
The Activation of Reticulophagy by ER Stress through the ATF4-MAP1LC3A-CCPG1 Pathway in Ovarian Granulosa Cells Is Linked to Apoptosis and Necroptosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
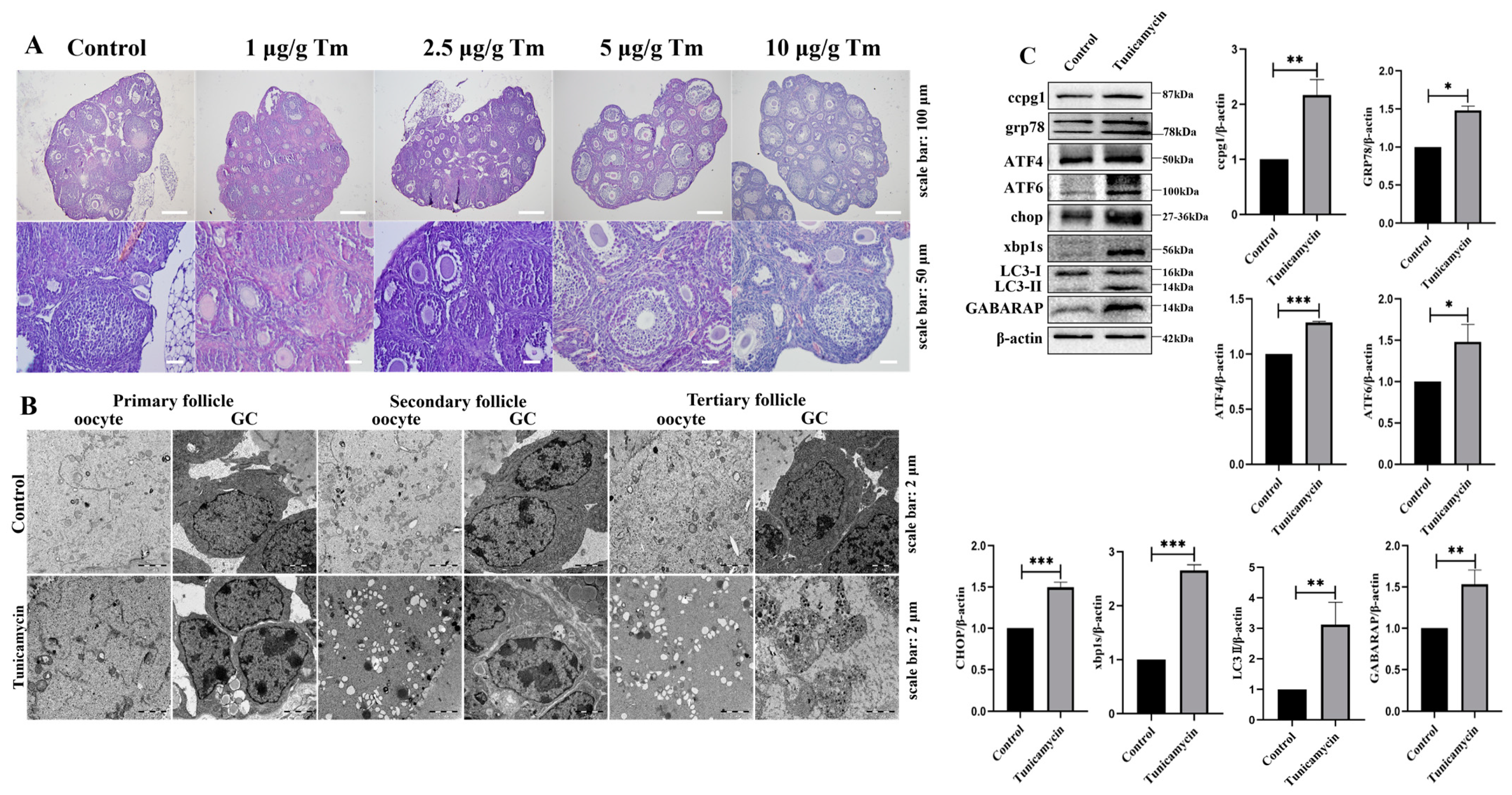
2.1. The Activation of Reticulophagy in the ER by the Stress Inducer Tunicamycin Causes POF
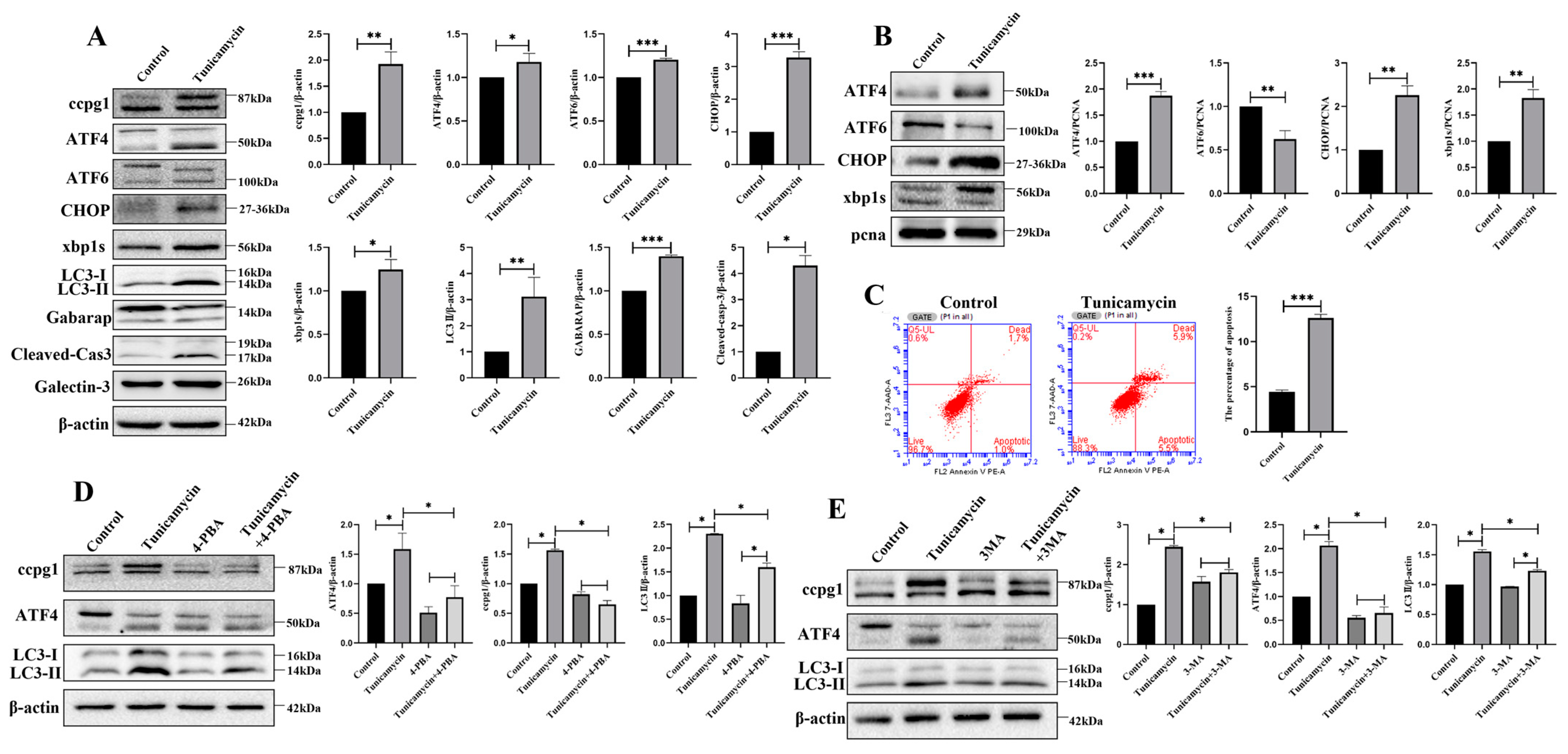
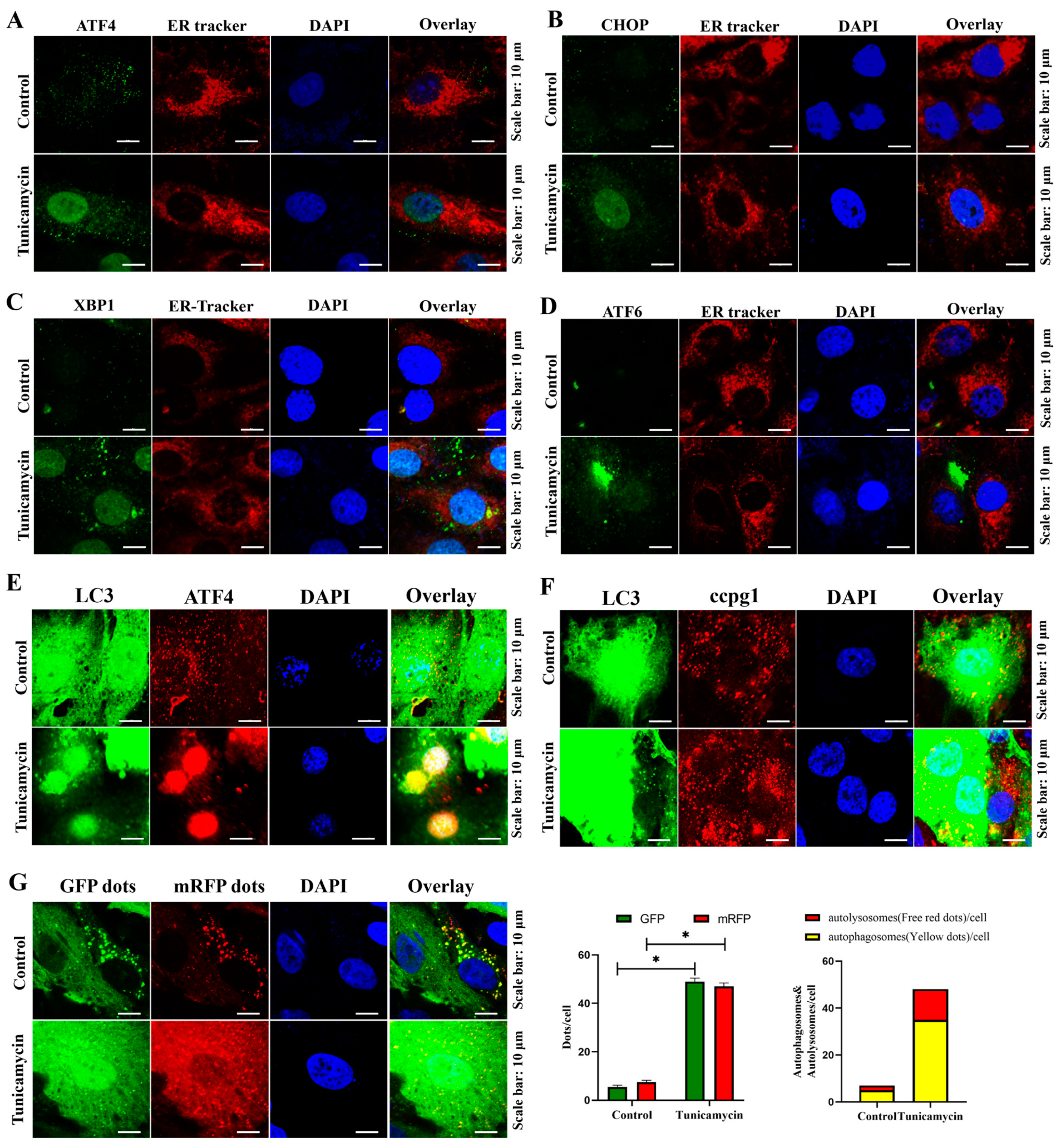
2.2. ER Stress Triggers Reticulophagy and Apoptosis of Granulosa Cells
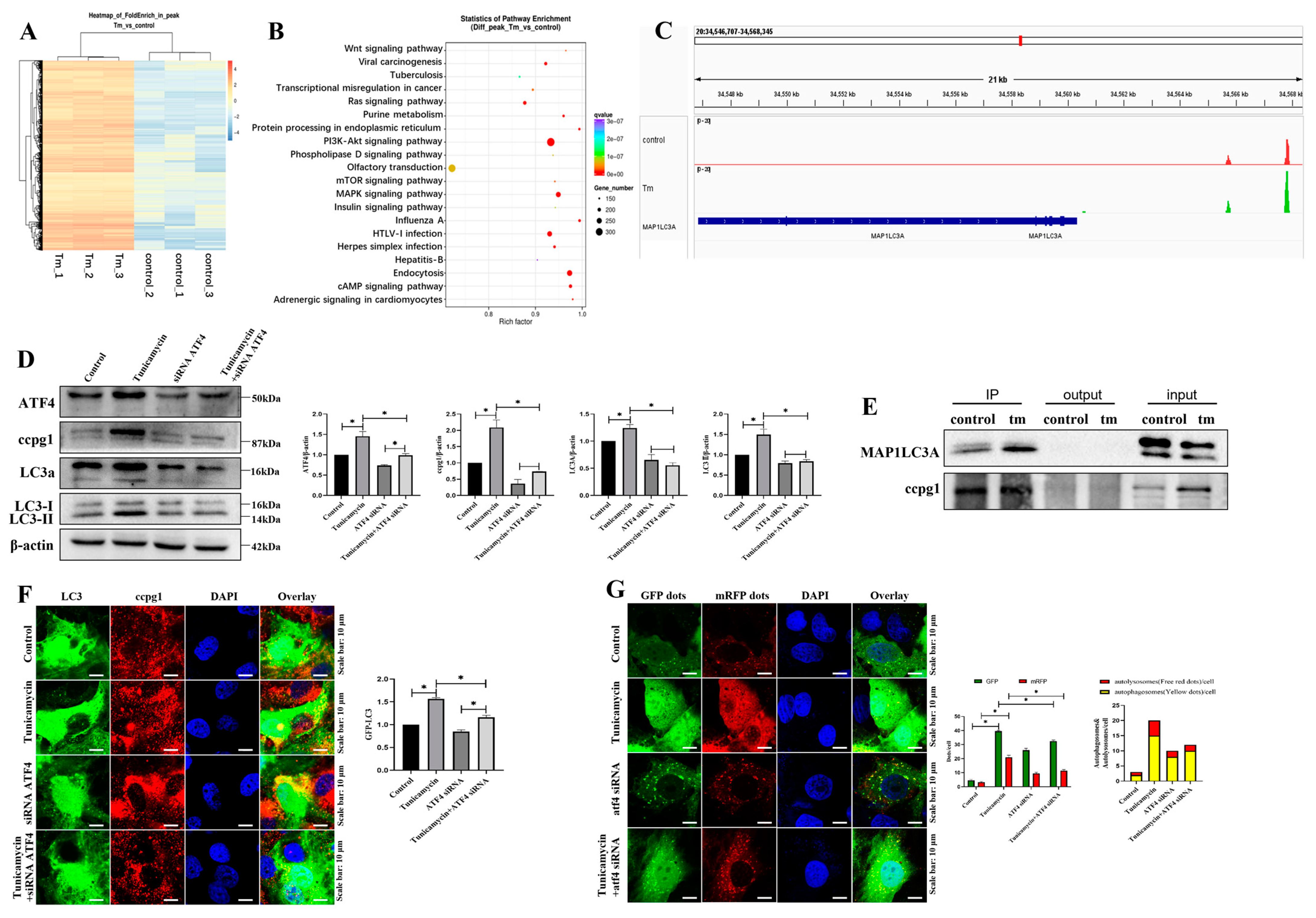
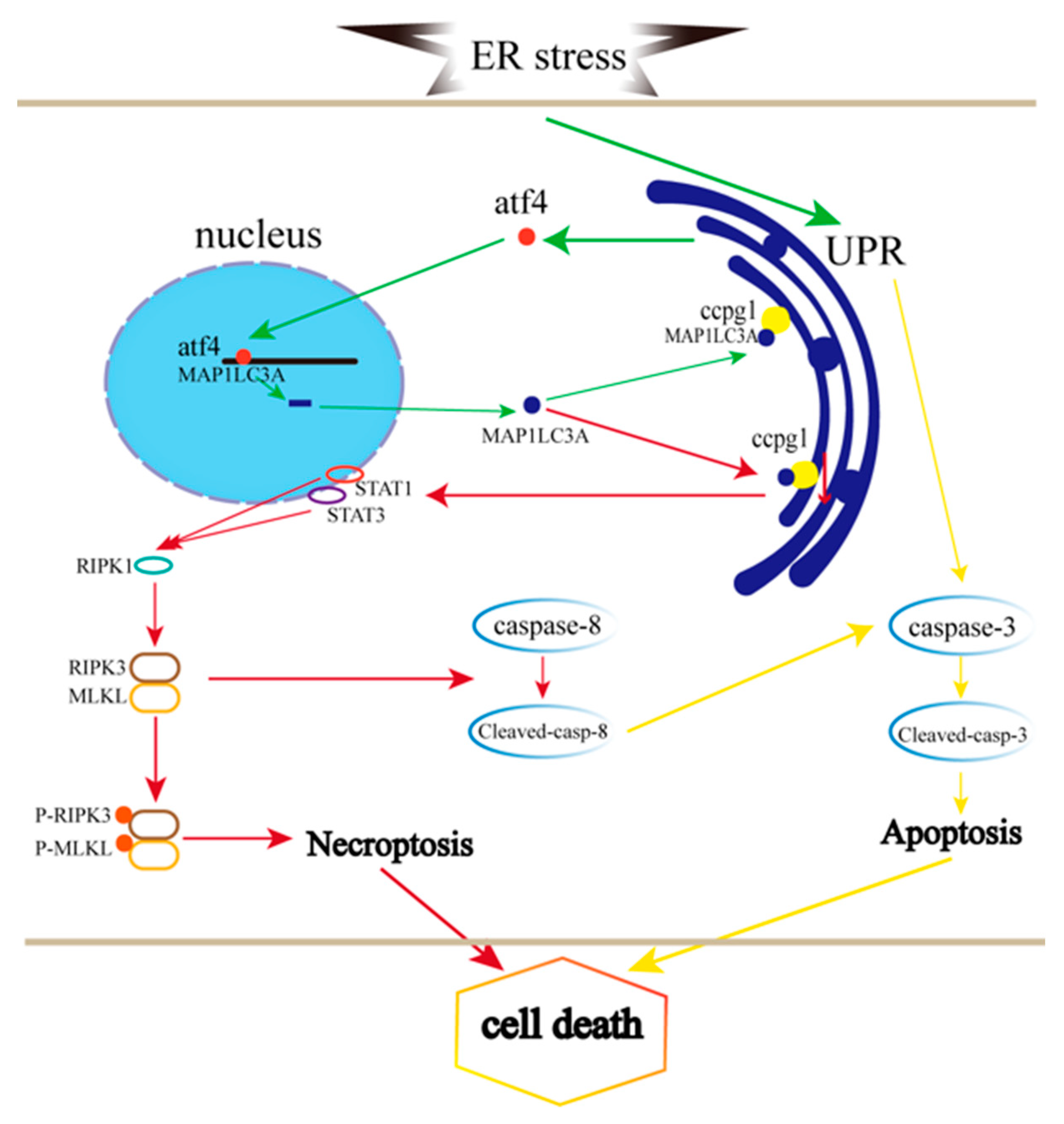
2.3. ATF4 Targets MAP1LC3A to Activate the Reticulophagy Receptor CCPG1
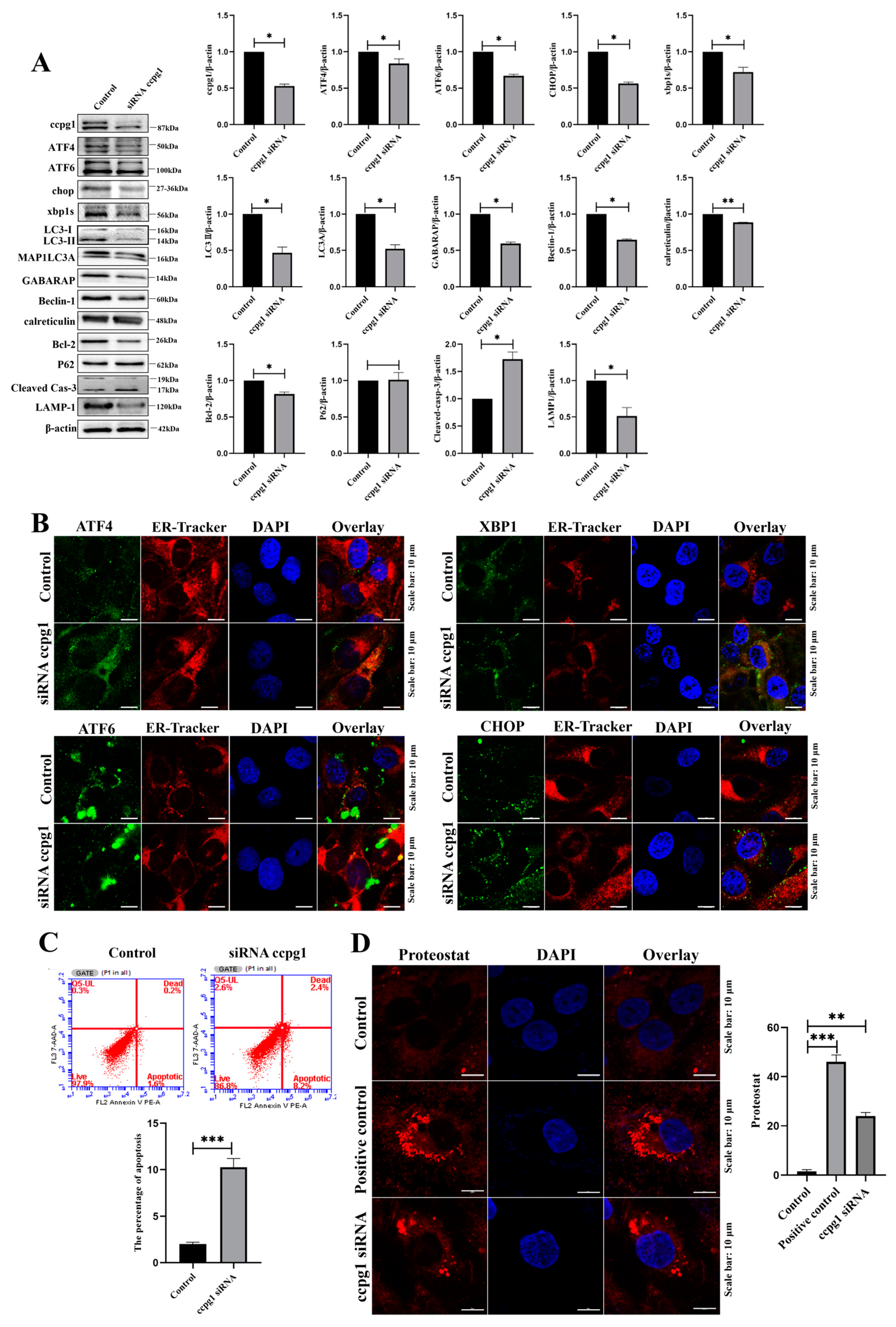
2.4. Impaired ER Proteostasis in CCPG1-Deficient and CCPG1-Overexpressing Cells
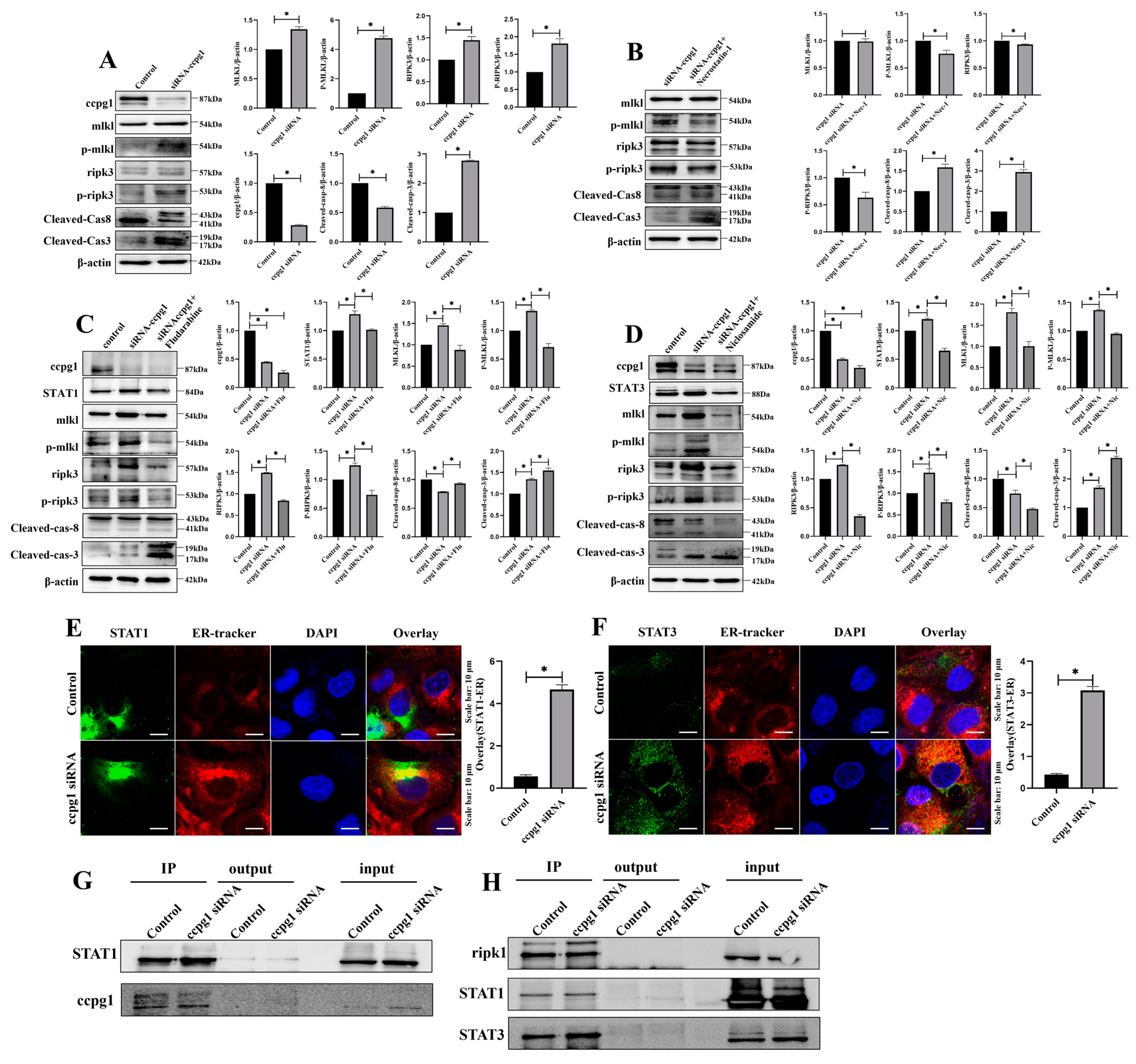
2.5. The Up- and Downregulation of CCPG1 Triggers Necroptosis of Granulosa Cells
2.6. CCPG1 Triggers Necroptosis of Granulosa Cells via STAT1 and STAT3
2.7. ER Stress Induced Apoptosis Negatively with Necroptosis
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Inhibitors and Concentration
4.3. Knockdown of ATF4 and CCPG1 in Granulosa Cells
4.4. Western Blot
4.5. ChIP Sequencing
4.6. Proteomic Profiling
4.7. Immunofluorescence
4.8. Coimmunoprecipitation
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kovanci, E.; Schutt, A.K. Premature ovarian failure: Clinical presentation and treatment. Obs. Gynecol. Clin. N. Am. 2015, 42, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Persani, L.; Rossetti, R.; Cacciatore, C. Genes involved in human premature ovarian failure. J. Mol. Endocrinol. 2010, 45, 257–279. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.X.; Wang, Z.B.; Lin, F.; Zhang, C.H.; Sun, H.M.; Zhou, L.; Zhou, Q.; Schatten, H.; Odile, F.C.; Brigitte, B.; et al. Ablation of beta subunit of protein kinase CK2 in mouse oocytes causes follicle atresia and premature ovarian failure. Cell Death Dis. 2018, 9, 508. [Google Scholar] [CrossRef]
- Yang, Y.; Lin, P.; Chen, F.; Wang, A.; Lan, X.; Song, Y.; Jin, Y. Luman recruiting factor regulates endoplasmic reticulum stress in mouse ovarian granulosa cell apoptosis. Theriogenology 2013, 79, 633–639.e3. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes Dev. 1999, 13, 1211–1233. [Google Scholar] [CrossRef] [PubMed]
- Koumenis, C. ER stress, hypoxia tolerance and tumor progression. Curr. Mol. Med. 2006, 6, 55–69. [Google Scholar] [CrossRef]
- Yang, E.S.; Bae, J.Y.; Kim, T.H.; Kim, Y.S.; Suk, K.; Bae, Y.C. Involvement of endoplasmic reticulum stress response in orofacial inflammatory pain. Exp. Neurobiol. 2014, 23, 372–380. [Google Scholar] [CrossRef]
- Yorimitsu, T.; Klionsky, D.J. Eating the endoplasmic reticulum: Quality control by autophagy. Trends Cell Biol. 2007, 17, 279–285. [Google Scholar] [CrossRef]
- Lemus, L.; Goder, V. Regulation of Endoplasmic Reticulum-Associated Protein Degradation (ERAD) by Ubiquitin. Cells 2014, 3, 824–847. [Google Scholar] [CrossRef]
- Yang, Y.; Pei, X.; Jin, Y.; Wang, Y.; Zhang, C. The roles of endoplasmic reticulum stress response in female mammalian reproduction. Cell Tissue Res 2016, 363, 589–597. [Google Scholar] [CrossRef]
- Liu, K.S.; Peng, Z.H.; Cheng, W.J.; Dai, C.F.; Tong, H. Endoplasmic reticulum stress-induced apoptosis in the development of reproduction. Reprod. Contracept. 2016, 27, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Guzel, E.; Arlier, S.; Guzeloglu-Kayisli, O.; Tabak, M.S.; Ekiz, T.; Semerci, N.; Larsen, K.; Schatz, F.; Lockwood, C.J.; Kayisli, U.A. Endoplasmic Reticulum Stress and Homeostasis in Reproductive Physiology and Pathology. Int. J. Mol. Sci. 2017, 18, 792. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Tan, J.; Miao, Y.; Zhang, Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J. Cell. Physiol. 2018, 233, 3867–3874. [Google Scholar] [CrossRef] [PubMed]
- Stephani, M.; Picchianti, L.; Gajic, A.; Beveridge, R.; Skarwan, E.; Sanchez de Medina Hernandez, V.; Mohseni, A.; Clavel, M.; Zeng, Y.; Naumann, C.; et al. A cross-kingdom conserved ER-phagy receptor maintains endoplasmic reticulum homeostasis during stress. eLife 2020, 9, e58396. [Google Scholar] [CrossRef] [PubMed]
- Schuck, S.; Gallagher, C.M.; Walter, P. ER-phagy mediates selective degradation of endoplasmic reticulum independently of the core autophagy machinery. J. Cell Sci. 2014, 127, 4078–4088. [Google Scholar] [CrossRef]
- Eldeeb, M.A.; Zorca, C.E.; Ragheb, M.A.; Rashidi, F.B.; Salah El-Din, D.S. Fine-tuning ER-phagy by post-translational modifications. Bioessays 2021, 43, e2000212. [Google Scholar] [CrossRef]
- Ferro-Novick, S.; Reggiori, F.; Brodsky, J.L. ER-Phagy, ER Homeostasis, and ER Quality Control: Implications for Disease. Trends Biochem. Sci. 2021, 46, 630–639. [Google Scholar] [CrossRef]
- Grumati, P.; Dikic, I.; Stolz, A. ER-phagy at a glance. J. Cell Sci. 2018, 131, jcs208884. [Google Scholar] [CrossRef]
- Smith, M.D.; Harley, M.E.; Kemp, A.J.; Wills, J.; Lee, M.; Arends, M.; von Kriegsheim, A.; Behrends, C.; Wilkinson, S. CCPG1 Is a Non-canonical Autophagy Cargo Receptor Essential for ER-Phagy and Pancreatic ER Proteostasis. Dev. Cell 2018, 44, 217–232.e211. [Google Scholar] [CrossRef]
- Smith, M.D.; Wilkinson, S. CCPG1, a cargo receptor required for reticulophagy and endoplasmic reticulum proteostasis. Autophagy 2018, 14, 1090–1091. [Google Scholar] [CrossRef]
- Smith, M.D.; Wilkinson, S. CCPG1, an unconventional cargo receptor for ER-phagy, maintains pancreatic acinar cell health. Mol. Cell Oncol. 2018, 5, e1441631. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Lee, J.A. Role of the mammalian ATG8/LC3 family in autophagy: Differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep. 2016, 49, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Zielke, S.; Kardo, S.; Zein, L.; Mari, M.; Covarrubias-Pinto, A.; Kinzler, M.N.; Meyer, N.; Stolz, A.; Fulda, S.; Reggiori, F.; et al. ATF4 links ER stress with reticulophagy in glioblastoma cells. Autophagy 2021, 17, 2432–2448. [Google Scholar] [CrossRef] [PubMed]
- Sardana, R.; Emr, S.D. Membrane Protein Quality Control Mechanisms in the Endo-Lysosome System. Trends Cell Biol. 2021, 31, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Smith, M.; Wilkinson, S. ER homeostasis and autophagy. Essays Biochem. 2017, 61, 625–635. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, L. Autophagy is a double-edged sword in the therapy of colorectal cancer. Oncol. Lett. 2021, 21, 378. [Google Scholar] [CrossRef]
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M.K. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight 2019, 4, e128834. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Chan, F.K.; Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. 2017, 12, 103–130. [Google Scholar] [CrossRef]
- Lindholm, D.; Korhonen, L.; Eriksson, O.; Kõks, S. Recent Insights into the Role of Unfolded Protein Response in ER Stress in Health and Disease. Front. Cell Dev. Biol. 2017, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Walter, P.; Yen, T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef] [PubMed]
- Chino, H.; Mizushima, N. ER-Phagy: Quality Control and Turnover of Endoplasmic Reticulum. Trends Cell Biol. 2020, 30, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef]
- Lin, P.; Yang, Y.; Li, X.; Chen, F.; Cui, C.; Hu, L.; Li, Q.; Liu, W.; Jin, Y. Endoplasmic reticulum stress is involved in granulosa cell apoptosis during follicular atresia in goat ovaries. Mol. Reprod Dev. 2012, 79, 423–432. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, T.; Guo, X.; Zhang, R.; Jia, Y.; Shang, C.; Wang, A.; Jin, Y.; Lin, P. Ufmylation regulates granulosa cell apoptosis via ER stress but not oxidative stress during goat follicular atresia. Theriogenology 2021, 169, 47–55. [Google Scholar] [CrossRef]
- Huang, N.; Yu, Y.; Qiao, J. Dual role for the unfolded protein response in the ovary: Adaption and apoptosis. Protein Cell 2017, 8, 14–24. [Google Scholar] [CrossRef]
- Liang, X.; Yan, Z.; Ma, W.; Qian, Y.; Zou, X.; Cui, Y.; Liu, J.; Meng, Y. Peroxiredoxin 4 protects against ovarian ageing by ameliorating D-galactose-induced oxidative damage in mice. Cell Death Dis. 2020, 11, 1053. [Google Scholar] [CrossRef]
- Tang, X.; Dong, H.; Fang, Z.; Li, J.; Yang, Q.; Yao, T.; Pan, Z. Ubiquitin-like modifier 1 ligating enzyme 1 relieves cisplatin-induced premature ovarian failure by reducing endoplasmic reticulum stress in granulosa cells. Reprod. Biol. Endocrinol. 2022, 20, 84. [Google Scholar] [CrossRef]
- Urra, H.; Dufey, E.; Lisbona, F.; Rojas-Rivera, D.; Hetz, C. When ER stress reaches a dead end. Biochim. Biophys. Acta 2013, 1833, 3507–3517. [Google Scholar] [CrossRef]
- Cai, Y.; Arikkath, J.; Yang, L.; Guo, M.L.; Periyasamy, P.; Buch, S. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 2016, 12, 225–244. [Google Scholar] [CrossRef] [PubMed]
- Fujita, E.; Kouroku, Y.; Isoai, A.; Kumagai, H.; Misutani, A.; Matsuda, C.; Hayashi, Y.K.; Momoi, T. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: Ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum. Mol. Genet. 2007, 16, 618–629. [Google Scholar] [CrossRef]
- Lim, Y.; Kim, S.; Kim, E.K. Palmitate reduces starvation-induced ER stress by inhibiting ER-phagy in hypothalamic cells. Mol. Brain 2021, 14, 65. [Google Scholar] [CrossRef]
- Loi, M.; Raimondi, A.; Morone, D.; Molinari, M. ESCRT-III-driven piecemeal micro-ER-phagy remodels the ER during recovery from ER stress. Nat. Commun. 2019, 10, 5058. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, S.; Wang, Z.; Ding, M.; Li, X.; Guo, J.; Han, G.; Zhao, P. Dexmedetomidine Alleviated Endoplasmic Reticulum Stress via Inducing ER-phagy in the Spinal Cord of Neuropathic Pain Model. Front. Neurosci. 2020, 14, 90. [Google Scholar] [CrossRef] [PubMed]
- Popelka, H.; Klionsky, D.J. Analysis of the native conformation of the LIR/AIM motif in the Atg8/LC3/GABARAP-binding proteins. Autophagy 2015, 11, 2153–2159. [Google Scholar] [CrossRef] [PubMed]
- Birgisdottir, Å.B.; Lamark, T.; Johansen, T. The LIR motif—Crucial for selective autophagy. J. Cell Sci. 2013, 126, 3237–3247. [Google Scholar] [CrossRef]
- Lahiri, V.; Klionsky, D.J. CCPG1 is a noncanonical autophagy cargo receptor essential for reticulophagy and pancreatic ER proteostasis. Autophagy 2018, 14, 1107–1109. [Google Scholar] [CrossRef]
- Loi, M.; Fregno, I.; Guerra, C.; Molinari, M. Eat it right: ER-phagy and recovER-phagy. Biochem. Soc. Trans. 2018, 46, 699–706. [Google Scholar] [CrossRef]
- Jin, M.; Liu, X.; Klionsky, D.J. SnapShot: Selective autophagy. Cell 2013, 152, 368–368.e362. [Google Scholar] [CrossRef]
- He, L.; Qian, X.; Cui, Y. Advances in ER-Phagy and Its Diseases Relevance. Cells 2021, 10, 2328. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, G.R.; Yadav, P.K.; Yadav, A.K.; Tiwari, M.; Gupta, A.; Sharma, A.; Pandey, A.N.; Pandey, A.K.; Chaube, S.K. Necroptosis in stressed ovary. J. Biomed. Sci. 2019, 26, 11. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, G.R.; Yadav, P.K.; Yadav, A.K.; Tiwari, M.; Gupta, A.; Sharma, A.; Sahu, K.; Pandey, A.N.; Pandey, A.K.; Chaube, S.K. Necrosis and necroptosis in germ cell depletion from mammalian ovary. J. Cell Physiol. 2019, 234, 8019–8027. [Google Scholar] [CrossRef]
- Du, Y.; Bagnjuk, K.; Lawson, M.S.; Xu, J.; Mayerhofer, A. Acetylcholine and necroptosis are players in follicular development in primates. Sci. Rep. 2018, 8, 6166. [Google Scholar] [CrossRef]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef]
- Lin, T.; Lee, J.E.; Kang, J.W.; Shin, H.Y.; Lee, J.B.; Jin, D.I. Endoplasmic Reticulum (ER) Stress and Unfolded Protein Response (UPR) in Mammalian Oocyte Maturation and Preimplantation Embryo Development. Int. J. Mol. Sci. 2019, 20, 409. [Google Scholar] [CrossRef]
- Khatun, H.; Wada, Y.; Konno, T.; Tatemoto, H.; Yamanaka, K.I. Endoplasmic reticulum stress attenuation promotes bovine oocyte maturation in vitro. Reproduction 2020, 159, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Takehara, I.; Igarashi, H.; Kawagoe, J.; Matsuo, K.; Takahashi, K.; Nishi, M.; Nagase, S. Impact of endoplasmic reticulum stress on oocyte aging mechanisms. Mol. Hum. Reprod. 2020, 26, 567–575. [Google Scholar] [CrossRef]
- Wu, J.; Chen, S.; Liu, H.; Zhang, Z.; Ni, Z.; Chen, J.; Yang, Z.; Nie, Y.; Fan, D. Tunicamycin specifically aggravates ER stress and overcomes chemoresistance in multidrug-resistant gastric cancer cells by inhibiting N-glycosylation. J. Exp. Clin. Cancer Res. 2018, 37, 272. [Google Scholar] [CrossRef]
- Wu, Y.; Ma, C.; Zhao, H.; Zhou, Y.; Chen, Z.; Wang, L. Alleviation of endoplasmic reticulum stress protects against cisplatin-induced ovarian damage. Reprod. Biol. Endocrinol. 2018, 16, 85. [Google Scholar] [CrossRef]
- Xian, Y.; Li, B.; Pan, P.; Wang, Y.; Pei, X.; Yang, Y. Role of Autophagy in Ovarian Cryopreservation by Vitrification. Cryo Lett. 2018, 39, 201–210. [Google Scholar]
- Tonnus, W.; Meyer, C.; Steinebach, C.; Belavgeni, A.; von Mässenhausen, A.; Gonzalez, N.Z.; Maremonti, F.; Gembardt, F.; Himmerkus, N.; Latk, M.; et al. Dysfunction of the key ferroptosis-surveilling systems hypersensitizes mice to tubular necrosis during acute kidney injury. Nat. Commun. 2021, 12, 4402. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Tsou, P.S.; Coit, P.; Gensterblum-Miller, E.; Renauer, P.; Rohraff, D.M.; Kilian, N.C.; Schonfeld, M.; Sawalha, A.H. Hypomethylation of STAT1 and HLA-DRB1 is associated with type-I interferon-dependent HLA-DRB1 expression in lupus CD8+ T cells. Ann. Rheum. Dis. 2019, 78, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Luo, M.; Rong, Q.X.; Zhang, H.; Chen, Z.; Wang, F.; Zhao, H.Y.; Fu, L.W. Niclosamide, an antihelmintic drug, enhances efficacy of PD-1/PD-L1 immune checkpoint blockade in non-small cell lung cancer. J. Immunother. Cancer 2019, 7, 245. [Google Scholar] [CrossRef]
- Zong, Y.; Feng, S.; Cheng, J.; Yu, C.; Lu, G. Up-Regulated ATF4 Expression Increases Cell Sensitivity to Apoptosis in Response to Radiation. Cell Physiol. Biochem. 2017, 41, 784–794. [Google Scholar] [CrossRef]
- Chino, H.; Hatta, T.; Natsume, T.; Mizushima, N. Intrinsically Disordered Protein TEX264 Mediates ER-phagy. Mol. Cell 2019, 74, 909–921.e906. [Google Scholar] [CrossRef] [PubMed]
- Jiajie, T.; Yanzhou, Y.; Hoi-Hung, A.C.; Zi-Jiang, C.; Wai-Yee, C. Conserved miR-10 family represses proliferation and induces apoptosis in ovarian granulosa cells. Sci. Rep. 2017, 7, 41304. [Google Scholar] [CrossRef]
- He, Q.; Gu, L.; Lin, Q.; Ma, Y.; Liu, C.; Pei, X.; Li, P.A.; Yang, Y. The Immp2l Mutation Causes Ovarian Aging Through ROS-Wnt/β-Catenin-Estrogen Pathway: Preventive Effect of Melatonin. Endocrinology 2020, 161, bqaa119. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Jing, Y.; Qu, X.; Yang, J.; Pan, P.; Liu, X.; Gao, H.; Pei, X.; Zhang, C.; Yang, Y. The Activation of Reticulophagy by ER Stress through the ATF4-MAP1LC3A-CCPG1 Pathway in Ovarian Granulosa Cells Is Linked to Apoptosis and Necroptosis. Int. J. Mol. Sci. 2023, 24, 2749. https://doi.org/10.3390/ijms24032749
Li H, Jing Y, Qu X, Yang J, Pan P, Liu X, Gao H, Pei X, Zhang C, Yang Y. The Activation of Reticulophagy by ER Stress through the ATF4-MAP1LC3A-CCPG1 Pathway in Ovarian Granulosa Cells Is Linked to Apoptosis and Necroptosis. International Journal of Molecular Sciences. 2023; 24(3):2749. https://doi.org/10.3390/ijms24032749
Chicago/Turabian StyleLi, Huiduo, Yanan Jing, Xiaoya Qu, Jinyi Yang, Pengge Pan, Xinrui Liu, Hui Gao, Xiuying Pei, Cheng Zhang, and Yanzhou Yang. 2023. "The Activation of Reticulophagy by ER Stress through the ATF4-MAP1LC3A-CCPG1 Pathway in Ovarian Granulosa Cells Is Linked to Apoptosis and Necroptosis" International Journal of Molecular Sciences 24, no. 3: 2749. https://doi.org/10.3390/ijms24032749
APA StyleLi, H., Jing, Y., Qu, X., Yang, J., Pan, P., Liu, X., Gao, H., Pei, X., Zhang, C., & Yang, Y. (2023). The Activation of Reticulophagy by ER Stress through the ATF4-MAP1LC3A-CCPG1 Pathway in Ovarian Granulosa Cells Is Linked to Apoptosis and Necroptosis. International Journal of Molecular Sciences, 24(3), 2749. https://doi.org/10.3390/ijms24032749