
Deletion of UCP1 in Tg2576 Mice Increases Body Temperature and Exacerbates Alzheimer’s Disease-Related Pathologies

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
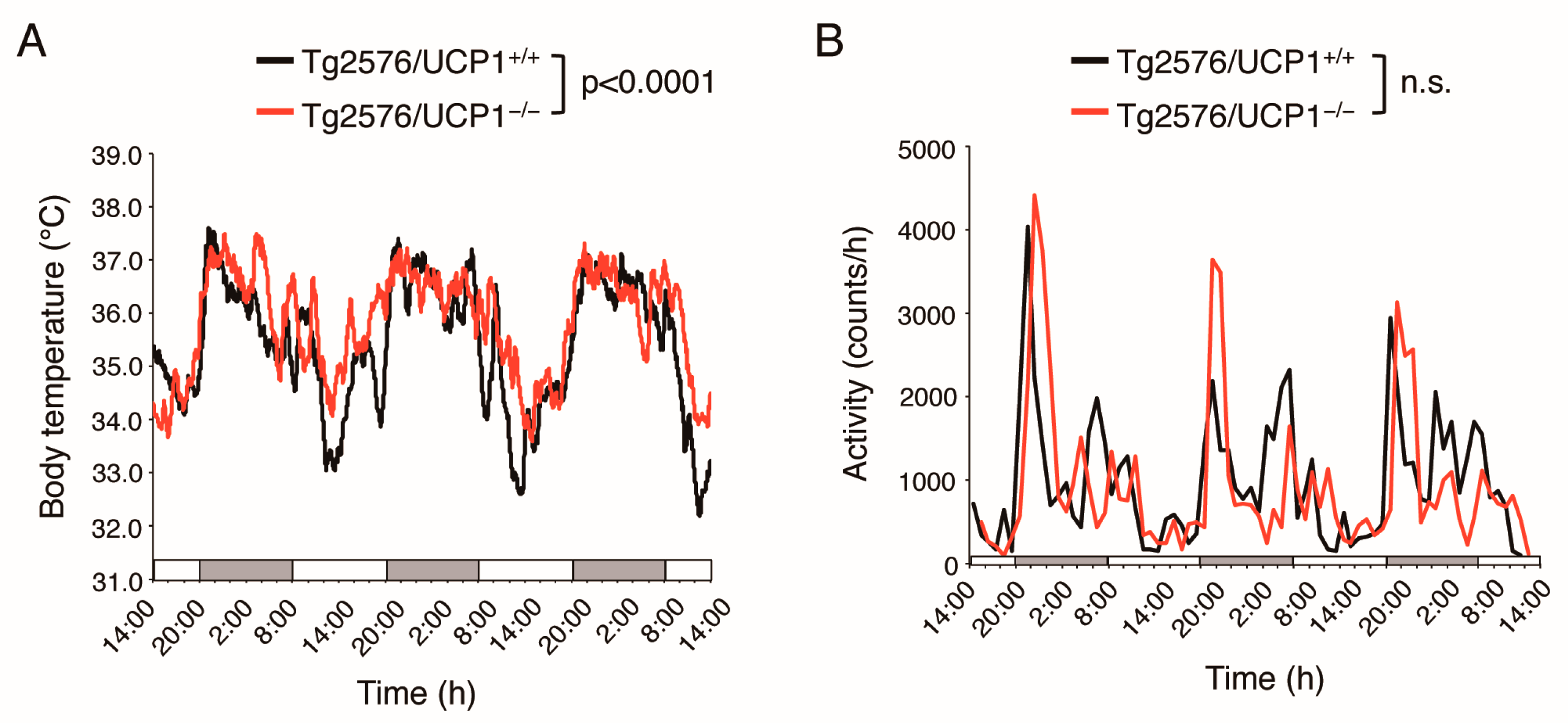
2.1. Effects of UCP1 Deletion in Tg2576 Mice on Core Body Temperature
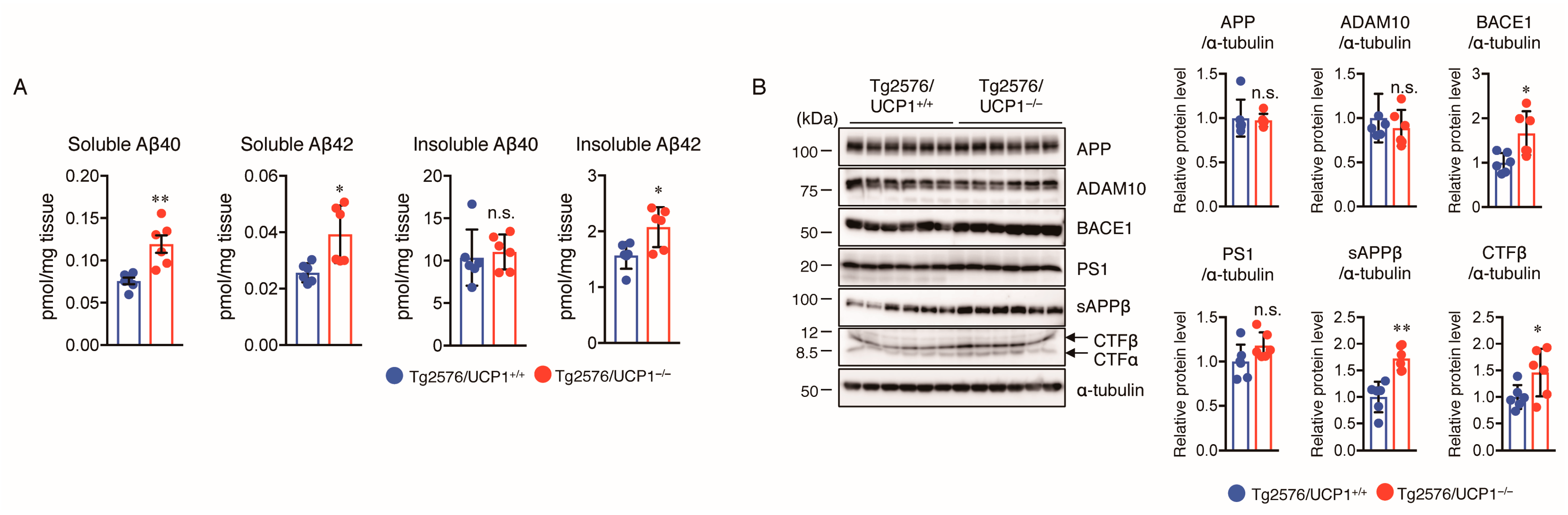
2.2. Effect of UCP1 Deletion in Tg2576 Mice on Aβ Generation
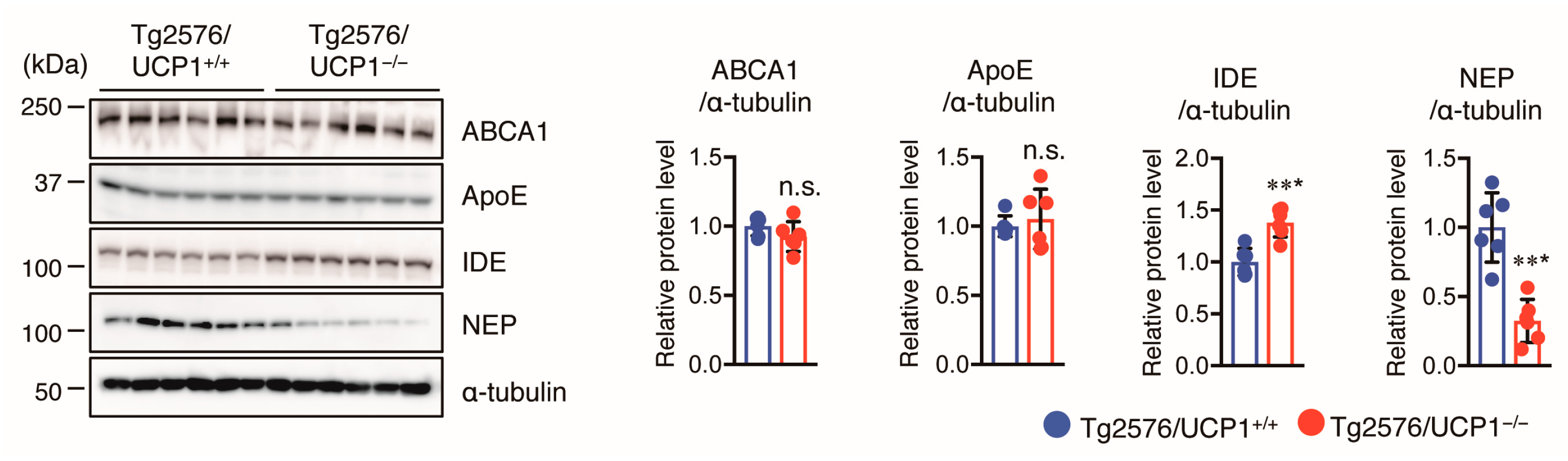
2.3. Effect of UCP1 Deletion in Tg2576 Mice on Aβ Degradation and Clearance
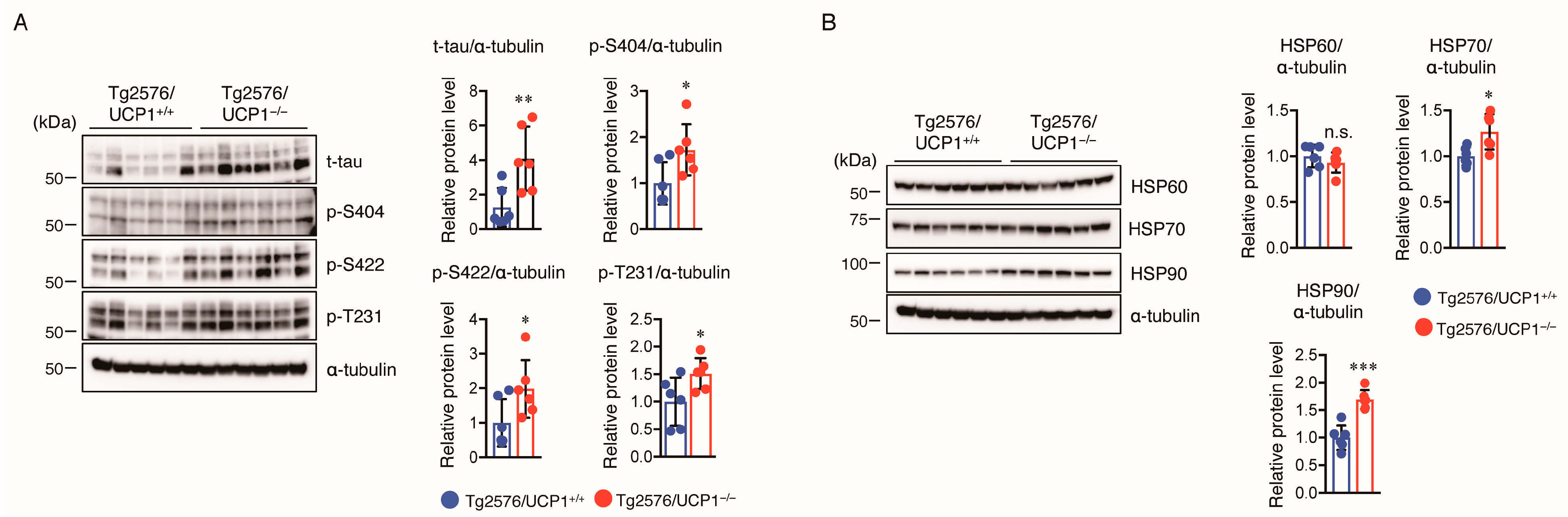
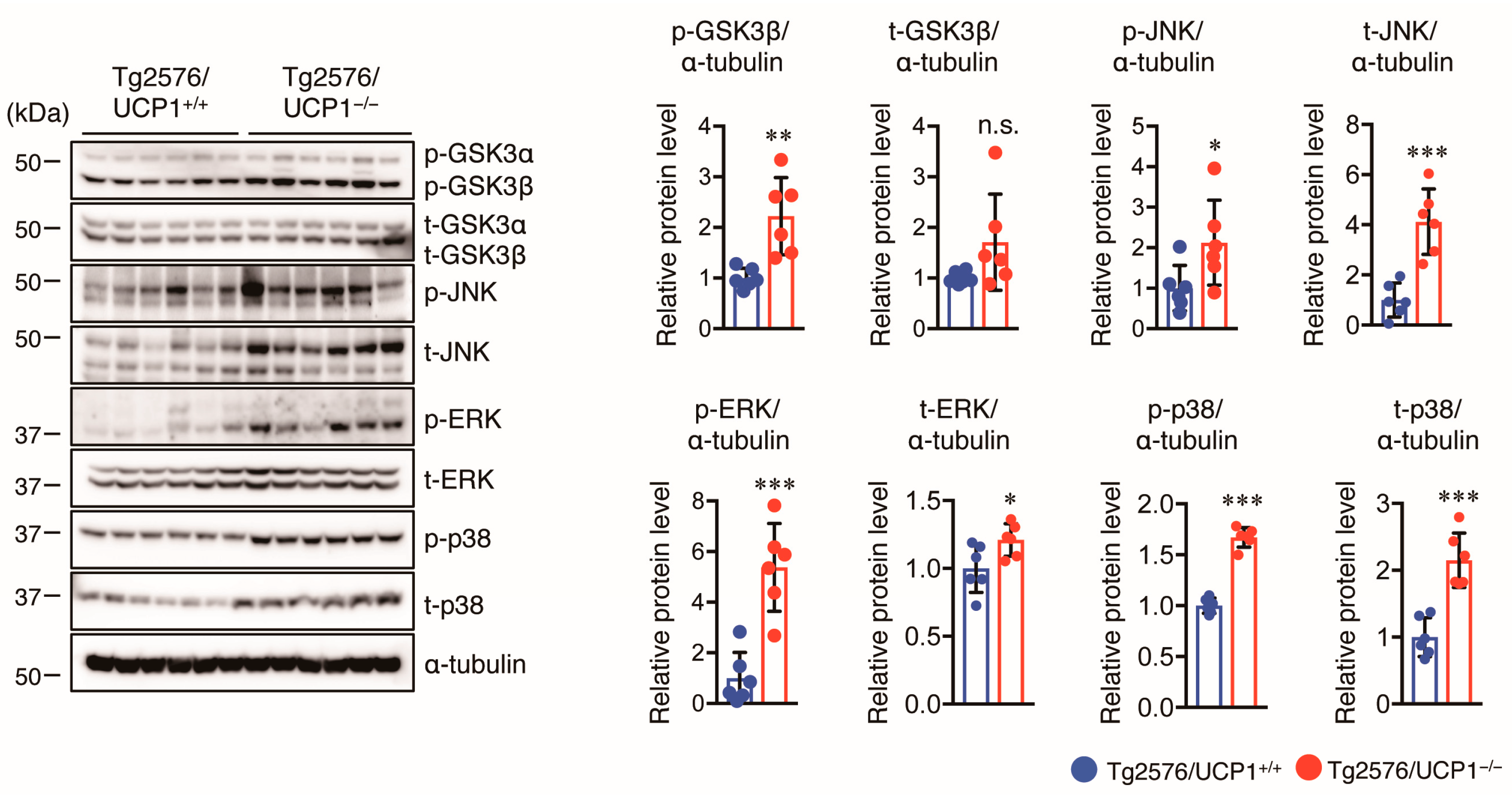
2.4. Effect of UCP1 Deletion in Tg2576 Mice on Tau Pathology
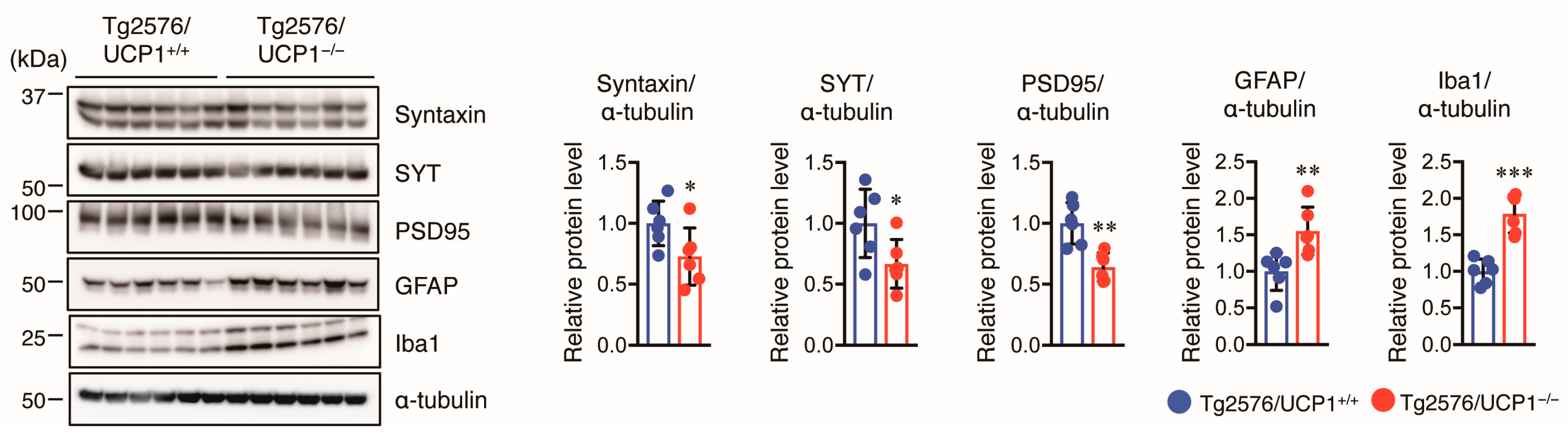
2.5. Effect of UCP1 Deletion in Tg2576 Mice on Glial Cell Activation and Synaptic Protein Levels
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. Biotelemetry
4.3. Aβ ELISA
4.4. Western Blot Analysis
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, N.; Tomita, T.; Hayashi, I.; Tsuruoka, M.; Niimura, M.; Takahashi, Y.; Thinakaran, G.; Iwatsubo, T. The role of presenilin cofactors in the gamma-secretase complex. Nature 2003, 422, 438–441. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 1998, 21, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Binder, L.I.; Guillozet-Bongaarts, A.L.; Garcia-Sierra, F.; Berry, R.W. Tau, tangles, and Alzheimer’s disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2005, 1739, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Assal, F.; Cummings, J.L. Neuropsychiatric symptoms in the dementias. Curr. Opin. Neurol. 2002, 15, 445–450. [Google Scholar] [CrossRef]
- Bombois, S.; Derambure, P.; Pasquier, F.; Monaca, C. Sleep disorders in aging and dementia. J. Nutr. Health Aging 2010, 14, 212–217. [Google Scholar] [CrossRef]
- Finkel, S.I. Behavioral and psychologic symptoms of dementia. Clin. Geriatr. Med. 2003, 19, 799–824. [Google Scholar] [CrossRef]
- Klaffke, S.; Staedt, J. Sundowning and circadian rhythm disorders in dementia. Acta Neurol. Belg. 2006, 106, 168–175. [Google Scholar]
- Degroot, D.W.; Kenney, W.L. Impaired defense of core temperature in aged humans during mild cold stress. Am. J. Physiol. -Regul. Integr. Comp. Physiol. 2007, 292, R103–R108. [Google Scholar] [CrossRef]
- Gomolin, I.H.; Aung, M.M.; Wolf-Klein, G.; Auerbach, C. Older is colder: Temperature range and variation in older people. J. Am. Geriatr. Soc. 2005, 53, 2170–2172. [Google Scholar] [CrossRef] [PubMed]
- Carrettiero, D.C.; Santiago, F.E.; Motzko-Soares, A.C.; Almeida, M.C. Temperature and toxic Tau in Alzheimer’s disease: New insights. Temperature 2015, 2, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Tournissac, M.; Vandal, M.; Francois, A.; Planel, E.; Calon, F. Old age potentiates cold-induced tau phosphorylation: Linking thermoregulatory deficit with Alzheimer’s disease. Neurobiol. Aging 2017, 50, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Whittington, R.A.; Papon, M.A.; Chouinard, F.; Planel, E. Hypothermia and Alzheimer’s disease neuropathogenic pathways. Curr. Alzheimer Res. 2010, 7, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Klegeris, A.; Schulzer, M.; Harper, D.G.; McGeer, P.L. Increase in core body temperature of Alzheimer’s disease patients as a possible indicator of chronic neuroinflammation: A meta-analysis. Gerontology 2007, 53, 7–11. [Google Scholar] [CrossRef]
- Mishima, K.; Okawa, M.; Satoh, K.; Shimizu, T.; Hozumi, S.; Hishikawa, Y. Different manifestations of circadian rhythms in senile dementia of Alzheimer’s type and multi-infarct dementia. Neurobiol. Aging 1997, 18, 105–109. [Google Scholar] [CrossRef]
- Okawa, M.; Mishima, K.; Hishikawa, Y.; Hozumi, S.; Hori, H.; Takahashi, K. Circadian rhythm disorders in sleep-waking and body temperature in elderly patients with dementia and their treatment. Sleep 1991, 14, 478–485. [Google Scholar] [CrossRef]
- Satlin, A.; Volicer, L.; Stopa, E.G.; Harper, D. Circadian locomotor activity and core-body temperature rhythms in Alzheimer’s disease. Neurobiol. Aging 1995, 16, 765–771. [Google Scholar] [CrossRef]
- Touitou, Y.; Reinberg, A.; Bogdan, A.; Auzeby, A.; Beck, H.; Touitou, C. Age-related changes in both circadian and seasonal rhythms of rectal temperature with special reference to senile dementia of Alzheimer type. Gerontology 1986, 32, 110–118. [Google Scholar] [CrossRef]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Enerback, S.; Jacobsson, A.; Simpson, E.M.; Guerra, C.; Yamashita, H.; Harper, M.E.; Kozak, L.P. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature 1997, 387, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Kozak, L.P.; Harper, M.E. Mitochondrial uncoupling proteins in energy expenditure. Annu. Rev. Nutr. 2000, 20, 339–363. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Rossmeisl, M.; McClaine, J.; Riachi, M.; Harper, M.E.; Kozak, L.P. Paradoxical resistance to diet-induced obesity in UCP1-deficient mice. J. Clin. Investig. 2003, 111, 399–407. [Google Scholar] [CrossRef]
- Wang, Y.; Kimura, K.; Inokuma, K.; Saito, M.; Kontani, Y.; Kobayashi, Y.; Mori, N.; Yamashita, H. Potential contribution of vasoconstriction to suppression of heat loss and homeothermic regulation in UCP1-deficient mice. Pflügers Arch. 2006, 452, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Kontani, Y.; Wang, Y.; Kimura, K.; Inokuma, K.I.; Saito, M.; Suzuki-Miura, T.; Wang, Z.; Sato, Y.; Mori, N.; Yamashita, H. UCP1 deficiency increases susceptibility to diet-induced obesity with age. Aging Cell 2005, 4, 147–155. [Google Scholar] [CrossRef]
- Jung, C.G.; Kato, R.; Zhou, C.; Abdelhamid, M.; Shaaban, E.I.A.; Yamashita, H.; Michikawa, M. Sustained high body temperature exacerbates cognitive function and Alzheimer’s disease-related pathologies. Sci. Rep. 2022, 12, 12273. [Google Scholar] [CrossRef]
- Blair, L.J.; Nordhues, B.A.; Hill, S.E.; Scaglione, K.M.; O’Leary, J.C., 3rd; Fontaine, S.N.; Breydo, L.; Zhang, B.; Li, P.; Wang, L.; et al. Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Investig. 2013, 123, 4158–4169. [Google Scholar] [CrossRef]
- Dickey, C.A.; Kamal, A.; Lundgren, K.; Klosak, N.; Bailey, R.M.; Dunmore, J.; Ash, P.; Shoraka, S.; Zlatkovic, J.; Eckman, C.B.; et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Investig. 2007, 117, 648–658. [Google Scholar] [CrossRef]
- Jinwal, U.K.; Akoury, E.; Abisambra, J.F.; O’Leary, J.C., 3rd; Thompson, A.D.; Blair, L.J.; Jin, Y.; Bacon, J.; Nordhues, B.A.; Cockman, M.; et al. Imbalance of Hsp70 family variants fosters tau accumulation. FASEB J. 2013, 27, 1450–1459. [Google Scholar] [CrossRef]
- Zhu, X.; Lee, H.G.; Raina, A.K.; Perry, G.; Smith, M.A. The role of mitogen-activated protein kinase pathways in Alzheimer’s disease. Neurosignals 2002, 11, 270–281. [Google Scholar] [CrossRef]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef] [PubMed]
- Ukropec, J.; Anunciado, R.P.; Ravussin, Y.; Hulver, M.W.; Kozak, L.P. UCP1-independent thermogenesis in white adipose tissue of cold-acclimated Ucp1-/- mice. J. Biol. Chem. 2006, 281, 31894–31908. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, H.; Cheung, B.S.; Hyman, B.T.; Irizarry, M.C. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 2002, 59, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Taslima, F.; Abdelhamid, M.; Kim, S.W.; Akatsu, H.; Michikawa, M.; Jung, C.G. Beta-Amyloid Increases the Expression Levels of Tid1 Responsible for Neuronal Cell Death and Amyloid Beta Production. Mol. Neurobiol. 2020, 57, 1099–1114. [Google Scholar] [CrossRef]
- Bates, K.A.; Verdile, G.; Li, Q.X.; Ames, D.; Hudson, P.; Masters, C.L.; Martins, R.N. Clearance mechanisms of Alzheimer’s amyloid-beta peptide: Implications for therapeutic design and diagnostic tests. Mol. Psychiatry 2009, 14, 469–486. [Google Scholar] [CrossRef]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Watanabe, K.; Sekiguchi, M.; Hosoki, E.; Kawashima-Morishima, M.; Lee, H.J.; Hama, E.; Sekine-Aizawa, Y.; et al. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat. Med. 2000, 6, 143–150. [Google Scholar] [CrossRef]
- Wang, D.S.; Lipton, R.B.; Katz, M.J.; Davies, P.; Buschke, H.; Kuslansky, G.; Verghese, J.; Younkin, S.G.; Eckman, C.; Dickson, D.W. Decreased neprilysin immunoreactivity in Alzheimer disease, but not in pathological aging. J. Neuropathol. Exp. Neurol. 2005, 64, 378–385. [Google Scholar] [CrossRef]
- Avila, J.; Lucas, J.J.; Perez, M.; Hernandez, F. Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef]
- Jinwal, U.K.; O’Leary, J.C., 3rd; Borysov, S.I.; Jones, J.R.; Li, Q.; Koren, J., 3rd; Abisambra, J.F.; Vestal, G.D.; Lawson, L.Y.; Johnson, A.G.; et al. Hsc70 rapidly engages tau after microtubule destabilization. J. Biol. Chem. 2010, 285, 16798–16805. [Google Scholar] [CrossRef]
- Abisambra, J.; Jinwal, U.K.; Miyata, Y.; Rogers, J.; Blair, L.; Li, X.; Seguin, S.P.; Wang, L.; Jin, Y.; Bacon, J.; et al. Allosteric heat shock protein 70 inhibitors rapidly rescue synaptic plasticity deficits by reducing aberrant tau. Biol. Psychiatry 2013, 74, 367–374. [Google Scholar] [CrossRef]
- Luo, W.; Dou, F.; Rodina, A.; Chip, S.; Kim, J.; Zhao, Q.; Moulick, K.; Aguirre, J.; Wu, N.; Greengard, P.; et al. Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc. Natl. Acad. Sci. USA 2007, 104, 9511–9516. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef] [PubMed]
- Meraz-Rios, M.A.; Toral-Rios, D.; Franco-Bocanegra, D.; Villeda-Hernandez, J.; Campos-Pena, V. Inflammatory process in Alzheimer’s Disease. Front. Integr. Neurosci. 2013, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Nisticò, R.; Pignatelli, M.; Piccinin, S.; Mercuri, N.B.; Collingridge, G. Targeting synaptic dysfunction in Alzheimer’s disease therapy. Mol. Neurobiol. 2012, 46, 572–587. [Google Scholar] [CrossRef]
- Mango, D.; Saidi, A.; Cisale, G.Y.; Feligioni, M.; Corbo, M.; Nisticò, R. Targeting Synaptic Plasticity in Experimental Models of Alzheimer’s Disease. Front. Pharmacol. 2019, 10, 778. [Google Scholar] [CrossRef]
- Arendt, T. Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 167–179. [Google Scholar] [CrossRef]
- Calon, F.; Lim, G.P.; Yang, F.; Morihara, T.; Teter, B.; Ubeda, O.; Rostaing, P.; Triller, A.; Salem, N., Jr.; Ashe, K.H.; et al. Docosahexaenoic acid protects from dendritic pathology in an Alzheimer’s disease mouse model. Neuron 2004, 43, 633–645. [Google Scholar] [CrossRef]
- Benarroch, E.E. Glutamatergic synaptic plasticity and dysfunction in Alzheimer disease: Emerging mechanisms. Neurology 2018, 91, 125–132. [Google Scholar] [CrossRef]
- Callahan, L.M.; Vaules, W.A.; Coleman, P.D. Quantitative decrease in synaptophysin message expression and increase in cathepsin D message expression in Alzheimer disease neurons containing neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 1999, 58, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Kawarabayashi, T.; Younkin, L.H.; Saido, T.C.; Shoji, M.; Ashe, K.H.; Younkin, S.G. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2001, 21, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Westerman, M.A.; Cooper-Blacketer, D.; Mariash, A.; Kotilinek, L.; Kawarabayashi, T.; Younkin, L.H.; Carlson, G.A.; Younkin, S.G.; Ashe, K.H. The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 2002, 22, 1858–1867. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, C.-G.; Yamashita, H.; Kato, R.; Zhou, C.; Matsushita, H.; Takeuchi, T.; Abdelhamid, M.; Chen, Y.; Michikawa, M. Deletion of UCP1 in Tg2576 Mice Increases Body Temperature and Exacerbates Alzheimer’s Disease-Related Pathologies. Int. J. Mol. Sci. 2023, 24, 2741. https://doi.org/10.3390/ijms24032741
Jung C-G, Yamashita H, Kato R, Zhou C, Matsushita H, Takeuchi T, Abdelhamid M, Chen Y, Michikawa M. Deletion of UCP1 in Tg2576 Mice Increases Body Temperature and Exacerbates Alzheimer’s Disease-Related Pathologies. International Journal of Molecular Sciences. 2023; 24(3):2741. https://doi.org/10.3390/ijms24032741
Chicago/Turabian StyleJung, Cha-Gyun, Hitoshi Yamashita, Reiko Kato, Chunyu Zhou, Hiroaki Matsushita, Tamaki Takeuchi, Mona Abdelhamid, Yuxin Chen, and Makoto Michikawa. 2023. "Deletion of UCP1 in Tg2576 Mice Increases Body Temperature and Exacerbates Alzheimer’s Disease-Related Pathologies" International Journal of Molecular Sciences 24, no. 3: 2741. https://doi.org/10.3390/ijms24032741
APA StyleJung, C.-G., Yamashita, H., Kato, R., Zhou, C., Matsushita, H., Takeuchi, T., Abdelhamid, M., Chen, Y., & Michikawa, M. (2023). Deletion of UCP1 in Tg2576 Mice Increases Body Temperature and Exacerbates Alzheimer’s Disease-Related Pathologies. International Journal of Molecular Sciences, 24(3), 2741. https://doi.org/10.3390/ijms24032741