
- Gray, J.D.; Rubin, T.G.; Hunter, R.G.; McEwen, B.S. Hippocampal gene expression changes underlying stress sensitization and recovery. Mol. Psychiatry 2014, 19, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Horikawa, Y.; Iizuka, K.; Sakurai, N.; Tanaka, T.; Shihara, N.; Oshima, A.; Takeda, J.; Mikuni, M. Large-scale analysis of glucocorticoid target genes in rat hypothalamus. J. Neurochem. 2008, 106, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Datson, N.A.; van den Oever, J.M.; Korobko, O.B.; Magarinos, A.M.; de Kloet, E.R.; McEwen, B.S. Previous history of chronic stress changes the transcriptional response to glucocorticoid challenge in the dentate gyrus region of the male rat hippocampus. Endocrinology 2013, 154, 3261–3272. [Google Scholar] [CrossRef]
- Datson, N.A.; Polman, J.A.; de Jonge, R.T.; van Boheemen, P.T.; van Maanen, E.M.; Welten, J.; McEwen, B.S.; Meiland, H.C.; Meijer, O.C. Specific regulatory motifs predict glucocorticoid responsiveness of hippocampal gene expression. Endocrinology 2011, 152, 3749–3757. [Google Scholar] [CrossRef]
- Polman, J.A.; Hunter, R.G.; Speksnijder, N.; van den Oever, J.M.; Korobko, O.B.; McEwen, B.S.; de Kloet, E.R.; Datson, N.A. Glucocorticoids modulate the mTOR pathway in the hippocampus: Differential effects depending on stress history. Endocrinology 2012, 153, 4317–4327. [Google Scholar] [CrossRef] [PubMed]
- Datson, N.A.; van der Perk, J.; de Kloet, E.R.; Vreugdenhil, E. Identification of corticosteroid-responsive genes in rat hippocampus using serial analysis of gene expression. Eur. J. Neurosci. 2001, 14, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.C.; Blalock, E.M.; Curran-Rauhut, M.A.; Kadish, I.; Blalock, S.J.; Brewer, L.; Porter, N.M.; Landfield, P.W. Glucocorticoid-dependent hippocampal transcriptome in male rats: Pathway-specific alterations with aging. Endocrinology 2013, 154, 2807–2820. [Google Scholar] [CrossRef]
- Alfonso, J.; Aguero, F.; Sanchez, D.O.; Flugge, G.; Fuchs, E.; Frasch, A.C.; Pollevick, G.D. Gene expression analysis in the hippocampal formation of tree shrews chronically treated with cortisol. J. Neurosci. Res. 2004, 78, 702–710. [Google Scholar] [CrossRef]
- Slezak, M.; Korostynski, M.; Gieryk, A.; Golda, S.; Dzbek, J.; Piechota, M.; Wlazlo, E.; Bilecki, W.; Przewlocki, R. Astrocytes are a neural target of morphine action via glucocorticoid receptor-dependent signaling. Glia 2013, 61, 623–635. [Google Scholar] [CrossRef]
- Peffer, M.E.; Chandran, U.R.; Luthra, S.; Volonte, D.; Galbiati, F.; Garabedian, M.J.; Monaghan, A.P.; DeFranco, D.B. Caveolin-1 regulates genomic action of the glucocorticoid receptor in neural stem cells. Mol. Cell. Biol. 2014, 34, 2611–2623. [Google Scholar] [CrossRef] [PubMed]
- Morsink, M.C.; Steenbergen, P.J.; Vos, J.B.; Karst, H.; Joels, M.; De Kloet, E.R.; Datson, N.A. Acute activation of hippocampal glucocorticoid receptors results in different waves of gene expression throughout time. J. Neuroendocrinol. 2006, 18, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Morsink, M.C.; Joels, M.; Sarabdjitsingh, R.A.; Meijer, O.C.; De Kloet, E.R.; Datson, N.A. The dynamic pattern of glucocorticoid receptor-mediated transcriptional responses in neuronal PC12 cells. J. Neurochem. 2006, 99, 1282–1298. [Google Scholar] [CrossRef]
- Jenkins, S.I.; Pickard, M.R.; Khong, M.; Smith, H.L.; Mann, C.L.; Emes, R.D.; Chari, D.M. Identifying the cellular targets of drug action in the central nervous system following corticosteroid therapy. ACS Chem. Neurosci. 2014, 5, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, K.; Morita, T.; Mayanagi, T.; Tanokashira, D.; Yoshida, T.; Sakai, A.; Sobue, K. Detrimental effects of glucocorticoids on neuronal migration during brain development. Mol. Psychiatry 2009, 14, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Anacker, C.; Cattaneo, A.; Musaelyan, K.; Zunszain, P.A.; Horowitz, M.; Molteni, R.; Luoni, A.; Calabrese, F.; Tansey, K.; Gennarelli, M.; et al. Role for the kinase SGK1 in stress, depression, and glucocorticoid effects on hippocampal neurogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 8708–8713. [Google Scholar] [CrossRef] [PubMed]
- Salaria, S.; Chana, G.; Caldara, F.; Feltrin, E.; Altieri, M.; Faggioni, F.; Domenici, E.; Merlo-Pich, E.; Everall, I.P. Microarray analysis of cultured human brain aggregates following cortisol exposure: Implications for cellular functions relevant to mood disorders. Neurobiol. Dis. 2006, 23, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, A.M.; Goscik, J.; Majewska, A.; Swiergiel, A.H.; Juszczak, G.R. The Effect of Acute and Chronic Social Stress on the Hippocampal Transcriptome in Mice. PLoS ONE 2015, 10, e0142195. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, A.M.; Goscik, J.; Swiergiel, A.H.; Majewska, A.; Wieczorek, M.; Juszczak, G.R.; Lisowski, P. Social stress increases expression of hemoglobin genes in mouse prefrontal cortex. BMC Neurosci. 2014, 15, 130. [Google Scholar] [CrossRef] [PubMed]
- Roszkowski, M.; Manuella, F.; von Ziegler, L.; Duran-Pacheco, G.; Moreau, J.L.; Mansuy, I.M.; Bohacek, J. Rapid stress-induced transcriptomic changes in the brain depend on beta-adrenergic signaling. Neuropharmacology 2016, 107, 329–338. [Google Scholar] [CrossRef]
- Bozas, E.; Tritos, N.; Phillipidis, H.; Stylianopoulou, F. At least three neurotransmitter systems mediate a stress-induced increase in c-fos mRNA in different rat brain areas. Cell. Mol. Neurobiol. 1997, 17, 157–169. [Google Scholar] [CrossRef]
- Allaman, I.; Pellerin, L.; Magistretti, P.J. Glucocorticoids modulate neurotransmitter-induced glycogen metabolism in cultured cortical astrocytes. J. Neurochem. 2004, 88, 900–908. [Google Scholar] [CrossRef]
- Schwabe, L.; Tegenthoff, M.; Hoffken, O.; Wolf, O.T. Simultaneous glucocorticoid and noradrenergic activity disrupts the neural basis of goal-directed action in the human brain. J. Neurosci. 2012, 32, 10146–10155. [Google Scholar] [CrossRef]
- Van Loo, P.L.; Mol, J.A.; Koolhaas, J.M.; Van Zutphen, B.F.; Baumans, V. Modulation of aggression in male mice: Influence of group size and cage size. Physiol. Behav. 2001, 72, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Mondragon, R.; Mayagoitia, L.; Lopez-Lujan, A.; Diaz, J.L. Social structure features in three inbred strains of mice, C57Bl/6J, Balb/cj, and NIH: A comparative study. Behav. Neural. Biol. 1987, 47, 384–391. [Google Scholar] [CrossRef]
- Hunt, C.; Hambly, C. Faecal corticosterone concentrations indicate that separately housed male mice are not more stressed than group housed males. Physiol. Behav. 2006, 87, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Okutsu, M.; Lira, V.A.; Higashida, K.; Peake, J.; Higuchi, M.; Suzuki, K. Corticosterone accelerates atherosclerosis in the apolipoprotein E-deficient mouse. Atherosclerosis 2014, 232, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Howell, K.R.; Kutiyanawalla, A.; Pillai, A. Long-term continuous corticosterone treatment decreases VEGF receptor-2 expression in frontal cortex. PLoS ONE 2011, 6, e20198. [Google Scholar] [CrossRef]
- Karatsoreos, I.N.; Bhagat, S.M.; Bowles, N.P.; Weil, Z.M.; Pfaff, D.W.; McEwen, B.S. Endocrine and physiological changes in response to chronic corticosterone: A potential model of the metabolic syndrome in mouse. Endocrinology 2010, 151, 2117–2127. [Google Scholar] [CrossRef]
- Bowles, N.P.; Hill, M.N.; Bhagat, S.M.; Karatsoreos, I.N.; Hillard, C.J.; McEwen, B.S. Chronic, noninvasive glucocorticoid administration suppresses limbic endocannabinoid signaling in mice. Neuroscience 2012, 204, 83–89. [Google Scholar] [CrossRef]
- Jene, T.; Gassen, N.C.; Opitz, V.; Endres, K.; Muller, M.B.; van der Kooij, M.A. Temporal profiling of an acute stress-induced behavioral phenotype in mice and role of hippocampal DRR1. Psychoneuroendocrinology 2018, 91, 149–158. [Google Scholar] [CrossRef]
- Engler, H.; Bailey, M.T.; Engler, A.; Stiner-Jones, L.M.; Quan, N.; Sheridan, J.F. Interleukin-1 receptor type 1-deficient mice fail to develop social stress-associated glucocorticoid resistance in the spleen. Psychoneuroendocrinology 2008, 33, 108–117. [Google Scholar] [CrossRef]
- Jaszczyk, A.; Stankiewicz, A.M.; Juszczak, G.R. Dissection of Mouse Hippocampus with Its Dorsal, Intermediate and Ventral Subdivisions Combined with Molecular Validation. Brain Sci. 2022, 12, 799. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.D.; Wang, S.; Chen, M.; Girard, L.; Minna, J.D.; Xie, Y.; Xiao, G. Probe mapping across multiple microarray platforms. Brief Bioinform. 2012, 13, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Lim, N.; Tesar, S.; Belmadani, M.; Poirier-Morency, G.; Mancarci, B.O.; Sicherman, J.; Jacobson, M.; Leong, J.; Tan, P.; Pavlidis, P. Curation of over 10 000 transcriptomic studies to enable data reuse. Database 2021, 2021, baab006. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Limma: Linear models for microarray data.editors. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Gentleman, R., Carey, V., Dudoit, S., Irizarry, R., Huber, W., Eds.; Springer: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- Ritchie, M.E.; Silver, J.; Oshlack, A.; Holmes, M.; Diyagama, D.; Holloway, A.; Smyth, G.K. A comparison of background correction methods for two-colour microarrays. Bioinformatics 2007, 23, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K.; Speed, T. Normalization of cDNA microarray data. Methods 2003, 31, 265–273. [Google Scholar] [CrossRef]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.D.; Ritchie, M.E.; Smyth, G.K. Microarray background correction: Maximum likelihood estimation for the normal-exponential convolution. Biostatistics 2009, 10, 352–363. [Google Scholar] [CrossRef]
- Smyth, G.K.; Altman, N.S. Separate-channel analysis of two-channel microarrays: Recovering inter-spot information. BMC Bioinform. 2013, 14, 165. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Andersen, C.L.; Jensen, J.L.; Orntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Hancock, A.A.; Bush, E.N.; Stanisic, D.; Kyncl, J.J.; Lin, C.T. Data normalization before statistical analysis: Keeping the horse before the cart. Trends Pharmacol. Sci. 1988, 9, 29–32. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total number of microarray probes | 62,976 |
| Number of technical/control probes | 3671 |
| Number of unique probes detecting mouse genes and printed in 1 copy on the microarray | 56,305 |
| Number of unique probes detecting mouse genes and printed in 10 copies on the microarray | 300 |
| Total number of unique probes detecting mouse genes (without copies of probes) | 56,605 |
| Total number of probes detecting mouse genes (including copies of probes) | 59,305 |
| Gene Name | Forward or Reverse Primer | Primer Sequence | Annealing Temperature | Efficiency |
|---|---|---|---|---|
| Sult1a1 | F | GATGGGAAAGTGTCCTATGGGT | 60 °C | 98.8% |
| R | TGAAGGATGTGTGGTGAACAATTA | |||
| Lao1 | F | ACAACGCTATCGTGCCTCAG | 60 °C | 95% |
| R | CATCAGGTAAGCCTTGGTGGA | |||
| Etnppl | F | TTGGTGAAGGACCGTGAGAAA | 60 °C | 108.6% |
| R | AACTTTGCATCGTCTTCCGTG | |||
| Apoc3 | F | ATGGAACAAGCCTCCAAGACG | 60 °C | 111.7% |
| R | TTGCTCCAGTAGCCTTTCAGG | |||
| Tbp | F | GCAGTGCCCAGCATCACTATT | 60 °C | 108.3% |
| R | AAGCCCTGAGCATAAGGTGG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaszczyk, A.; Stankiewicz, A.M.; Goscik, J.; Majewska, A.; Jezierski, T.; Juszczak, G.R. Overnight Corticosterone and Gene Expression in Mouse Hippocampus: Time Course during Resting Period. Int. J. Mol. Sci. 2023, 24, 2828. https://doi.org/10.3390/ijms24032828
Jaszczyk A, Stankiewicz AM, Goscik J, Majewska A, Jezierski T, Juszczak GR. Overnight Corticosterone and Gene Expression in Mouse Hippocampus: Time Course during Resting Period. International Journal of Molecular Sciences. 2023; 24(3):2828. https://doi.org/10.3390/ijms24032828
Chicago/Turabian StyleJaszczyk, Aneta, Adrian M. Stankiewicz, Joanna Goscik, Alicja Majewska, Tadeusz Jezierski, and Grzegorz R. Juszczak. 2023. "Overnight Corticosterone and Gene Expression in Mouse Hippocampus: Time Course during Resting Period" International Journal of Molecular Sciences 24, no. 3: 2828. https://doi.org/10.3390/ijms24032828
APA StyleJaszczyk, A., Stankiewicz, A. M., Goscik, J., Majewska, A., Jezierski, T., & Juszczak, G. R. (2023). Overnight Corticosterone and Gene Expression in Mouse Hippocampus: Time Course during Resting Period. International Journal of Molecular Sciences, 24(3), 2828. https://doi.org/10.3390/ijms24032828