


Giant Fern Genomes Show Complex Evolution Patterns: A Comparative Analysis in Two Species of Tmesipteris (Psilotaceae)
 ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Results
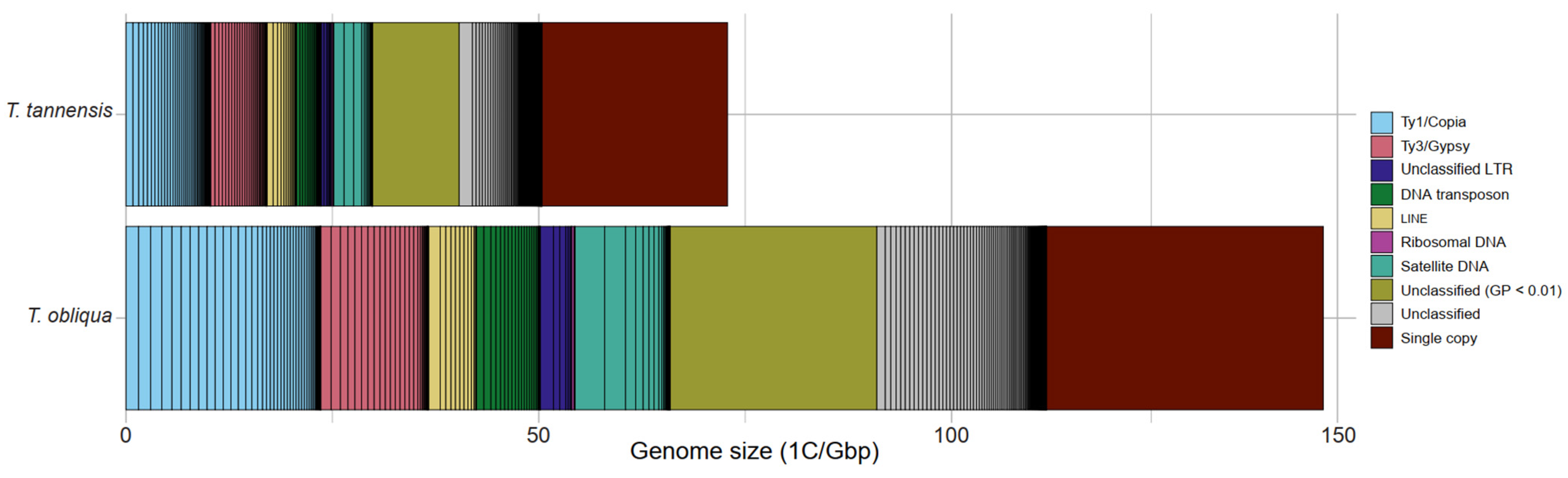
2.1. Repeat Composition in Tmesipteris tannensis and Tmesipteris obliqua
2.2. Heterogeneity among Copies of Repetitive Elements
2.3. Average Insertion Times of LTR-Retrotransposons
3. Discussion
3.1. TEs Largely Contribute to the Giant Genomes of Tmesipteris
3.2. The Occurrence of Dark Matter in the Giant Genomes of Tmesipteris
3.3. Evolutionary Mechanisms Underpinning Repeat Dynamics in the Evolution of the Giant Tmesipteris Genomes
4. Materials and Methods
4.1. Plant Material and DNA Sequencing
4.2. Graph-Based Clustering in RepeatExplorer2
4.3. Repeat Heterogeneity Analysis and Average LTR Insertion Times
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dodsworth, S.; Leitch, A.R.; Leitch, I.J. Genome Size Diversity in Angiosperms and Its Influence on Gene Space. Curr. Opin. Genet. Dev. 2015, 35, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Novák, P.; Guignard, M.S.; Neumann, P.; Kelly, L.J.; Mlinarec, J.; Koblížková, A.; Dodsworth, S.; Kovařík, A.; Pellicer, J.; Wang, W.; et al. Repeat-Sequence Turnover Shifts Fundamentally in Species with Large Genomes. Nat. Plants 2020, 6, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ruano, F.J.; Navarro-Domínguez, B.; Camacho, J.P.M.; Garrido-Ramos, M.A. Transposable Element Landscapes Illuminate Past Evolutionary Events in the Endangered Fern Vandenboschia Speciosa. Genome 2022, 65, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Marchant, D.B.; Chen, G.; Cai, S.; Chen, F.; Schafran, P.; Jenkins, J.; Shu, S.; Plott, C.; Webber, J.; Lovell, J.T.; et al. Dynamic Genome Evolution in a Model Fern. Nat. Plants 2022, 8, 1038–1051. [Google Scholar] [CrossRef]
- Li, F.-W.; Brouwer, P.; Carretero-Paulet, L.; Cheng, S.; de Vries, J.; Delaux, P.-M.; Eily, A.; Koppers, N.; Kuo, L.-Y.; Li, Z.; et al. Fern Genomes Elucidate Land Plant Evolution and Cyanobacterial Symbioses. Nat. Plants 2018, 4, 460–472. [Google Scholar] [CrossRef]
- Schneider, H.; Liu, H.; Clark, J.; Hidalgo, O.; Pellicer, J.; Zhang, S.; Kelly, L.J.; Fay, M.F.; Leitch, I.J. Are the Genomes of Royal Ferns Really Frozen in Time? Evidence for Coinciding Genome Stability and Limited Evolvability in the Royal Ferns. New Phytol. 2015, 207, 10–13. [Google Scholar] [CrossRef]
- Bomfleur, B.; McLoughlin, S.; Vajda, V. Fossilized Nuclei and Chromosomes Reveal 180 Million Years of Genomic Stasis in Royal Ferns. Science 2014, 343, 1376–1377. [Google Scholar] [CrossRef]
- Wagner, W.H.; Wagner, F.S. Polyploidy in Pteridophytes. In Basic Life Sciences; Lewis, W.H., Ed.; Springer: Boston, MA, USA, 1979; Volume 13, pp. 199–214. ISBN 978-1-4613-3069-1. [Google Scholar]
- Clark, J.; Hidalgo, O.; Pellicer, J.; Liu, H.; Marquardt, J.; Robert, Y.; Christenhusz, M.; Zhang, S.; Gibby, M.; Leitch, I.J.; et al. Genome Evolution of Ferns: Evidence for Relative Stasis of Genome Size across the Fern Phylogeny. New Phytol. 2016, 210, 1072–1082. [Google Scholar] [CrossRef]
- Pellicer, J.; Hidalgo, O.; Dodsworth, S.; Leitch, I.J. Genome Size Diversity and Its Impact on the Evolution of Land Plants. Genes 2018, 9, 88. [Google Scholar] [CrossRef]
- Fang, Y.; Qin, X.; Liao, Q.; Du, R.; Luo, X.; Zhou, Q.; Li, Z.; Chen, H.; Jin, W.; Yuan, Y.; et al. The Genome of Homosporous Maidenhair Fern Sheds Light on the Euphyllophyte Evolution and Defences. Nat. Plants 2022, 8, 1024–1037. [Google Scholar] [CrossRef]
- Wang, X.; Morton, J.A.; Pellicer, J.; Leitch, I.J.; Leitch, A.R. Genome Downsizing after Polyploidy: Mechanisms, Rates and Selection Pressures. Plant J. 2021, 107, 1003–1015. [Google Scholar] [CrossRef]
- Dodsworth, S.; Chase, M.W.; Leitch, A.R. Is Post-Polyploidization Diploidization the Key to the Evolutionary Success of Angiosperms? Bot. J. Linn. Soc. 2016, 180, 1–5. [Google Scholar] [CrossRef]
- Wolf, P.G.; Sessa, E.B.; Marchant, D.B.; Li, F.W.; Rothfels, C.J.; Sigel, E.M.; Gitzendanner, M.A.; Visger, C.J.; Banks, J.A.; Soltis, D.E.; et al. An Exploration into Fern Genome Space. Genome Biol. Evol. 2015, 7, 2533–2544. [Google Scholar] [CrossRef]
- Huang, X.; Wang, W.; Gong, T.; Wickell, D.; Kuo, L.-Y.; Zhang, X.; Wen, J.; Kim, H.; Lu, F.; Zhao, H.; et al. The Flying Spider-Monkey Tree Fern Genome Provides Insights into Fern Evolution and Arborescence. Nat. Plants 2022, 8, 500–512. [Google Scholar] [CrossRef]
- Novák, P.; Neumann, P.; Pech, J.; Steinhaisl, J.; Macas, J. RepeatExplorer: A Galaxy-Based Web Server for Genome-Wide Characterization of Eukaryotic Repetitive Elements from Next-Generation Sequence Reads. Bioinformatics 2013, 29, 792–793. [Google Scholar] [CrossRef]
- Marchant, D.B.; Sessa, E.B.; Wolf, P.G.; Heo, K.; Barbazuk, W.B.; Soltis, P.S.; Soltis, D.E. The C-Fern (Ceratopteris richardii) Genome: Insights into Plant Genome Evolution with the First Partial Homosporous Fern Genome Assembly. Sci. Rep. 2019, 9, 18181. [Google Scholar] [CrossRef]
- Wang, D.; Zheng, Z.; Li, Y.; Hu, H.; Wang, Z.; Du, X.; Zhang, S.; Zhu, M.; Dong, L.; Ren, G.; et al. Which Factors Contribute Most to Genome Size Variation within Angiosperms? Ecol. Evol. 2021, 11, 2660–2668. [Google Scholar] [CrossRef]
- Pellicer, J.; Fernández, P.; Fay, M.F.; Michálková, E.; Leitch, I.J. Genome Size Doubling Arises from the Differential Repetitive DNA Dynamics in the Genus Heloniopsis (Melanthiaceae). Front. Genet. 2021, 12, 726211. [Google Scholar] [CrossRef]
- Perrie, L.R.; Brownsey, P.J.; Lovis, J.D. Tmesipteris horomaka, a New Octoploid Species from Banks Peninsula. New Zealand J. Bot. 2010, 48, 15–29. [Google Scholar] [CrossRef]
- PPG, I. A Community-Derived Classification for Extant Lycophytes and Ferns. J. Syst. Evol. 2016, 54, 563–603. [Google Scholar] [CrossRef]
- Brownsey, P.J.; Lovis, J.D. Chromosome Numbers for the New Zealand Species of Psilotum and Tmesipteris, and the Phylogenetic Relationships of the Psilotales. New Zealand J. Bot. 1987, 25, 439–454. [Google Scholar] [CrossRef]
- Hidalgo, O.; Pellicer, J.; Christenhusz, M.J.M.; Schneider, H.; Leitch, I.J. Genomic Gigantism in the Whisk-Fern Family (Psilotaceae): Tmesipteris obliqua Challenges Record Holder Paris japonica. Bot. J. Linn. Soc. 2017, 183, 509–514. [Google Scholar] [CrossRef]
- Pellicer, J.; Fay, M.F.; Leitch, I.J. The Largest Eukaryotic Genome of Them All? Bot. J. Linn. Soc. 2010, 164, 10–15. [Google Scholar] [CrossRef]
- Kelly, L.J.; Renny-Byfield, S.; Pellicer, J.; Macas, J.; Novák, P.; Neumann, P.; Lysak, M.A.; Day, P.D.; Berger, M.; Fay, M.F.; et al. Analysis of the Giant Genomes of Fritillaria (Liliaceae) Indicates That a Lack of DNA Removal Characterizes Extreme Expansions in Genome Size. New Phytol. 2015, 208, 596–607. [Google Scholar] [CrossRef]
- Leroux, O.; Eeckhout, S.; Viane, R.L.L.; Popper, Z.A. Ceratopteris richardii (C-Fern): A Model for Investigating Adaptive Modification of Vascular Plant Cell Walls. Front. Plant Sci. 2013, 4, 367. [Google Scholar] [CrossRef]
- Kelly, L.J.; Leitch, A.R.; Fay, M.F.; Renny-Byfield, S.; Pellicer, J.; Macas, J.; Leitch, I.J. Why Size Really Matters When Sequencing Plant Genomes. Plant Ecol. Divers. 2012, 5, 415–425. [Google Scholar] [CrossRef]
- Novák, P.; Neumann, P.; Macas, J. Global Analysis of Repetitive DNA from Unassembled Sequence Reads Using RepeatExplorer2. Nat. Protoc. 2020, 15, 3745–3776. [Google Scholar] [CrossRef]
- Maumus, F.; Quesneville, H. Deep Investigation of Arabidopsis thaliana Junk DNA Reveals a Continuum between Repetitive Elements and Genomic Dark Matter. PLoS ONE 2014, 9, e94101. [Google Scholar] [CrossRef]
- Pellicer, J.; Leitch, I.J. The Plant DNA C-Values Database (Release 7.1): An Updated Online Repository of Plant Genome Size Data for Comparative Studies. New Phytol. 2020, 226, 301–305. [Google Scholar] [CrossRef]
- Pelosi, J.A.; Kim, E.H.; Barbazuk, W.B.; Sessa, E.B. Phylotranscriptomics Illuminates the Placement of Whole Genome Duplications and Gene Retention in Ferns. Front. Plant Sci. 2022, 13, 882441. [Google Scholar] [CrossRef]
- Leebens-Mack, J.H.; Barker, M.S.; Carpenter, E.J.; Deyholos, M.K.; Gitzendanner, M.A.; Graham, S.W.; Grosse, I.; Li, Z.; Melkonian, M.; Mirarab, S.; et al. One Thousand Plant Transcriptomes and the Phylogenomics of Green Plants. Nature 2019, 574, 679–685. [Google Scholar] [CrossRef]
- Fujiwara, T.; Liu, H.; Meza-Torres, E.I.; Morero, R.E.; Vega, A.J.; Liang, Z.; Ebihara, A.; Leitch, I.J.; Schneider, H. Evolution of Genome Space Occupation in Ferns: Linking Genome Diversity and Species Richness. Ann. Bot. 2021, 2021, mcab094. [Google Scholar] [CrossRef] [PubMed]
- Leitch, A.R.; Leitch, I.J. Ecological and Genetic Factors Linked to Contrasting Genome Dynamics in Seed Plants. New Phytol. 2012, 194, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Wendel, J.F.; Schnabel, A.; Seelanan, T. Bidirectional Interlocus Concerted Evolution Following Allopolyploid Speciation in Cotton (Gossypium). Proc. Natl. Acad. Sci. USA 1995, 92, 280–284. [Google Scholar] [CrossRef]
- Haufler, C.H. Ever since Klekowski: Testing a Set of Radical Hypotheses Revives the Genetics of Ferns and Lycophytes. Am. J. Bot. 2014, 101, 2036–2042. [Google Scholar] [CrossRef]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway Spruce Genome Sequence and Conifer Genome Evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef]
- Freeling, M.; Woodhouse, M.R.; Subramaniam, S.; Turco, G.; Lisch, D.; Schnable, J.C. Fractionation Mutagenesis and Similar Consequences of Mechanisms Removing Dispensable or Less-Expressed DNA in Plants. Curr. Opin. Plant Biol. 2012, 15, 131–139. [Google Scholar] [CrossRef]
- Soltis, P.S.; Marchant, D.B.; Van de Peer, Y.; Soltis, D.E. Polyploidy and Genome Evolution in Plants. Curr. Opin. Genet. Dev. 2015, 35, 119–125. [Google Scholar] [CrossRef]
- Li, Z.; McKibben, M.T.W.; Finch, G.S.; Blischak, P.D.; Sutherland, B.L.; Barker, M.S. Patterns and Processes of Diploidization in Land Plants. Annu. Rev. Plant Biol. 2021, 72, 387–410. [Google Scholar] [CrossRef]
- Michael, T.P. Plant Genome Size Variation: Bloating and Purging DNA. Brief. Funct. Genom. 2014, 13, 308–317. [Google Scholar] [CrossRef]
- Baniaga, A.E.; Barker, M.S. Nuclear Genome Size Is Positively Correlated with Median LTR-RT Insertion Time in Fern and Lycophyte Genomes. Am. Fern J. 2019, 109, 248–266. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A Rapid Total DNA Preparation Procedure for Fresh Plant Tissue. Focus 1990, 12, 13–15. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Neumann, P.; Novák, P.; Hoštáková, N.; Macas, J. Systematic Survey of Plant LTR-Retrotransposons Elucidates Phylogenetic Relationships of Their Polyprotein Domains and Provides a Reference for Element Classification. Mob. DNA 2019, 10, 1. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2; Use R! Springer International Publishing: Cham, Switzerland, 2016; ISBN 978-3-319-24275-0. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2019. [Google Scholar]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Kimura, M. A Simple Method for Estimating Evolutionary Rates of Base Substitutions through Comparative Studies of Nucleotide Sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Khan, M.A.; Elias, I.; Sjölund, E.; Nylander, K.; Guimera, R.V.; Schobesberger, R.; Schmitzberger, P.; Lagergren, J.; Arvestad, L. Fastphylo: Fast Tools for Phylogenetics. BMC Bioinform. 2013, 14, 334. [Google Scholar] [CrossRef]
- Barker, M.S. Evolutionary Genomic Analyses of Ferns Reveal That High Chromosome Numbers Are a Product of High Retention and Fewer Rounds of Polyploidy Relative to Angiosperms. Am. Fern J. 2009, 99, 136–141. [Google Scholar]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Tindale, M.D.; Roy, S.K. A cytotaxonomic survey of the Pteridophyta of Australia. Australian Syst. Bot. 2002, 15, 839–937. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Proportion (GP) | |||||
|---|---|---|---|---|---|
| T. tannensis | T. obliqua | ||||
| Repeat Type | Lineage | [%] | [Gbp] | [%] | [Gbp] |
| Class 1 TE | |||||
| Ty1/Copia | 14.13 | 20.77 | 16.45 | 12.01 | |
| SIRE | 0.11 | 0.17 | 0.06 | 0.05 | |
| Ale | 1.45 | 2.13 | 1.40 | 1.02 | |
| Ivana | 5.00 | 7.34 | 4.51 | 3.29 | |
| Tork | 4.54 | 6.67 | 7.99 | 5.84 | |
| Other | 3.03 | 4.45 | 2.48 | 1.81 | |
| Ty3/Gypsy | 9.36 | 13.76 | 9.07 | 6.62 | |
| Tekay | 0.26 | 0.38 | 0.10 | 0.07 | |
| Athila | 8.79 | 12.92 | 8.01 | 5.85 | |
| Reina | 0.00 | 0.00 | 0.04 | 0.03 | |
| Other | 0.32 | 0.46 | 0.93 | 0.68 | |
| LTR-unclassified | 1.86 | 2.73 | 2.65 | 1.94 | |
| Class 2 TE | |||||
| LINE | 4.91 | 7.22 | 4.04 | 2.95 | |
| DNA transposons | 3.39 | 4.99 | 4.27 | 3.12 | |
| TIR/EnSpm-CACTA | 1.62 | 2.39 | 1.65 | 1.21 | |
| TIR/haT | 1.68 | 2.47 | 2.62 | 1.91 | |
| Helitron | 0.09 | 0.13 | 0.00 | 0.00 | |
| Other repeats | |||||
| Tandem repeats | |||||
| rDNA | 0.23 | 0.17 | 0.27 | 0.20 | |
| Satellite | 6.47 | 9.78 | 8.04 | 5.87 | |
| Unclassified | 14.61 | 21.48 | 15.49 | 11.31 | |
| Small unclassified clusters (GP < 0.01%) | 14.31 | 21.03 | 16.97 | 12.39 | |
| Total repeats | 69.27 | 101.83 | 77.26 | 56.40 | |
| Single copy | 30.73 | 45.17 | 22.74 | 16.60 | |
| Repetitive Elements (T. obliqua/T. tannensis) | Slope | R2 | p-Value |
|---|---|---|---|
| Ty1/Copia | 2.131 | 0.914 | <2 × 10−16 |
| Ty3/Gypsy | 1.785 | 0.943 | <2 × 10−16 |
| All | 1.665 | 0.605 | <2 × 10−16 |
| All classified | 1.904 | 0.886 | <2 × 10−16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández, P.; Leitch, I.J.; Leitch, A.R.; Hidalgo, O.; Christenhusz, M.J.M.; Pokorny, L.; Pellicer, J. Giant Fern Genomes Show Complex Evolution Patterns: A Comparative Analysis in Two Species of Tmesipteris (Psilotaceae). Int. J. Mol. Sci. 2023, 24, 2708. https://doi.org/10.3390/ijms24032708
Fernández P, Leitch IJ, Leitch AR, Hidalgo O, Christenhusz MJM, Pokorny L, Pellicer J. Giant Fern Genomes Show Complex Evolution Patterns: A Comparative Analysis in Two Species of Tmesipteris (Psilotaceae). International Journal of Molecular Sciences. 2023; 24(3):2708. https://doi.org/10.3390/ijms24032708
Chicago/Turabian StyleFernández, Pol, Ilia J. Leitch, Andrew R. Leitch, Oriane Hidalgo, Maarten J. M. Christenhusz, Lisa Pokorny, and Jaume Pellicer. 2023. "Giant Fern Genomes Show Complex Evolution Patterns: A Comparative Analysis in Two Species of Tmesipteris (Psilotaceae)" International Journal of Molecular Sciences 24, no. 3: 2708. https://doi.org/10.3390/ijms24032708
APA StyleFernández, P., Leitch, I. J., Leitch, A. R., Hidalgo, O., Christenhusz, M. J. M., Pokorny, L., & Pellicer, J. (2023). Giant Fern Genomes Show Complex Evolution Patterns: A Comparative Analysis in Two Species of Tmesipteris (Psilotaceae). International Journal of Molecular Sciences, 24(3), 2708. https://doi.org/10.3390/ijms24032708