
Functions of Steroid Hormones in the Male Reproductive Tract as Revealed by Mouse Models


Abstract
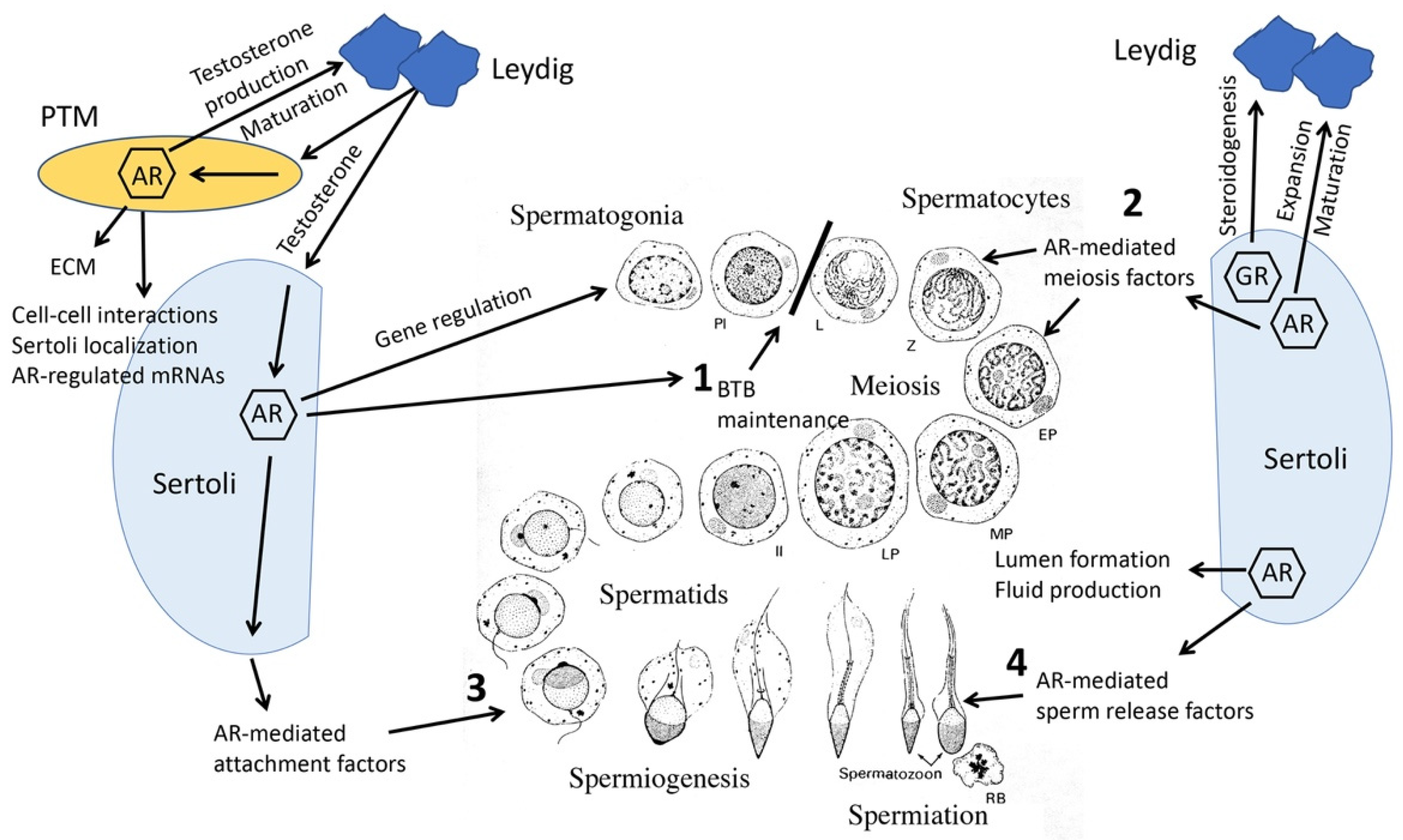
1. Androgen Actions in the Male Reproductive Tract
2. Global Elimination of ARs
2.1. ARKO Mice
2.2. Inducible AR Knock Out (iARKO)
3. Sertoli Cell-Specific Mouse Models
3.1. Sertoli Cell-Specific Knockout (SCARKO)
3.2. Hypomorph SCARKO Mice
3.3. Gain of Function, TgSCAR
3.4. Specificity-Affecting AR Knock-In (SPARKI)
3.5. Glucocorticoid Receptor Knock Out
4. AR-Regulated, Differentially Expressed Genes (DEGs) Identified by Mouse Models
4.1. SCARKO Models
4.2. Ribotag Mouse Models
4.3. Single Cell RNA Sequencing (scRNA-Seq) Analysis of SCARKO Mice
5. Regulation of Germ Cell Meiosis by AR Activities in Sertoli Cells
6. Androgen Pathway Selective Transgenic Mice
7. Peritubular Myoid (PTM), Leydig and Germ Cell-Specific AR Knockout Models
7.1. PTM Cells
7.2. Leydig Cells
7.3. Germ Cells
8. Knockout Mouse Models Reveal Collaborative Interrelationships between Sertoli, PTM, Leydig and Germ Cells
9. Knockout of AR in Accessory Sex Organs
9.1. Prostate
9.2. Seminal Vesicles
9.3. Epididymis
10. Estrogen Actions in the Male
10.1. Discovery of Estrogens and Their Presence in the Male
10.2. Estrogen Administration to Males Produces Deleterious Male Reproductive Effects
10.3. Estrogens Are Present in Males and Estrogen Receptors Are Widely Distributed in Males
10.4. Use of the Estrogen Receptor 1 Knockout Mouse to Establish Roles for Estrogen in Reproductive Tract Development and Function and Fertility in Male Mice
10.5. Normal Estrogen Actions in the Male Reproductive Tract Require Membrane Estrogen Receptor 1
11. 17α-E2, a Stereoisomer of 17β-E2, Increases Longevity in Male Mice
12. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chang, C.; Lee, S.O.; Wang, R.S.; Yeh, S.; Chang, T.M. Androgen receptor (AR) physiological roles in male and female reproductive systems: Lessons learned from AR-knockout mice lacking AR in selective cells. Biol. Reprod. 2013, 89, 21. [Google Scholar] [CrossRef] [PubMed]
- Lyon, M.F.; Hawkes, S.G. X-linked gene for testicular feminization in the mouse. Nature 1970, 227, 1217–1219. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.; Tsai, M.Y.; Xu, Q.; Mu, X.M.; Lardy, H.; Huang, K.E.; Lin, H.; Yeh, S.D.; Altuwaijri, S.; Zhou, X.; et al. Generation and characterization of androgen receptor knockout (ARKO) mice: An in vivo model for the study of androgen functions in selective tissues. Proc. Natl. Acad. Sci. USA 2002, 99, 13498–13503. [Google Scholar] [CrossRef]
- De Gendt, K.; Swinnen, J.V.; Saunders, P.T.; Schoonjans, L.; Dewerchin, M.; Devos, A.; Tan, K.; Atanassova, N.; Claessens, F.; Lecureuil, C.; et al. A Sertoli cell-selective knockout of the androgen receptor causes spermatogenic arrest in meiosis. Proc. Natl. Acad. Sci. USA 2004, 101, 1327–1332. [Google Scholar] [CrossRef]
- Smith, L.B.; Walker, W.H. The regulation of spermatogenesis by androgens. Semin. Cell Dev. Biol. 2014, 30, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Willems, A.; De Gendt, K.; Deboel, L.; Swinnen, J.V.; Verhoeven, G. The development of an inducible androgen receptor knockout model in mouse to study the postmeiotic effects of androgens on germ cell development. Spermatogenesis 2011, 1, 341–353. [Google Scholar] [CrossRef]
- Tan, K.A.; De Gendt, K.; Atanassova, N.; Walker, M.; Sharpe, R.M.; Saunders, P.T.; Denolet, E.; Verhoeven, G. The role of androgens in Sertoli cell proliferation and functional maturation: Studies in mice with total or Sertoli cell-selective ablation of the androgen receptor. Endocrinology 2005, 146, 2674–2683. [Google Scholar] [CrossRef]
- Tsai, M.Y.; Yeh, S.D.; Wang, R.S.; Yeh, S.; Zhang, C.; Lin, H.Y.; Tzeng, C.R.; Chang, C. Differential effects of spermatogenesis and fertility in mice lacking androgen receptor in individual testis cells. Proc. Natl. Acad. Sci. USA 2006, 103, 18975–18980. [Google Scholar] [CrossRef]
- Holdcraft, R.W.; Braun, R.E. Androgen receptor function is required in Sertoli cells for the terminal differentiation of haploid spermatids. Development 2004, 131, 459–467. [Google Scholar] [CrossRef]
- Hazra, R.; Corcoran, L.; Robson, M.; McTavish, K.J.; Upton, D.; Handelsman, D.J.; Allan, C.M. Temporal role of Sertoli cell androgen receptor expression in spermatogenic development. Mol. Endocrinol. 2013, 27, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Schauwaers, K.; De Gendt, K.; Saunders, P.T.; Atanassova, N.; Haelens, A.; Callewaert, L.; Moehren, U.; Swinnen, J.V.; Verhoeven, G.; Verrijdt, G.; et al. Loss of androgen receptor binding to selective androgen response elements causes a reproductive phenotype in a knockin mouse model. Proc. Natl. Acad. Sci. USA 2007, 104, 4961–4966. [Google Scholar] [CrossRef] [PubMed]
- Hazra, R.; Jimenez, M.; Desai, R.; Handelsman, D.J.; Allan, C.M. Sertoli cell androgen receptor expression regulates temporal fetal and adult Leydig cell differentiation, function, and population size. Endocrinology 2013, 154, 3410–3422. [Google Scholar] [CrossRef] [PubMed]
- Sanz, E.; Evanoff, R.; Quintana, A.; Evans, E.; Miller, J.A.; Ko, C.; Amieux, P.S.; Griswold, M.D.; McKnight, G.S. RiboTag analysis of actively translated mRNAs in Sertoli and Leydig cells in vivo. PLoS ONE 2013, 8, e66179. [Google Scholar] [CrossRef]
- De Gendt, K.; Verhoeven, G.; Amieux, P.S.; Wilkinson, M.F. Genome-wide identification of AR-regulated genes translated in Sertoli cells in vivo using the RiboTag approach. Mol. Endocrinol. 2014, 28, 575–591. [Google Scholar] [CrossRef]
- Cooke, P.S.; Walker, W.H. Male fertility in mice requires classical and nonclassical androgen signaling. Cell Rep. 2021, 36, 109557. [Google Scholar] [CrossRef] [PubMed]
- Welsh, M.; Saunders, P.T.; Atanassova, N.; Sharpe, R.M.; Smith, L.B. Androgen action via testicular peritubular myoid cells is essential for male fertility. FASEB J. 2009, 23, 4218–4230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yeh, S.; Chen, Y.T.; Wu, C.C.; Chuang, K.H.; Lin, H.Y.; Wang, R.S.; Chang, Y.J.; Mendis-Handagama, C.; Hu, L.; et al. Oligozoospermia with normal fertility in male mice lacking the androgen receptor in testis peritubular myoid cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17718–17723. [Google Scholar] [CrossRef]
- Xu, Q.; Lin, H.Y.; Yeh, S.D.; Yu, I.C.; Wang, R.S.; Chen, Y.T.; Zhang, C.; Altuwaijri, S.; Chen, L.M.; Chuang, K.H.; et al. Infertility with defective spermatogenesis and steroidogenesis in male mice lacking androgen receptor in Leydig cells. Endocrine 2007, 32, 96–106. [Google Scholar] [CrossRef]
- Simanainen, U.; Allan, C.M.; Lim, P.; McPherson, S.; Jimenez, M.; Zajac, J.D.; Davey, R.; Handelsman, D.J. Disruption of prostate epithelial androgen receptor impedes prostate lobe-specific growth and function. Endocrinology 2007, 148, 2264–2272. [Google Scholar] [CrossRef]
- Wu, C.T.; Altuwaijri, S.; Ricke, W.A.; Huang, S.P.; Yeh, S.; Zhang, C.; Niu, Y.; Tsai, M.-Y.; Chang, C. Increased prostate cell proliferation and loss of cell differentiation in mice lacking prostate epithelial androgen receptor. Proc. Natl. Acad. Sci. USA 2007, 104, 12679–12684. [Google Scholar] [CrossRef]
- Yu, S.; Zhang, C.; Lin, C.-C.; Niu, Y.; Lai, K.-P.; Chang, H.-C.; Yeh, S.-D.; Chang, C.; Yeh, S. Altered prostate epithelial development and IGF-1 signal in mice lacking the androgen receptor in stromal smooth muscle cells. Prostate 2011, 71, 517–524. [Google Scholar] [CrossRef]
- Yu, S.; Yeh, C.R.; Niu, Y.; Chang, H.C.; Tsai, Y.C.; Moses, H.L.; Shyr, C.-R.; Chang, C.; Yeh, S. Altered prostate epithelial development in mice lacking the androgen receptor in stromal fibroblasts. Prostate 2012, 72, 437–449. [Google Scholar] [CrossRef]
- Lai, K.P.; Yamashita, S.; Vitkus, S.; Shyr, C.R.; Yeh, S.; Chang, C. Suppressed prostate epithelial development with impaired branching morphogenesis in mice lacking stromal fibromuscular androgen receptor. Mol. Endocrinol. 2012, 26, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Krutskikh, A.; De Gendt, K.; Sharp, V.; Verhoeven, G.; Poutanen, M.; Huhtaniemi, I. Targeted inactivation of the androgen receptor gene in murine proximal epididymis causes epithelial hypotrophy and obstructive azoospermia. Endocrinology 2011, 152, 689–696. [Google Scholar] [CrossRef]
- O’Hara, L.; Welsh, M.; Saunders, P.T.; Smith, L.B. Androgen receptor expression in the caput epididymal epithelium is essential for development of the initial segment and epididymal spermatozoa transit. Endocrinology 2011, 152, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Hess, R.A.; Bunick, D.; Lee, K.-H.; Bahr, J.; Taylor, J.A.; Korach, K.S.; Lubahn, D.B. A role for oestrogens in the male reproductive system. Nature 1997, 390, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Adlanmerini, M.; Solinhac, R.; Abot, A.; Fabre, A.; Raymond-Letron, I.; Guihot, A.L.; Boudou, F.; Sautier, L.; Vessieres, E.; Kim, S.H.; et al. Mutation of the palmitoylation site of estrogen receptor α in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc. Natl. Acad. Sci. USA 2014, 111, E283–E290. [Google Scholar] [CrossRef] [PubMed]
- Pedram, A.; Razandi, M.; Lewis, M.; Hammes, S.; Levin, E.R. Membrane-localized estrogen receptor α is required for normal organ development and function. Dev. Cell 2014, 29, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Willems, A.; Batlouni, S.R.; Esnal, A.; Swinnen, J.V.; Saunders, P.T.; Sharpe, R.M.; França, L.R.; De Gendt, K.; Verhoeven, G. Selective ablation of the androgen receptor in mouse sertoli cells affects sertoli cell maturation, barrier formation and cytoskeletal development. PLoS ONE 2010, 5, e14168. [Google Scholar] [CrossRef]
- O’Shaughnessy, P.J.; Verhoeven, G.; De Gendt, K.; Monteiro, A.; Abel, M.H. Direct action through the sertoli cells is essential for androgen stimulation of spermatogenesis. Endocrinology 2010, 151, 2343–2348. [Google Scholar] [CrossRef]
- Verhoeven, G.; Willems, A.; Denolet, E.; Swinnen, J.V.; De Gendt, K. Androgens and spermatogenesis: Lessons from transgenic mouse models. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 1537–1556. [Google Scholar] [CrossRef] [PubMed]
- Eacker, S.M.; Shima, J.E.; Connolly, C.M.; Sharma, M.; Holdcraft, R.W.; Griswold, M.D.; Braun, R.E. Transcriptional profiling of androgen receptor (AR) mutants suggests instructive and permissive roles of AR signaling in germ cell development. Mol. Endocrinol. 2007, 21, 895–907. [Google Scholar] [CrossRef]
- Cao, C.; Ma, Q.; Mo, S.; Shu, G.; Liu, Q.; Ye, J.; Gui, Y. Single-Cell RNA Sequencing Defines the Regulation of Spermatogenesis by Sertoli-Cell Androgen Signaling. Front. Cell Dev. Biol. 2021, 9, 763267. [Google Scholar] [CrossRef]
- Larose, H.; Kent, T.; Ma, Q.; Shami, A.N.; Harerimana, N.; Li, J.Z.; Hammoud, S.S.; Handel, M.A. Regulation of meiotic progression by Sertoli-cell androgen signaling. Mol. Biol. Cell 2020, 31, 2841–2862. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-R.; Hao, X.-X.; Zhang, Y.; Deng, S.-L.; Wang, Z.-P.; Wang, Y.-Q.; Wang, X.-X.; Liu, Y.-X. Androgen receptor in Sertoli cells regulates DNA double-strand break repair and chromosomal synapsis of spermatocytes partially through intercellular EGF-EGFR signaling. Oncotarget 2016, 7, 18722–18735. [Google Scholar] [CrossRef] [PubMed]
- Bolcun-Filas, E.; Hall, E.; Speed, R.; Taggart, M.; Grey, C.; de Massy, B.; Benavente, R.; Cooke, H.J. Mutation of the mouse Syce1 gene disrupts synapsis and suggests a link between synaptonemal complex structural components and DNA repair. PLoS Genet. 2009, 5, e1000393. [Google Scholar] [CrossRef]
- De Vries, F.A.; de Boer, E.; van den Bosch, M.; Baarends, W.M.; Ooms, M.; Yuan, L.; Liu, J.-G.; van Zeeland, A.A.; Heyting, C.; Pastink, A. Mouse Sycp1 functions in synaptonemal complex assembly, meiotic recombination, and XY body formation. Genes Dev. 2005, 19, 1376–1389. [Google Scholar] [CrossRef]
- Hamer, G.; Novak, I.; Kouznetsova, A.; Hoog, C. Disruption of pairing and synapsis of chromosomes causes stage-specific apoptosis of male meiotic cells. Theriogenology 2008, 69, 333–339. [Google Scholar] [CrossRef]
- Schramm, S.; Fraune, J.; Naumann, R.; Hernandez-Hernandez, A.; Hoog, C.; Cooke, H.J.; Alsheimer, M.; Benavente, R. A novel mouse synaptonemal complex protein is essential for loading of central element proteins, recombination, and fertility. PLoS Genet. 2011, 7, e1002088. [Google Scholar] [CrossRef]
- Walker, W.H.; Easton, E.; Moreci, R.S.; Toocheck, C. Anamthathmakula, and P. Jeyasuria. Restoration of spermatogenesis and male fertility using an androgen receptor transgene. PLoS ONE 2015, 10, e0120783. [Google Scholar] [CrossRef]
- Welsh, M.; Moffat, L.; Belling, K.; de Franca, L.R.; Segatelli, T.M.; Saunders, P.T.; Sharpe, R.M.; Smith, L.B. Androgen receptor signalling in peritubular myoid cells is essential for normal differentiation and function of adult Leydig cells. Int. J. Androl. 2012, 35, 25–40. [Google Scholar] [CrossRef]
- Xu, C.; Graf, L.F.; Fazli, L.; Coleman, I.M.; Mauldin, D.E.; Li, D.; Nelson, P.S.; Gleave, M.; Plymate, S.R.; Cox, M.E.; et al. Regulation of global gene expression in the bone marrow microenvironment by androgen: Androgen ablation increases insulin-like growth factor binding protein-5 expression. Prostate 2007, 67, 1621–1629. [Google Scholar] [CrossRef]
- De Gendt, K.; Atanassova, N.; Tan, K.A.; de Franca, L.R.; Parreira, G.G.; McKinnell, C.; Sharpe, R.M.; Saunders, P.T.; Mason, J.I.; Hartung, S.; et al. Development and function of the adult generation of Leydig cells in mice with Sertoli cell-selective or total ablation of the androgen receptor. Endocrinology 2005, 146, 4117–4126. [Google Scholar] [CrossRef]
- O’Shaughnessy, P.J.; Johnston, H.; Willerton, L.; Baker, P.J. Failure of normal adult Leydig cell development in androgen-receptor-deficient mice. J. Cell Sci. 2002, 115, 3491–3496. [Google Scholar] [CrossRef]
- O’Shaughnessy, P.J.; Monteiro, A.; Abel, M. Testicular development in mice lacking receptors for follicle stimulating hormone and androgen. PLoS ONE 2012, 7, e35136. [Google Scholar] [CrossRef]
- O’Shaughnessy, P.J.; Mitchell, R.T.; Monteiro, A.; O’Hara, L.; Cruickshanks, L.; der Grinten, H.C.; Brown, P.; Abel, M.; Smith, L.B. Androgen receptor expression is required to ensure development of adult Leydig cells and to prevent development of steroidogenic cells with adrenal characteristics in the mouse testis. BMC Dev. Biol. 2019, 19, 8. [Google Scholar] [CrossRef]
- Welsh, M.; Moffat, L.; McNeilly, A.; Brownstein, D.; Saunders, P.T.; Sharpe, R.M.; Smith, L.B. Smooth muscle cell-specific knockout of androgen receptor: A new model for prostatic disease. Endocrinology 2011, 152, 3541–3551. [Google Scholar] [CrossRef]
- Simanainen, U.; McNamara, K.; Davey, R.A.; Zajac, J.D.; Handelsman, D.J. Severe subfertility in mice with androgen receptor inactivation in sex accessory organs but not in testis. Endocrinology 2008, 149, 3330–3338. [Google Scholar] [CrossRef]
- Welsh, M.; Moffat, L.; Jack, L.; McNeilly, A.; Brownstein, D.; Saunders, P.T.; Sharpe, R.M.; Smith, L.B. Deletion of androgen receptor in the smooth muscle of the seminal vesicles impairs secretory function and alters its responsiveness to exogenous testosterone and estradiol. Endocrinology 2010, 151, 3374–3385. [Google Scholar] [CrossRef]
- Santen, R.J.; Simpson, E. History of estrogen: Its purification, structure, synthesis, biologic actions, and clinical implications. Endocrinology 2019, 160, 605–625. [Google Scholar] [CrossRef]
- Zondek, B. Mass excretion of oestrogenic hormone in the urine of the stallion. Nature 1934, 133, 209–210. [Google Scholar] [CrossRef]
- Burrows, H. Carcinoma mammae occurring in a male mouse under continued treatment with oestrin. Am. J. Cancer 1935, 24, 613–616. [Google Scholar] [PubMed]
- Cooke, P.S.; Nanjappa, M.K.; Ko, C.; Prins, G.S.; Hess, R.A. Estrogens in male physiology. Physiol. Rev. 2017, 97, 995–1043. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer. J. Clin. 1972, 22, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Herbst, A.L.; Ulfelder, H.; Poskanzer, D.C. Adenocarcinoma of the vagina. Association of maternal stilbestrol therapy with tumor appearance in young women. N. Engl. J. Med. 1971, 284, 878–881. [Google Scholar] [CrossRef]
- Lubahn, D.B.; Moyer, J.S.; Golding, T.S.; Couse, J.F.; Korach, K.S.; Smithies, O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc. Natl. Acad. Sci. USA 1993, 90, 11162–11166. [Google Scholar] [CrossRef]
- Hess, R.A.; Cooke, P.S. Estrogen in the male: A historical perspective. Biol. Reprod. 2018, 99, 27–44. [Google Scholar] [CrossRef]
- Yellayi, S.; Teuscher, C.; Woods, J.A.; Welsh, T.H., Jr.; Tung, K.S.; Nakai, M.; Rosenfeld, C.S.; Lubahn, D.B.; Cooke, P.S. Normal development of thymus in male and female mice requires estrogen/estrogen receptor-α signaling pathway. Endocrine 2000, 12, 207–213. [Google Scholar] [CrossRef]
- Heine, P.A.; Taylor, J.A.; Iwamoto, G.A.; Lubahn, D.B.; Cooke, P.S. Increased adipose tissue in male and female estrogen receptor-α knockout mice. Proc. Natl. Acad. Sci. USA 2000, 97, 12729–12734. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F.; Lange, C.A.; Levin, E.R. Membrane-Initiated Estrogen, Androgen, and Progesterone Receptor Signaling in Health and Disease. Endocr. Rev. 2022, 43, 720–742. [Google Scholar] [CrossRef]
- Nanjappa, M.K.; Hess, R.A.; Medrano, T.I.; Locker, S.H.; Levin, E.R.; Cooke, P.S. Membrane-localized estrogen receptor 1 is required for normal male reproductive development and function in mice. Endocrinology 2016, 157, 2909–2919. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Strong, R.; Allison, D.B.; Ames, B.N.; Astle, C.M.; Atamna, H.; Fernandez, E.; Flurkey, K.; Javors, M.A.; Nadon, N.L.; et al. Acarbose, 17-α-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging Cell 2014, 13, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Strong, R.; Miller, R.A.; Antebi, A.; Astle, C.M.; Bogue, M.; Denzel, M.S.; Fernandez, E.; Flurkey, K.; Hamilton, K.L.; Lamming, D.W.; et al. Longer lifespan in male mice treated with a weakly estrogenic agonist, an antioxidant, an α-glucosidase inhibitor or a Nrf2-inducer. Aging Cell 2016, 15, 872–884. [Google Scholar] [CrossRef]
- Harrison, D.E.; Strong, R.; Reifsnyder, P.; Kumar, N.; Fernandez, E.; Flurkey, K.; Javors, M.A.; Lopez-Cruzan, M.; Macchiarini, F.; Nelson, J.F.; et al. 17-α-estradiol late in life extends lifespan in aging UM-HET3 male mice; nicotinamide riboside and three other drugs do not affect lifespan in either sex. Aging Cell 2021, 20, e13328. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.-A. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor β. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.H.; Williams, M.; Upchurch, S.; Eriksson, H.; Helton, E.; Markaverich, B.M. Effects of estradiol-17α on nuclear occupancy of the estrogen receptor, stimulation of nuclear type II sites and uterine growth. J. Steroid Biochem. 1982, 16, 323–328. [Google Scholar] [CrossRef]
- Toran-Allerand, C.D. Estrogen and the brain: Beyond ER-α and ER-β. Exp. Gerontol. 2004, 39, 1579–1586. [Google Scholar] [CrossRef]
- Toran-Allerand, C.D.; Tinnikov, A.A.; Singh, R.J.; Nethrapalli, I.S. 17α-estradiol: A brain-active estrogen? Endocrinology 2005, 146, 3843–3850. [Google Scholar] [CrossRef]
- Mann, S.N.; Hadad, N.; Nelson Holte, M.; Rothman, A.R.; Sathiaseelan, R.; Ali Mondal, S.; Agbaga, M.P.; Unnikrishnan, A.; Subramaniam, M.; Hawse, J.; et al. Health benefits attributed to 17α-estradiol, a lifespan-extending compound, are mediated through estrogen receptor α. eLife 2020, 9, e59616. [Google Scholar] [CrossRef]


{kind=link}
| Mouse Model | Cell Type | Mutation/Activity | References |
|---|---|---|---|
| ARKO, T-AR−/y | Global | Ar knock out | [3,4] |
| iARKO | Global | Inducible Ar knock out | [6] |
| SCARKO, S-AR−/y | Sertoli | Ar knock out | [7,8] |
| Arflox(ex1−neo)/Y | Sertoli | Ar hypomorph | [9] |
| TgSCAR | Global | Ar gain of function | [10] |
| SPARKI | Global | Gr replacement of Ar exon 3 | [11] |
| SCGRKO | Sertoli | Gr knock out | [12] |
| Ribotag | Leydig, Sertoli | Detects translated mRNAs | [13] |
| RiboTag-SCARKO | Sertoli | Detects translated mRNAs in Ar knockout | [14] |
| AR-C | Sertoli, Global | Classical AR activity | [15] |
| AR-NC | Sertoli, Global | Nonclassical AR activity | [15] |
| (PTM)-ARKO | Smooth Muscle (SM) | Ar knock out | [16] |
| PM-AR−/y | SM | Ar knock out | [17] |
| L-AR−/Y | Leydig | Ar knock out | [18] |
| G-AR−/y | Germ | Ar knock out | [8] |
| PEARKO | Prostate Epithelium | Ar knock out | [19] |
| Pes-ARKO | Prostate Epithelium | Ar knock out | [20] |
| SM-ARKO | SM | Ar knock out | [21] |
| FSP1-ARKO | Fibroblast | Ar knock out | [22] |
| dARKO | Fibroblast + SM | Ar knock out | [23] |
| ProxE-ARKO | Proximal Epididymis | Ar knock out | [24] |
| CEARKO | Principal (Epididymis) | Ar knock out | [25] |
| Esr1KO | Global | Esr1 knock out | [26] |
| NOER | Global | Membrane Esr1 knock out | [27,28] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walker, W.H.; Cooke, P.S. Functions of Steroid Hormones in the Male Reproductive Tract as Revealed by Mouse Models. Int. J. Mol. Sci. 2023, 24, 2748. https://doi.org/10.3390/ijms24032748
Walker WH, Cooke PS. Functions of Steroid Hormones in the Male Reproductive Tract as Revealed by Mouse Models. International Journal of Molecular Sciences. 2023; 24(3):2748. https://doi.org/10.3390/ijms24032748
Chicago/Turabian StyleWalker, William H., and Paul S. Cooke. 2023. "Functions of Steroid Hormones in the Male Reproductive Tract as Revealed by Mouse Models" International Journal of Molecular Sciences 24, no. 3: 2748. https://doi.org/10.3390/ijms24032748
APA StyleWalker, W. H., & Cooke, P. S. (2023). Functions of Steroid Hormones in the Male Reproductive Tract as Revealed by Mouse Models. International Journal of Molecular Sciences, 24(3), 2748. https://doi.org/10.3390/ijms24032748