
From Classical to Modern Computational Approaches to Identify Key Genetic Regulatory Components in Plant Biology

 , ,
, ,  and
and 
Abstract
1. Introduction
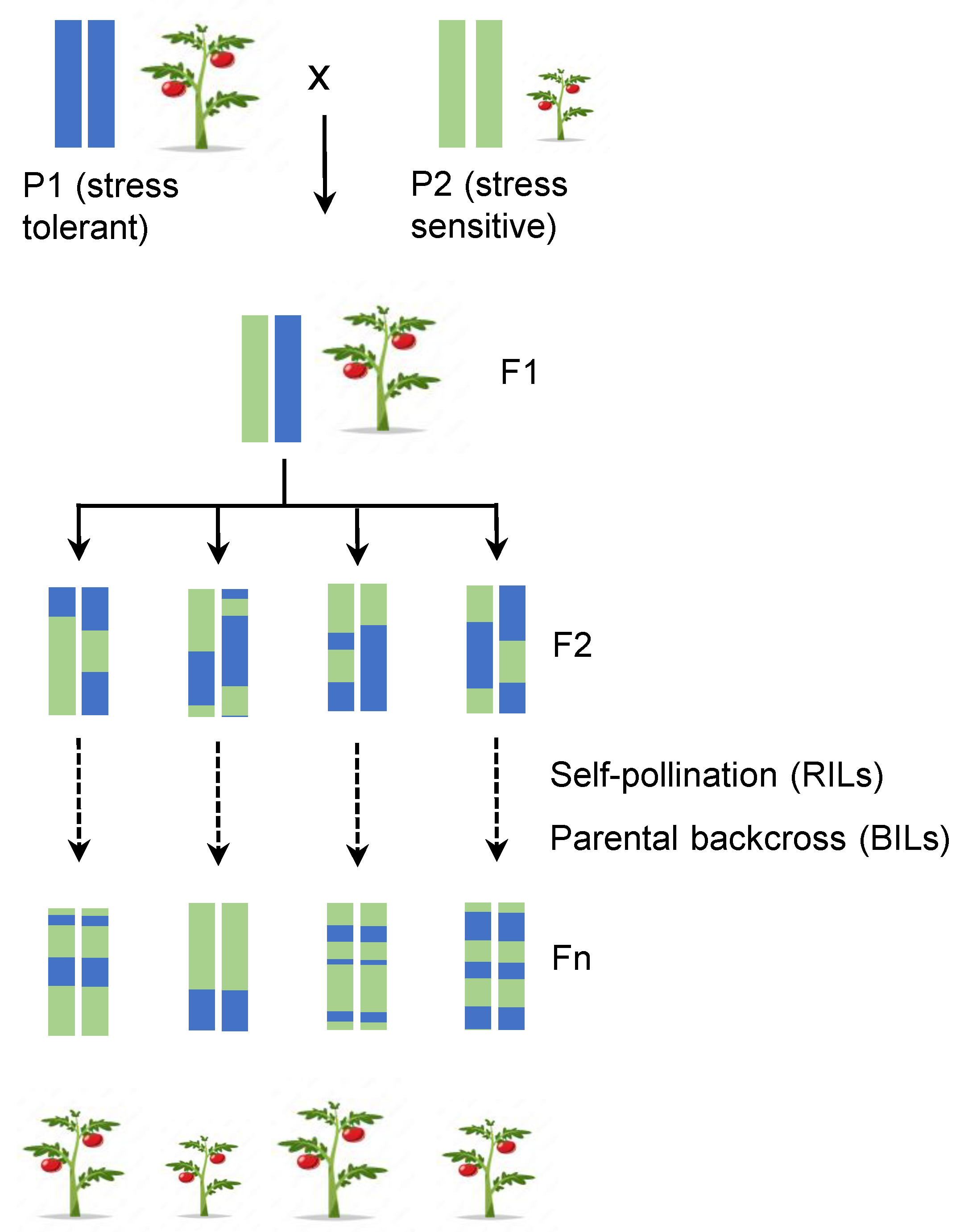
2. Classical Approaches to Identify Regulatory Components: Introduction to Marker-Assisted Breeding
2.1. Where It All Began: Quantitative Trait Loci (QTLs)

2.2. Marked Assisted Selection
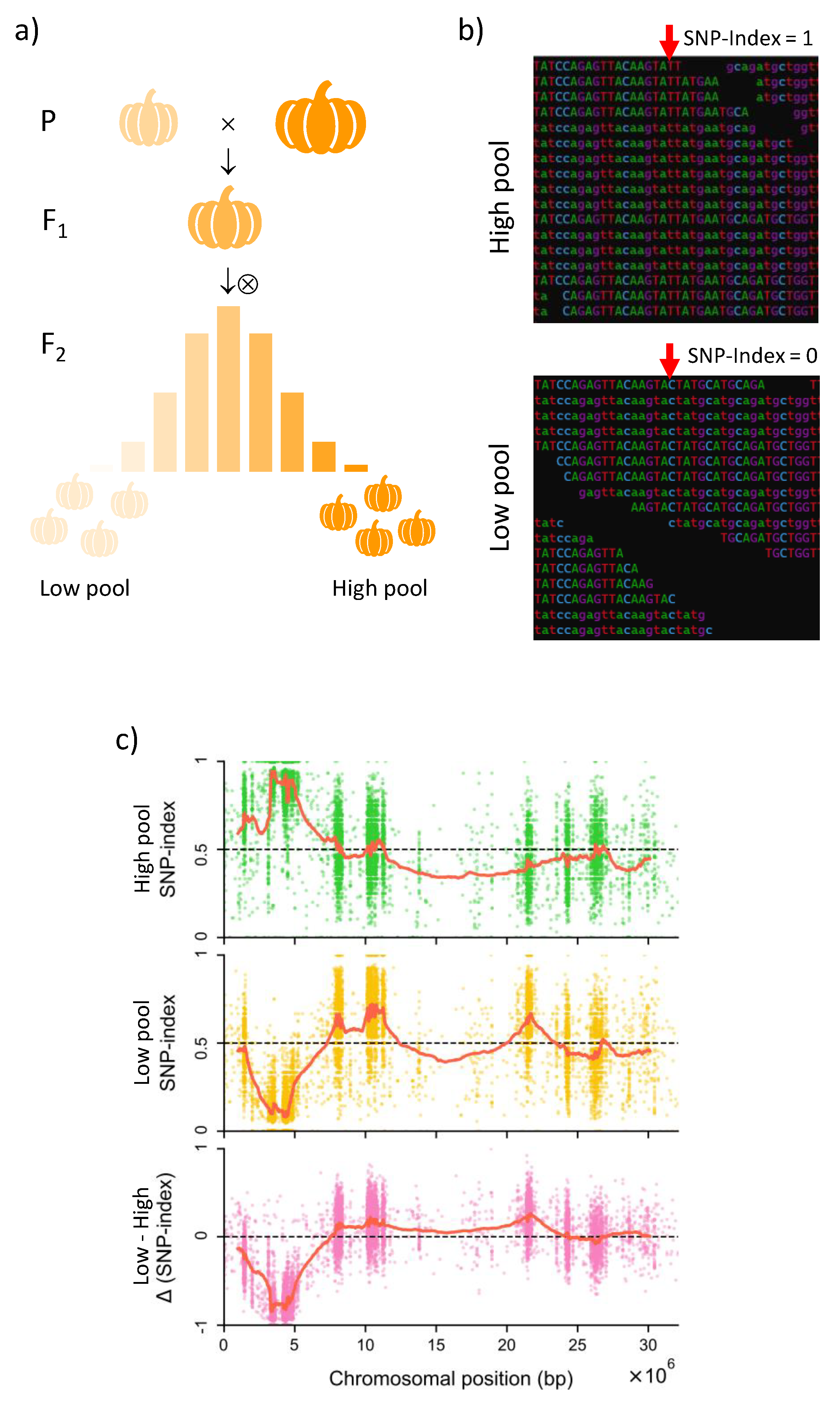
3. Annotation of Genes: Role of NGS and Comparative Genomics
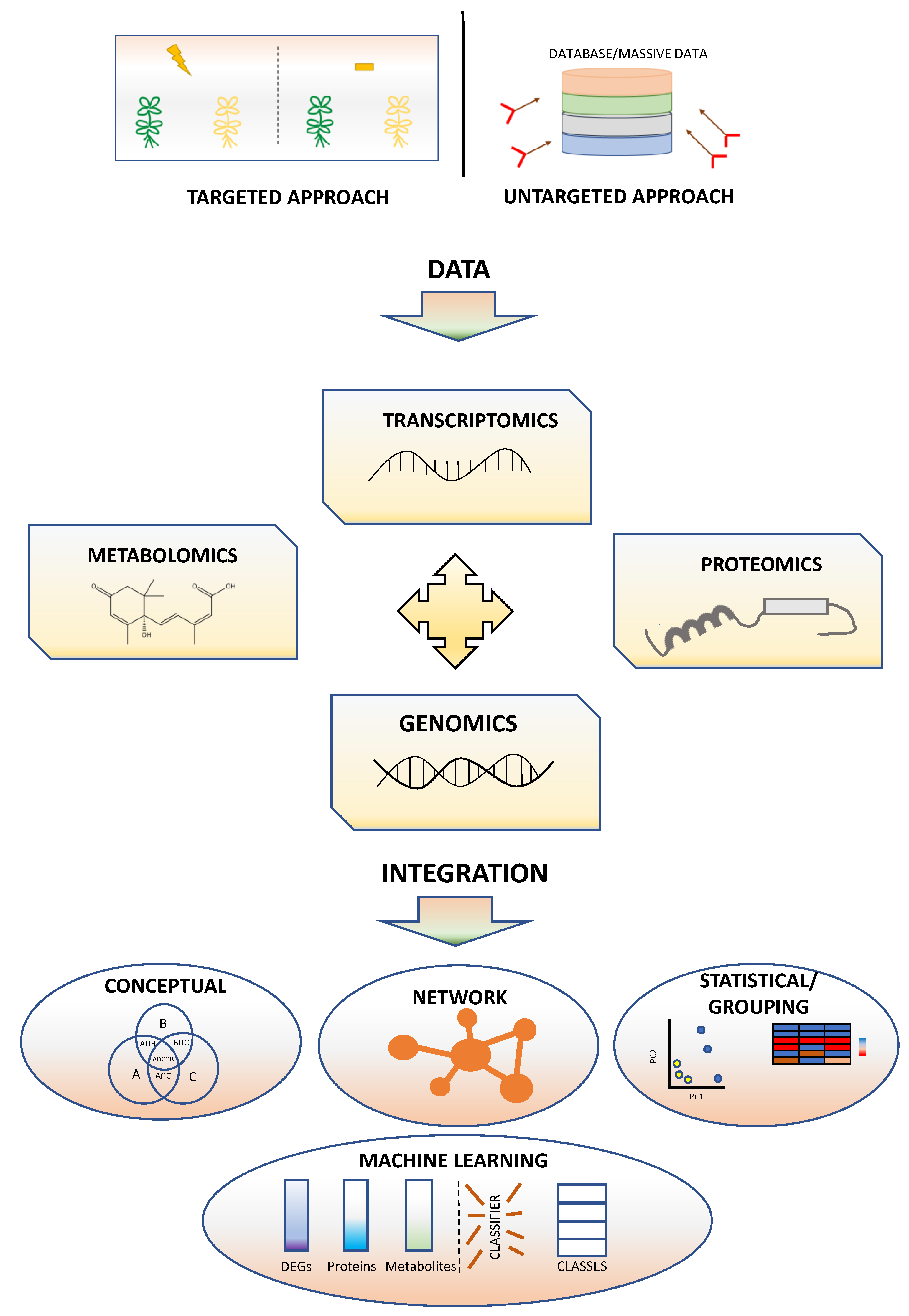
4. Correlation of Genes and Traits Using Omics Technologies
4.1. Glossary of Network Analysis
- Co-expression network: This refers to a set of (more or less) densely interconnected variables in which the degree of connectivity is linked to similarity in expression profiles, abundance or intensity of a given variable throughout the samples (genotypes, conditions, time series, etc.). These usually express gene expression data and metabolite or protein accumulation.
- Edges and nodes: In a network, the variables are nodes or vertices and are usually depicted as points. The connections between the nodes are referred to as edges and are usually depicted as lines between points.
- Module: A cluster of highly interconnected (showing high absolute correlation, either positive or negative) variables (genes, metabolites, proteins, etc.) that potentially reflects functional similarities among cluster members. Modules can be further refined by applying GO or pathway enrichment criteria.
- Connectivity: The correlation existing between pairs of variables, inferred from correlation- or mutual-information-based methods.
- Module eigengene E: Defined as the first principal component of a given module, it is a representation of the variable expression profiles in a module. These values can be correlated to an external trait (e.g., phenotype). It is also related to the module membership by correlation of the variable expression level with the module eigengene E; values close to 1 or -1 indicate positive membership.
- Hub: It is generally defined as a “highly connected gene or protein” which is a member inside co-expression modules. The topology of a hub might reflect its role as a regulatory element.
- Module significance: Absolute average variable significance within a module, which is determined by correlating variable expression to an external trait (e.g., phenotype).
- log2-fold change between treated samples and controls, which facilitates identification of over- and down-regulated genes.
- Reads per kilobase of transcript per million mapped reads (RPKM) for single-end reads from RNA-seq experiments [67], which facilitates comparison of transcript levels within and between samples.
- Fragments per kilobase of transcript per million mapped fragments (FPKM) for RNA-seq experiments producing paired-end reads.
- Transcripts Per Million (TPM), which is similar to the former two, but the order of operations is inverted.
- Trimmed Mean of M-values (TMM) for genes meeting a corrected p-value and false discovery rate (FDR) lower than 0.05, which dramatically reduces the number of false discoveries due to different distribution of expressed transcripts [68]. This method assumes that the most genes are not differentially expressed.
4.2. Co-Expression Network Analysis
4.3. Construction of a Network
4.4. Module Selection
5. Applications in Plant Biology
6. Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eshed, Y.; Abu-Abied, M.; Saranga, Y.; Zamir, D. Lycopersicon esculentum lines containing small overlapping introgressions from L. pennellii. Theor. Appl. Genet. 1992, 83, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Eshed, Y.; Zamir, D. An introgression line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield- associated QTL. Genetics 1995, 141, 1147–1162. [Google Scholar] [CrossRef] [PubMed]
- Ofner, I.; Lashbrooke, J.; Pleban, T.; Aharoni, A.; Zamir, D. Solanum pennellii backcross inbred lines (BILs) link small genomic bins with tomato traits. Plant J. 2016, 87, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.B.; Kao, C.H.; Basten, C.J. Estimating the genetic architecture of quantitative traits. Genet. Res. 1999, 74, 279–289. [Google Scholar] [CrossRef]
- Remington, D.L.; Purugganan, M.D. Evolution of Functional Traits in Plants. Candidate Genes, Quantitative Trait Loci, and Functional Trait Evolution in Plants. Int. J. Plant Sci. 2003, 164, S7–S20. [Google Scholar] [CrossRef]
- Mackay, T.F.C. Complementing complexity. Nat. Genet. 2004, 36, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Roff, D.A. A centennial celebration for quantitative genetics. Evolution 2007, 61, 1017–1032. [Google Scholar] [CrossRef]
- Yano, M.; Harushima, Y.; Nagamura, Y.; Kurata, N.; Minobe, Y.; Sasaki, T. Identification of quantitative trait loci controlling heading date in rice using a high-density linkage map. Theor. Appl. Genet. 1997, 95, 1025–1032. [Google Scholar] [CrossRef]
- Yamamoto, T.; Kuboki, Y.; Lin, S.Y.; Sasaki, T.; Yano, M. Fine mapping of quantitative trait loci Hd-1, Hd-2 and Hd-3, controlling heading date of rice, as single Mendelian factors. Theor. Appl. Genet. 1998, 97, 37–44. [Google Scholar] [CrossRef]
- Yamamoto, T.; Hongxuan, L.; Sasaki, T.; Yano, M. Identification of heading date quantitative trait locus Hd6 and characterization of its epistatic interactions with Hd2 in rice using advanced backcross progeny. Genetics 2000, 154, 885–891. [Google Scholar] [CrossRef]
- Villalobos-López, M.A.; Arroyo-Becerra, A.; Quintero-Jiménez, A.; Iturriaga, G. Biotechnological Advances to Improve Abiotic Stress Tolerance in Crops. Int. J. Mol. Sci. 2022, 23, 12053. [Google Scholar] [CrossRef] [PubMed]
- Arisha, M.H.; Shah, S.N.M.; Gong, Z.H.; Jing, H.; Li, C.; Zhang, H.X. Ethyl methane sulfonate induced mutations in M2 generation and physiological variations in M1 generation of peppers (Capsicum annuum L.). Front. Plant Sci. 2015, 6, 399. [Google Scholar] [CrossRef] [PubMed]
- Candela, H.; Hake, S. The art and design of genetic screens: Maize. Nat. Rev. Genet. 2008, 9, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Kong, F.; Sun, K.; Wang, T.; Guo, T. From Classical Radiation to Modern Radiation: Past, Present, and Future of Radiation Mutation Breeding. Front. Public Health 2021, 9, 768071. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Shikazono, N.; Hase, Y. Studies on biological effects of ion beams on lethality, molecular nature of mutation, mutation rate, and spectrum of mutation phenotype for mutation breeding in higher plants. J. Radiat. Res. 2010, 51, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Behrouzi, P.; Wit, E.C. Detecting epistatic selection with partially observed genotype data by using copula graphical models. J. R. Stat. Soc. Ser. C Appl. Stat. 2019, 68, 141–160. [Google Scholar] [CrossRef]
- Kage, U.; Kumar, A.; Dhokane, D.; Karre, S.; Kushalappa, A.C. Functional molecular markers for crop improvement. Crit. Rev. Biotechnol. 2016, 36, 917–930. [Google Scholar] [CrossRef]
- Singh, R.; Kumar, K.; Bharadwaj, C.; Verma, P.K. Broadening the horizon of crop research: A decade of advancements in plant molecular genetics to divulge phenotype governing genes. Planta 2022, 255, 46. [Google Scholar] [CrossRef]
- Tanksley, S.D.; Ganal, M.W.; Martin, G.B. Chromosome landing: A paradigm for map-based gene cloning in plants with large genomes. Trends Genet. 1995, 11, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Jiao, W.B.; Schneeberger, K. The impact of third generation genomic technologies on plant genome assembly. Curr. Opin. Plant Biol. 2017, 36, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Michael, T.P.; VanBuren, R. Building near-complete plant genomes. Curr. Opin. Plant Biol. 2020, 54, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Shang, L.; Zhu, Q.H.; Fan, L.; Guo, L. Twenty years of plant genome sequencing: Achievements and challenges. Trends Plant Sci. 2022, 27, 391–401. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Schilbert, H.M.; Rempel, A.; Pucker, B. Comparison of read mapping and variant calling tools for the analysis of plant NGS data. Plants 2020, 9, 439. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente Cantó, C.; Vigouroux, Y. Evaluation of nine statistics to identify QTLs in bulk segregant analysis using next generation sequencing approaches. BMC Genom. 2022, 23, 490. [Google Scholar] [CrossRef]
- West, M.A.L.; Kim, K.; Kliebenstein, D.J.; Van Leeuwen, H.; Michelmore, R.W.; Doerge, R.W.; St. Clair, D.A. Global eQTL mapping reveals the complex genetic architecture of transcript-level variation in Arabidopsis. Genetics 2007, 175, 1441–1450. [Google Scholar] [CrossRef]
- Kliebenstein, D. Quantitative genomics: Analyzing intraspecific variation using global gene expression polymorphisms or eqtls. Annu. Rev. Plant Biol. 2009, 60, 93–114. [Google Scholar] [CrossRef]
- Holloway, B.; Li, B. Expression QTLs: Applications for crop improvement. Mol. Breed. 2010, 26, 381–391. [Google Scholar] [CrossRef]
- Li, L.; Petsch, K.; Shimizu, R.; Liu, S.; Xu, W.W.; Ying, K.; Yu, J.; Scanlon, M.J.; Schnable, P.S.; Timmermans, M.C.P.; et al. Mendelian and Non-Mendelian Regulation of Gene Expression in Maize. PLoS Genet. 2013, 9, e1003202. [Google Scholar] [CrossRef] [PubMed]
- Kliebenstein, D.J.; West, M.A.L.; van Leeuwen, H.; Loudet, O.; Doerge, R.W.; St. Clair, D.A. Identification of QTLs controlling gene expression networks defined a priori. BMC Bioinform. 2006, 7, 308. [Google Scholar] [CrossRef] [PubMed]
- Gusev, A.; Ko, A.; Shi, H.; Bhatia, G.; Chung, W.; Penninx, B.W.J.H.; Jansen, R.; De Geus, E.J.C.; Boomsma, D.I.; Wright, F.A.; et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016, 48, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Gur, A.; Zamir, D. Mendelizing all components of a pyramid of three yield QTL in Tomato. Front. Plant Sci. 2015, 6, 1096. [Google Scholar] [CrossRef]
- Sønderby, I.E.; Hansen, B.G.; Bjarnholt, N.; Ticconi, C.; Halkier, B.A.; Kliebenstein, D.J. A systems biology approach identifies a R2R3 MYB gene subfamily with distinct and overlapping functions in regulation of aliphatic glucosinolates. PLoS ONE 2007, 2, e1322. [Google Scholar] [CrossRef]
- Li, Z.; Wang, P.; You, C.; Yu, J.; Zhang, X.; Yan, F.; Ye, Z.; Shen, C.; Li, B.; Guo, K.; et al. Combined GWAS and eQTL analysis uncovers a genetic regulatory network orchestrating the initiation of secondary cell wall development in cotton. New Phytol. 2020, 226, 1738–1752. [Google Scholar] [CrossRef]
- Han, X.; Gao, C.; Liu, L.; Zhang, Y.; Jin, Y.; Yan, Q.; Yang, L.; Li, F.; Yang, Z. Integration of eQTL Analysis and GWAS Highlights Regulation Networks in Cotton under Stress Condition. Int. J. Mol. Sci. 2022, 23, 7564. [Google Scholar] [CrossRef]
- Michael, T.P. Plant genome size variation: Bloating and purging DNA. Brief. Funct. Genom. Proteom. 2014, 13, 308–317. [Google Scholar] [CrossRef]
- Salman-Minkov, A.; Sabath, N.; Mayrose, I. Whole-genome duplication as a key factor in crop domestication. Nat. Plants 2016, 2, 16115. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, H.; Long, Y.; Shu, Y.; Zhai, J. Plant Public RNA-seq Database: A comprehensive online database for expression analysis of ~45,000 plant public RNA-Seq libraries. Plant Biotechnol. J. 2022, 20, 806–808. [Google Scholar] [CrossRef]
- Marks, R.A.; Hotaling, S.; Frandsen, P.B.; VanBuren, R. Representation and participation across 20 years of plant genome sequencing. Nat. Plants 2021, 7, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Bolger, M.E.; Arsova, B.; Usadel, B. Plant genome and transcriptome annotations: From misconceptions to simple solutions. Brief. Bioinform. 2018, 19, 437–449. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Krishnakumar, V.; Chan, A.P.; Thibaud-Nissen, F.; Schobel, S.; Town, C.D. Araport11: A complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 2017, 89, 789–804. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Pan, S.; Cheng, S.; Zhang, B.; Mu, D.; Ni, P.; Zhang, G.; Yang, S.; Li, R.; Wang, J.; et al. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195. [Google Scholar] [CrossRef]
- Dohm, J.C.; Minoche, A.E.; Holtgräwe, D.; Capella-Gutiérrez, S.; Zakrzewski, F.; Tafer, H.; Rupp, O.; Sörensen, T.R.; Stracke, R.; Reinhardt, R.; et al. The genome of the recently domesticated crop plant sugar beet (Beta vulgaris). Nature 2014, 505, 546–549. [Google Scholar] [CrossRef]
- De Mendoza, A.; Sebé-Pedrós, A. Origin and evolution of eukaryotic transcription factors. Curr. Opin. Genet. Dev. 2019, 58–59, 25–32. [Google Scholar] [CrossRef]
- Schmitz, R.J.; Grotewold, E.; Stam, M. Cis-regulatory sequences in plants: Their importance, discovery, and future challenges. Plant Cell 2022, 34, 718–741. [Google Scholar] [CrossRef]
- Rao, X.; Dixon, R.A. Co-expression networks for plant biology: Why and how. Acta Biochim. Biophys. Sin. 2019, 51, 981–988. [Google Scholar] [CrossRef]
- Schaefer, R.J.; Michno, J.M.; Myers, C.L. Unraveling gene function in agricultural species using gene co-expression networks. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Depuydt, T.; De Rybel, B.; Vandepoele, K. Plant Science Charting plant gene functions in the multi-omics and single-cell era. Trends Plant Sci. 2022; in press. [Google Scholar] [CrossRef] [PubMed]
- Pardo-diaz, J.; Beguerisse-díaz, M.; Poole, P.S.; Deane, C.M.; Reinert, G. Extracting Information from Gene Coexpression Networks of Rhizobium leguminosarum. J. Comput. Biol. 2022, 29, 752–768. [Google Scholar] [CrossRef] [PubMed]
- Kodama, Y.; Shumway, M.; Leinonen, R. The sequence read archive: Explosive growth of sequencing data. Nucleic Acids Res. 2012, 40, 2011–2013. [Google Scholar] [CrossRef] [PubMed]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2020, 48, D440–D444. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Mutwil, M.; Klie, S.; Tohge, T.; Giorgi, F.M.; Wilkins, O.; Campbell, M.M.; Fernie, A.R.; Usadel, B.; Nikoloski, Z.; Persson, S. PlaNet: Combined sequence and expression comparisons across plant networks derived from seven species. Plant Cell 2011, 23, 895–910. [Google Scholar] [CrossRef]
- Arend, D.; Junker, A.; Scholz, U.; Schüler, D.; Wylie, J.; Lange, M. PGP repository: A Plant phenomics and genomics data publication infrastructure. Database 2016, 2016, baw033. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Nakaya, A. Thed KEGG databases at GenomeNet. Nucleic Acids Res. 2002, 30, 42–46. [Google Scholar] [CrossRef]
- Joshi-Tope, G.; Gillespie, M.; Vastrik, I.; D’Eustachio, P.; Schmidt, E.; de Bono, B.; Jassal, B.; Gopinath, G.R.; Wu, G.R.; Matthews, L.; et al. Reactome: A knowledgebase of biological pathways. Nucleic Acids Res. 2005, 33, 428–432. [Google Scholar] [CrossRef]
- Turinsky, A.L.; Razick, S.; Turner, B.; Donaldson, I.M.; Wodak, S.J. Navigating the Global Protein–Protein Interaction Landscape Using iRefWeb. Struct. Genom. 2013, 1091, 315–331. [Google Scholar]
- Zuberi, K.; Franz, M.; Rodriguez, H.; Montojo, J.; Lopes, C.T.; Bader, G.D.; Morris, Q. GeneMANIA prediction server 2013 update. Nucleic Acids Res. 2013, 41, 115–122. [Google Scholar] [CrossRef]
- Wong, D.C.J.; Matus, J.T. Constructing Integrated Networks for Identifying New Secondary Metabolic Pathway Regulators in Grapevine: Recent Applications and Future Opportunities. Front. Plant Sci. 2017, 8, 505. [Google Scholar] [CrossRef]
- Savoi, S.; Wong, D.C.J.; Degu, A.; Herrera, J.C.; Bucchetti, B.; Peterlunger, E.; Fait, A.; Mattivi, F.; Castellarin, S.D. Multi-Omics and Integrated Network Analyses Reveal New Insights into the Systems Relationships between Metabolites, Structural Genes, and Transcriptional Regulators in Developing Grape Berries (Vitis vinifera L.) Exposed to Water Deficit. Front. Plant Sci. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed]
- Rivarola Sena, A.C.; Andres-Robin, A.; Vialette, A.C.; Just, J.; Launay-Avon, A.; Borrega, N.; Dubreucq, B.; Scutt, C.P. Custom methods to identify conserved genetic modules applied to novel transcriptomic data from Amborella trichopoda. J. Exp. Bot. 2022, 73, 2487–2498. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.K.; Zheng, X.; Goh, J.C.; Mutwil, M. Exploiting plant transcriptomic databases: Resources, tools, and approaches. Plant Commun. 2022, 3, 100323. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Fiehn, O.; Wohlgemuth, G.; Scholz, M.; Kind, T.; Lee, D.Y.; Lu, Y.; Moon, S.; Nikolau, B. Quality control for plant metabolomics: Reporting MSI-compliant studies. Plant J. 2008, 53, 691–704. [Google Scholar] [CrossRef]
- Misra, B.B. Data normalization strategies in metabolomics: Current challenges, approaches, and tools. Eur. J. Mass Spectrom. 2020, 26, 165–174. [Google Scholar] [CrossRef]
- Hirai, M.Y.; Klein, M.; Fujikawa, Y.; Yano, M.; Goodenowe, D.B.; Yamazaki, Y.; Kanaya, S.; Nakamura, Y.; Kitayama, M.; Suzuki, H.; et al. Elucidation of gene-to-gene and metabolite-to-gene networks in arabidopsis by integration of metabolomics and transcriptomics. J. Biol. Chem. 2005, 280, 25590–25595. [Google Scholar] [CrossRef]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in1H NMR metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef]
- Johnson, K.A.; Krishnan, A. Robust normalization and transformation techniques for constructing gene coexpression networks from RNA-seq data. Genome Biol. 2022, 23, 1. [Google Scholar] [CrossRef]
- Correia, B.; Valledor, L.; Hancock, R.D.; Renaut, J.; Pascual, J.; Soares, A.M.V.M.; Pinto, G. Integrated proteomics and metabolomics to unlock global and clonal responses of Eucalyptus globulus recovery from water deficit. Metabolomics 2016, 12, 141. [Google Scholar] [CrossRef]
- Rauniyar, N.; Yates, J.R. Isobaric Labeling-Based Relative Quanti fi cation in Shotgun Proteomics. J. Proteome Res. 2014, 13, 5293–5309. [Google Scholar] [CrossRef]
- Stöckel, J.; Jacobs, J.M.; Elvitigala, T.R.; Liberton, M.; Welsh, E.A.; Polpitiya, A.D.; Gritsenko, M.A.; Nicora, C.D.; Koppenaal, D.W.; Smith, R.D.; et al. Diurnal rhythms result in significant changes in the cellular protein complement in the cyanobacterium Cyanothece 51142. PLoS ONE 2011, 6, e16680. [Google Scholar] [CrossRef]
- Minadakis, G.; Sokratous, K.; Spyrou, G.M. ProtExA: A tool for post-processing proteomics data providing differential expression metrics, co-expression networks and functional analytics. Comput. Struct. Biotechnol. J. 2020, 18, 1695–1703. [Google Scholar] [CrossRef]
- Cueff, G.; Rajjou, L.; Hoang, H.H.; Bailly, C.; Corbineau, F.; Leymarie, J. In-Depth Proteomic Analysis of the Secondary Dormancy Induction by Hypoxia or High Temperature in Barley Grains. Plant Cell Physiol. 2022, 63, 550–564. [Google Scholar] [CrossRef]
- Hall, R.D.; D’Auria, J.C.; Silva Ferreira, A.C.; Gibon, Y.; Kruszka, D.; Mishra, P.; van de Zedde, R. High-throughput plant phenotyping: A role for metabolomics? Trends Plant Sci. 2022, 27, 549–563. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef]
- Uppal, K.; Ma, C.; Go, Y.M.; Jones, D.P. XMWAS: A data-driven integration and differential network analysis tool. Bioinformatics 2018, 34, 701–702. [Google Scholar] [CrossRef]
- Tran, V.D.T.; Moretti, S.; Coste, A.T.; Amorim-Vaz, S.; Sanglard, D.; Pagni, M. Condition-specific series of metabolic sub-networks and its application for gene set enrichment analysis. Bioinformatics 2019, 35, 2258–2266. [Google Scholar] [CrossRef]
- Picart-Armada, S.; Fernández-Albert, F.; Vinaixa, M.; Yanes, O.; Perera-Lluna, A. FELLA: An R package to enrich metabolomics data. BMC Bioinform. 2018, 19, 538. [Google Scholar] [CrossRef]
- Cottret, L.; Frainay, C.; Chazalviel, M.; Cabanettes, F.; Gloaguen, Y.; Camenen, E.; Merlet, B.; Heux, S.; Portais, J.C.; Poupin, N.; et al. MetExplore: Collaborative edition and exploration of metabolic networks. Nucleic Acids Res. 2018, 46, W495–W502. [Google Scholar] [CrossRef]
- Zhou, G.; Xia, J. OmicsNet: A web-based tool for creation and visual analysis of biological networks in 3D space. Nucleic Acids Res. 2018, 46, W514–W522. [Google Scholar] [CrossRef]
- Zoppi, J.; Guillaume, J.F.; Neunlist, M.; Chaffron, S. MiBiOmics: An interactive web application for multi-omics data exploration and integration. BMC Bioinform. 2021, 22, 6. [Google Scholar] [CrossRef]
- Hinshaw, S.J.; Lee, A.H.Y.; Gill, E.E.; Hancock, R.E.W. MetaBridge: Enabling network-based integrative analysis via direct protein interactors of metabolites. Bioinformatics 2018, 34, 3225–3227. [Google Scholar] [CrossRef]
- Huang, J.; Vendramin, S.; Shi, L.; McGinnis, K.M. Construction and optimization of a large gene coexpression network in maize using RNA-seq data. Plant Physiol. 2017, 175, 568–583. [Google Scholar] [CrossRef]
- Emamjomeh, A.; Saboori Robat, E.; Zahiri, J.; Solouki, M.; Khosravi, P. Gene co-expression network reconstruction: A review on computational methods for inferring functional information from plant-based expression data. Plant Biotechnol. Rep. 2017, 11, 71–86. [Google Scholar] [CrossRef]
- Fukushima, A.; Kusano, M. A network perspective on nitrogen metabolism from model to crop plants using integrated “omics” approaches. J. Exp. Bot. 2014, 65, 5619–5630. [Google Scholar] [CrossRef]
- Bassel, G.W.; Gaudinier, A.; Brady, S.M.; Hennig, L.; Rhee, S.Y.; De Smet, I. Systems analysis of plant functional, transcriptional, physical interaction, and metabolic networks. Plant Cell 2012, 24, 3859–3875. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, J.; Yang, J.; Fennell, A.; Zhang, C.; Ma, Q. QUBIC: A bioconductor package for qualitative biclustering analysis of gene co-expression data. Bioinformatics 2017, 33, 450–452. [Google Scholar] [CrossRef]
- Liesecke, F.; Daudu, D.; De Bernonville, R.D.; Besseau, S.; Clastre, M.; Courdavault, V.; De Craene, J.O.; Crèche, J.; Giglioli-Guivarc’h, N.; Glévarec, G.; et al. Ranking genome-wide correlation measurements improves microarray and RNA-seq based global and targeted co-expression networks. Sci. Rep. 2018, 8, 10885. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Yip, A.M.; Horvath, S. Gene network interconnectedness and the generalized topological overlap measure. BMC Bioinform. 2007, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.J.R.; Shealy, B.T.; Greer, M.S.; Hadish, J.A.; McGowan, M.T.; Biggs, T.; Smith, M.C.; Feltus, F.A.; Ficklin, S.P. Addressing noise in co-expression network construction. Brief. Bioinform. 2022, 23, bbab495. [Google Scholar] [CrossRef]
- Su, G.; Morris, J.H.; Demchak, B.; Bader, G.D. Biological Network Exploration with Cytoscape 3. Curr. Protoc. Bioinform. 2014, 47, 8–13. [Google Scholar] [CrossRef]
- Auer, F.; Kramer, F. RCX—An R package adapting the Cytoscape Exchange format for biological networks. Bioinform. Adv. 2022, 2, vbac020. [Google Scholar] [CrossRef] [PubMed]
- Gustavsen, J.A.; Pai, S.; Isserlin, R.; Demchak, B.; Pico, A.R. Rcy3: Network biology using cytoscape from within R. F1000Research 2019, 8, 793166. [Google Scholar] [CrossRef]
- Hasbún, R.; Jesús, M.; Valledor, L. Chloroplast proteomics reveals transgenerational cross-stress priming in Pinus radiata. Environ. Exp. Bot. 2022, 202, 105009. [Google Scholar] [CrossRef]
- Singh, A.; Shannon, C.P.; Gautier, B.; Rohart, F.; Vacher, M.; Tebbutt, S.J.; Cao, K.A.L. DIABLO: An integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics 2019, 35, 3055–3062. [Google Scholar] [CrossRef] [PubMed]
- González, I.; Cao, K.A.L.; Davis, M.J.; Déjean, S. Visualising associations between paired “omics” data sets. BioData Min. 2012, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Rohart, F.; Eslami, A.; Matigian, N.; Bougeard, S.; Lê Cao, K.A. MINT: A multivariate integrative method to identify reproducible molecular signatures across independent experiments and platforms. BMC Bioinform. 2017, 18, 128. [Google Scholar] [CrossRef] [PubMed]
- Fait, A.; Batushansky, A.; Shrestha, V.; Yobi, A.; Angelovici, R. Can metabolic tightening and expansion of co-expression network play a role in stress response and tolerance? Plant Sci. 2020, 293, 110409. [Google Scholar] [CrossRef]
- Mo, Z.; Duan, L.; Pu, Y.; Tian, Z.; Ke, Y.; Luo, W.; Pi, K.; Huang, Y.; Nie, Q.; Liu, R. Proteomics and Co-expression Network Analysis Reveal the Importance of Hub Proteins and Metabolic Pathways in Nicotine Synthesis and Accumulation in Tobacco (Nicotiana tabacum L.). Front. Plant Sci. 2022, 13, 860455. [Google Scholar] [CrossRef]
- Mondal, R.; Madhurya, K.; Saha, P.; Chattopadhyay, S.K.; Antony, S.; Kumar, A.; Roy, S.; Roy, D. Expression profile, transcriptional and post-transcriptional regulation of genes involved in hydrogen sulphide metabolism connecting the balance between development and stress adaptation in plants: A data-mining bioinformatics approach. Plant Biol. 2022, 24, 602–617. [Google Scholar] [CrossRef]
- Xu, J.; Zhu, J.; Lin, Y.; Zhu, H.; Tang, L.; Wang, X.; Wang, X. Comparative transcriptome and weighted correlation network analyses reveal candidate genes involved in chlorogenic acid biosynthesis in sweet potato. Sci. Rep. 2022, 12, 2770. [Google Scholar] [CrossRef]
- Hu, J.; Zhuang, Y.; Li, X.; Li, X.; Sun, C.; Ding, Z.; Xu, R.; Zhang, D. Time-series transcriptome comparison reveals the gene regulation network under salt stress in soybean (Glycine max) roots. BMC Plant Biol. 2022, 22, 157. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, Y.; Shi, H.; Hu, H.; Yi, L.; Hou, J. Time-course transcriptome and WGCNA analysis revealed the drought response mechanism of two sunflower inbred lines. PLoS ONE 2022, 17, e0265447. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Zhang, S.; Li, W.; Chen, B.; Li, W. Gene-coexpression network analysis identifies specific modules and hub genes related to cold stress in rice. BMC Genom. 2022, 23, 251. [Google Scholar] [CrossRef]
- Li, X.; Huang, H.; Rizwan, M.; Wang, N.; Jiang, J.; She, W.; Zheng, G.; Pan, H.; Guo, Z.; Pan, D.; et al. Transcriptome Analysis Reveals Candidate Lignin-Related Pomelo ( Citrus maxima ). Genes 2022, 13, 845. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Wei, C.; Cheng, Y.; Shang, Z.; Guo, X.; Guan, J. RNA-Seq Analysis Identifies Transcription Factors Involved in Anthocyanin Biosynthesis of ‘Red Zaosu’ Pear Peel and Functional Study of PpPIF8. Int. J. Mol. Sci. 2022, 23, 4798. [Google Scholar] [CrossRef] [PubMed]
- Burbidge, C.A.; Ford, C.M.; Melino, V.J.; Wong, D.C.J.; Jia, Y.; Jenkins, C.L.D.; Soole, K.L.; Castellarin, S.D.; Darriet, P.; Rienth, M.; et al. Biosynthesis and Cellular Functions of Tartaric Acid in Grapevines. Front. Plant Sci. 2021, 12, 643024. [Google Scholar] [CrossRef]
- Giacomello, S. A new era for plant science: Spatial single-cell transcriptomics. Curr. Opin. Plant Biol. 2021, 60, 102041. [Google Scholar] [CrossRef]
- Clark, N.M.; Elmore, J.M.; Walley, J.W. To the proteome and beyond: Advances in single-cell omics profiling for plant systems. Plant Physiol. 2022, 188, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Feldberg, L.; Dong, Y.; Heinig, U.; Rogachev, I.; Aharoni, A. DLEMMA-MS-Imaging for Identification of Spatially Localized Metabolites and Metabolic Network Map Reconstruction. Anal. Chem. 2018, 90, 10231–10238. [Google Scholar] [CrossRef]
- Zhang, C.; Žukauskaitė, A.; Petřík, I.; Pěnčík, A.; Hönig, M.; Grúz, J.; Široká, J.; Novák, O.; Doležal, K. In situ characterisation of phytohormones from wounded Arabidopsis leaves using desorption electrospray ionisation mass spectrometry imaging. Analyst 2021, 146, 2653–2663. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Repository | Functionalities | Species | Web | Reference |
|---|---|---|---|---|
| SRA | Transcriptomics | All branches of life | https://www.ncbi.nlm.nih.gov/sra | [54] |
| Metabolights | Raw metabolomics | All branches of life | https://www.ebi.ac.uk/metabolights/ | [55] |
| STRingDB | Protein–protein interactions | All branches of life | https://string-db.org/ | [56] |
| PlaNet | Co-function networks | Photosynthetic organisms | http://aranet.mpimp-golm.mpg.de/ | [57] |
| PGP | Genomics and Phenomics | Chloroplastida 1 | https://edal-pgp.ipk-gatersleben.de/ | [58] |
| KEGG | Molecular networks | All branches of life | https://www.genome.jp/kegg/ | [59] |
| Reactome | Pathway knowledge | Animalia | https://reactome.org/ | [60] |
| iRefWeb | Protein-protein interactions | All branches of life | http://wodaklab.org/iRefWeb | [61] |
| GeneMANIA | Gene function | Animalia, Fungi and Plantae | http://genemania.org/ | [62] |
| Software | Omics 1 | Functionalities | Comments | Repositories | Reference |
|---|---|---|---|---|---|
| mixOmics ggmixOmics | T, P, M, R | Multivariant-based framework (PCA, CCA, PLS-DA, etc.) | Dimensions reduction, extraction of variable subgroups connected with traits and visualizations | R/CRAN | [80] |
| xMWAS | T, P, M | Multivariant- and network-based framework | Application for paired and unpaired study R/GitHub | R/GitHub | [81] |
| metaboGSE | T, M | Connection of network-based approaches and gene set enrichment analysis | Creation of subnetworks in the context of experimental condition | R/CRAN | [82] |
| FELLA | M | Network-based enrichment analysis of metabolites lists | Supporting KEGG database | R/BioC | [83] |
| MetExplore | T, P, M | Network-based analysis, pathway mapping, flux balance modeling and analysis | Easy way for network creation, visualization, curation and metabolite mapping | https://metexplore.toulouse.inrae.fr/index.html/ | [84] |
| OmicsNet | T, P, M, R | Network- and pathway-based approach | Building, visualization and exploration of biological networks in 3D space | https://www.omicsnet.ca/ | [85] |
| MiBiOmics | T, P, M | Correlation-based tool for creating, dimensionality reduction and exploration of networks | Provide the tools for data processing (filtration, normalization and transformation) | https://shiny-bird.univ-nantes.fr/app/Mibiomics | [86] |
| MetaBridge | T, M | Network-based pathway mapping | Identification of connections between metabolites and enzymes, visualization of data and results | https://metabridge.org/ | [87] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acién, J.M.; Cañizares, E.; Candela, H.; González-Guzmán, M.; Arbona, V. From Classical to Modern Computational Approaches to Identify Key Genetic Regulatory Components in Plant Biology. Int. J. Mol. Sci. 2023, 24, 2526. https://doi.org/10.3390/ijms24032526
Acién JM, Cañizares E, Candela H, González-Guzmán M, Arbona V. From Classical to Modern Computational Approaches to Identify Key Genetic Regulatory Components in Plant Biology. International Journal of Molecular Sciences. 2023; 24(3):2526. https://doi.org/10.3390/ijms24032526
Chicago/Turabian StyleAcién, Juan Manuel, Eva Cañizares, Héctor Candela, Miguel González-Guzmán, and Vicent Arbona. 2023. "From Classical to Modern Computational Approaches to Identify Key Genetic Regulatory Components in Plant Biology" International Journal of Molecular Sciences 24, no. 3: 2526. https://doi.org/10.3390/ijms24032526
APA StyleAcién, J. M., Cañizares, E., Candela, H., González-Guzmán, M., & Arbona, V. (2023). From Classical to Modern Computational Approaches to Identify Key Genetic Regulatory Components in Plant Biology. International Journal of Molecular Sciences, 24(3), 2526. https://doi.org/10.3390/ijms24032526