
Microvascular Thrombosis as a Critical Factor in Severe COVID-19
 , ,
, , 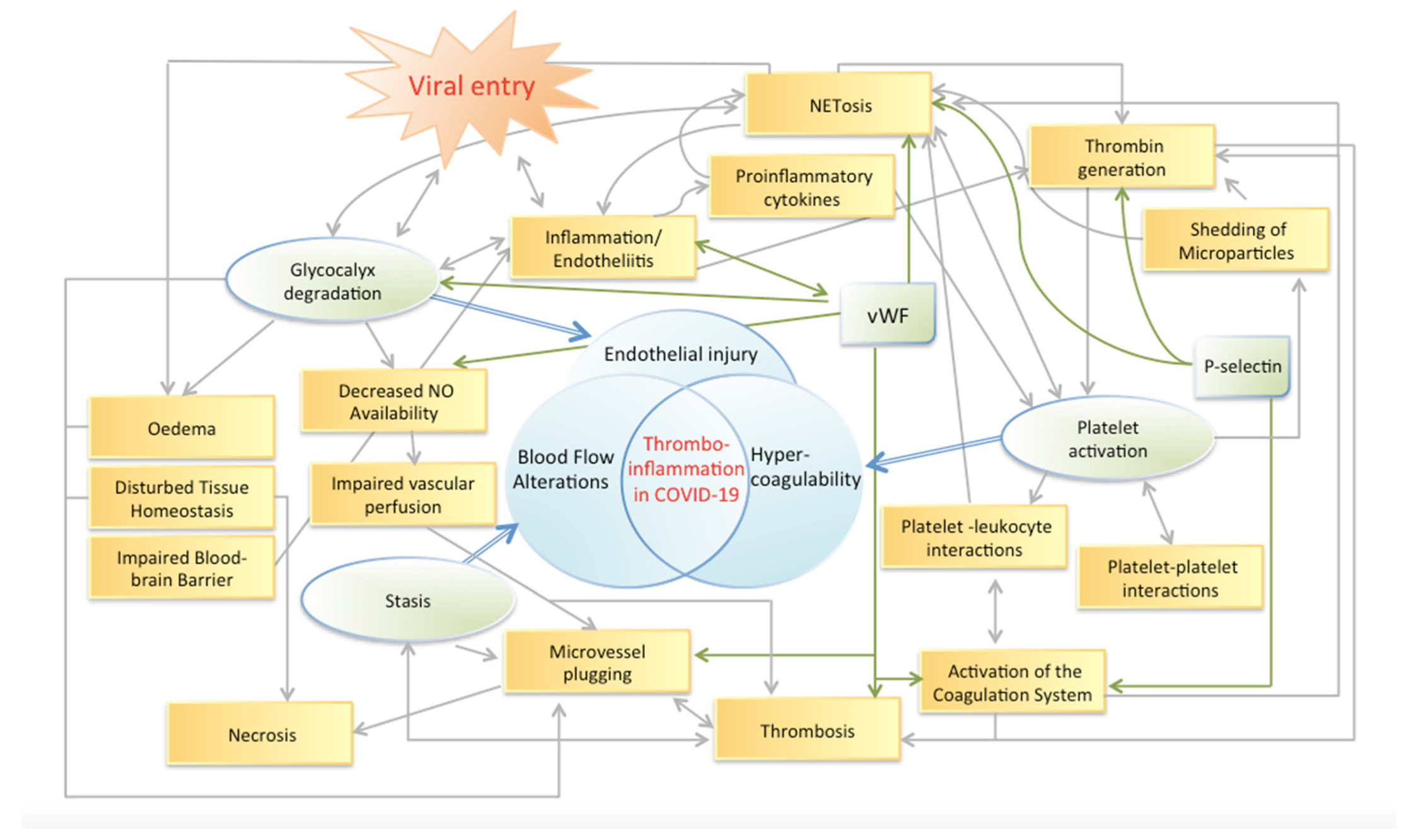{kind=link}
Abstract
Author Contributions
Funding
Conflicts of Interest
References
- Nadarajah, R.; Wu, J.; Hurdus, B.; Asma, S.; Bhatt, D.L.; Biondi-Zoccai, G.; Mehta, L.S.; Ram, C.V.S.; Ribeiro, A.L.P.; Van Spall, H.G.C.; et al. The collateral damage of COVID-19 to cardiovascular services: A meta-analysis. Eur. Heart J. 2022, 43, 3164–3178. [Google Scholar] [CrossRef]
- Arsenault, C.; Gage, A.; Kim, M.K.; Kapoor, N.R.; Akweongo, P.; Amponsah, F.; Aryal, A.; Asai, D.; Awoonor-Williams, J.K.; Ayele, W.; et al. COVID-19 and resilience of healthcare systems in ten countries. Nat. Med. 2022, 28, 1314–1324. [Google Scholar] [CrossRef] [PubMed]
- Wadowski, P.P.; Piechota-Polańczyk, A.; Andreas, M.; Kopp, C.W. Cardiovascular Disease Management in the Context of Global Crisis. Int. J. Environ. Res. Public Health 2022, 20, 689. [Google Scholar] [CrossRef] [PubMed]
- Mathew, D.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Greenplate, A.R.; Wu, J.E.; Alanio, C.; Kuri-Cervantes, L.; Pampena, M.B.; D’Andrea, K.; et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 2020, 369, eabc8511. [Google Scholar] [CrossRef]
- Jonigk, D.; Werlein, C.; Acker, T.; Aepfelbacher, M.; Amann, K.U.; Baretton, G.; Barth, P.; Bohle, R.M.; Büttner, A.; Büttner, R.; et al. Organ manifestations of COVID-19: What have we learned so far (not only) from autopsies? Virchows Arch. 2022, 481, 139–159. [Google Scholar] [CrossRef]
- Bonaventura, A.; Vecchié, A.; Dagna, L.; Martinod, K.; Dixon, D.L.; Van Tassell, B.W.; Dentali, F.; Montecucco, F.; Massberg, S.; Levi, M.; et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 2021, 21, 319–329. [Google Scholar] [CrossRef]
- Gonzalez-Gonzalez, F.J.; Ziccardi, M.R.; McCauley, M.D. Virchow’s Triad and the Role of Thrombosis in COVID-Related Stroke. Front. Physiol. 2021, 12, 769254. [Google Scholar] [CrossRef]
- Conway, E.M.; Mackman, N.; Warren, R.Q.; Wolberg, A.S.; Mosnier, L.O.; Campbell, R.A.; Gralinski, L.E.; Rondina, M.T.; van de Veerdonk, F.L.; Hoffmeister, K.M.; et al. Understanding COVID-19-associated coagulopathy. Nat. Rev. Immunol. 2022, 22, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Flaumenhaft, R.; Enjyoji, K.; Schmaier, A.A. Vasculopathy in COVID-19. Blood 2022, 140, 222–235. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Jilma, B.; Kopp, C.W.; Ertl, S.; Gremmel, T.; Koppensteiner, R. Glycocalyx as Possible Limiting Factor in COVID-19. Front. Immunol. 2021, 12, 607306. [Google Scholar] [CrossRef] [PubMed]
- Targosz-Korecka, M.; Kubisiak, A.; Kloska, D.; Kopacz, A.; Grochot-Przeczek, A.; Szymonski, M. Endothelial glycocalyx shields the interaction of SARS-CoV-2 spike protein with ACE2 receptors. Sci. Rep. 2021, 11, 12157. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Siripanthong, B.; Asatryan, B.; Hanff, T.C.; Chatha, S.R.; Khanji, M.Y.; Ricci, F.; Muser, D.; Ferrari, V.A.; Nazarian, S.; Santangeli, P.; et al. The Pathogenesis and Long-Term Consequences of COVID-19 Cardiac Injury. JACC Basic Transl. Sci. 2022, 7, 294–308. [Google Scholar] [CrossRef]
- Wagner, D.D.; Heger, L.A. Thromboinflammation: From Atherosclerosis to COVID-19. Arter. Thromb. Vasc. Biol. 2022, 42, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, D.; Kawakami, R.; Guagliumi, G.; Sakamoto, A.; Kawai, K.; Gianatti, A.; Nasr, A.; Kutys, R.; Guo, L.; Cornelissen, A.; et al. Microthrombi as a Major Cause of Cardiac Injury in COVID-19. Circulation 2021, 143, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Laurence, J.; Nuovo, G.; Racine-Brzostek, S.E.; Seshadri, M.; Elhadad, S.; Crowson, A.N.; Mulvey, J.J.; Harp, J.; Ahamed, J.; Magro, C. Premortem Skin Biopsy Assessing Microthrombi, Interferon Type I Antiviral and Regulatory Proteins, and Complement Deposition Correlates with Coronavirus Disease 2019 Clinical Stage. Am. J. Pathol. 2022, 192, 1282–1294. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Xu, J.; Xiao, W.; Liang, X.; Shi, L.; Zhang, P.; Wang, Y.; Wang, Y.; Yang, H. A meta-analysis on the risk factors adjusted association between cardiovascular disease and COVID-19 severity. BMC Public Health 2021, 21, 147. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Hülsmann, M.; Schörgenhofer, C.; Lang, I.M.; Wurm, R.; Gremmel, T.; Koppensteiner, R.; Steinlechner, B.; Schwameis, M.; Jilma, B. Sublingual functional capillary rarefaction in chronic heart failure. Eur. J. Clin. Investig. 2017, 48, e12869. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Steinlechner, B.; Zimpfer, D.; Schlöglhofer, T.; Schima, H.; Hülsmann, M.; Lang, I.M.; Gremmel, T.; Koppensteiner, R.; Zehetmayer, S.; et al. Functional capillary impairment in patients with ventricular assist devices. Sci. Rep. 2019, 9, 5909. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Schörgenhofer, C.; Rieder, T.; Ertl, S.; Pultar, J.; Serles, W.; Sycha, T.; Mayer, F.; Koppensteiner, R.; Gremmel, T.; et al. Microvascular rarefaction in patients with cerebrovascular events. Microvasc. Res. 2022, 140, 104300. [Google Scholar] [CrossRef]
- Ataga, K.I. Hypercoagulability and thrombotic complications in hemolytic anemias. Haematologica 2009, 94, 1481–1484. [Google Scholar] [CrossRef]
- Constantinescu-Bercu, A.; Wang, Y.A.; Woollard, K.J.; Mangin, P.; Vanhoorelbeke, K.; Crawley, J.T.; Salles-Crawley, I.I. The GPIbα intracellular tail-role in transducing VWF- and collagen/GPVI-mediated signaling. Haematologica 2021, 107, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Sadler, J. von Willebrand factor. J. Biol. Chem. 1991, 266, 22777–22780. [Google Scholar] [CrossRef] [PubMed]
- Philippe, A.; Gendron, N.; Bory, O.; Beauvais, A.; Mirault, T.; Planquette, B.; Sanchez, O.; Diehl, J.-L.; Chocron, R.; Smadja, D.M. Von Willebrand factor collagen-binding capacity predicts in-hospital mortality in COVID-19 patients: Insight from VWF/ADAMTS13 ratio imbalance. Angiogenesis 2021, 24, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Andrade, S.A.D.; de Souza, D.A.; Torres, A.L.; de Lima, C.F.G.; Ebram, M.C.; Celano, R.M.G.; Schattner, M.; Chudzinski-Tavassi, A.M. Patho-physiology of COVID-19: Critical Role of Hemostasis. Front. Cell. Infect. Microbiol. 2022, 12, 696. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Emsley, J. The organizing principle of the platelet glycoprotein Ib–IX–V complex. J. Thromb. Haemost. 2013, 11, 605–614. [Google Scholar] [CrossRef]
- Yun, S.-H.; Sim, E.-H.; Goh, R.-Y.; Park, J.-I.; Han, J.-Y. Platelet Activation: The Mechanisms and Potential Biomarkers. BioMed Res. Int. 2016, 2016, 9060143. [Google Scholar] [CrossRef]
- Dörmann, D.; Clemetson, K.J.; Kehrel, B.E. The GPIb thrombin-binding site is essential for thrombin-induced platelet procoagulant activity. Blood 2000, 96, 2469–2478. [Google Scholar] [CrossRef]
- Parker, W.A.; Orme, R.C.; Hanson, J.; Stokes, H.M.; Bridge, C.M.; Shaw, P.A.; Sumaya, W.; Thorneycroft, K.; Petrucci, G.; Porro, B.; et al. Very-low-dose twice-daily aspirin maintains platelet inhibition and improves haemostasis during dual-antiplatelet therapy for acute coronary syndrome. Platelets 2019, 30, 148–157. [Google Scholar] [CrossRef]
- Paul, B.Z.S.; Jin, J.; Kunapuli, S.P. Molecular Mechanism of Thromboxane A2-induced Platelet Aggregation: Essential role for p2t(ac) and alpha(2a) receptors. J. Biol. Chem. 1999, 274, 29108–29114. [Google Scholar] [CrossRef]
- Gremmel, T.; Frelinger, A.L., III; Michelson, A.D. Platelet Physiology. Semin. Thromb. Hemost. 2016, 42, 191–204. [Google Scholar] [CrossRef]
- Parker, W.A.E.; Storey, R.F. Long-term antiplatelet therapy following myocardial infarction: Implications of PEGASUS-TIMI 54. Heart 2016, 102, 783–789. [Google Scholar] [CrossRef]
- Storey, R.F.; Sanderson, H.M.; White, A.E.; May, J.A.; Cameron, K.E.; Heptinstall, S. The central role of the P2T receptor in amplification of human platelet activation, aggregation, secretion and procoagulant activity. Br. J. Haematol. 2000, 110, 925–934. [Google Scholar] [CrossRef]
- Storey, R.F.; Judge, H.M.; Wilcox, R.G.; Heptinstall, S. Inhibition of ADP-induced P-selectin Expression and Platelet-Leukocyte Conjugate Formation by Clopidogrel and the P2Y12 Receptor Antagonist AR-C69931MX but not As-pirin. Thromb. Haemost. 2002, 88, 488–494. [Google Scholar]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef]
- Michelson, A.D.; Barnard, M.R.; Hechtman, H.B.; MacGregor, H.; Connolly, R.J.; Loscalzo, J.; Valeri, C.R. In vivo tracking of platelets: Circulating degranulated platelets rapidly lose surface P-selectin but continue to circulate and function. Proc. Natl. Acad. Sci. USA 1996, 93, 11877–11882. [Google Scholar] [CrossRef]
- Garcia, C.; Duong, J.A.; Poëtte, M.; Ribes, A.; Payre, B.; Mémier, V.; Sié, P.; Minville, V.; Voisin, S.; Payrastre, B.; et al. Platelet activation and partial desensitization are associated with viral xenophagy in patients with severe COVID-19. Blood Adv. 2022, 6, 3884–3898. [Google Scholar] [CrossRef]
- Sanderson, H.M.; Heptinstall, S.; Vickers, J.; Lösche, W. Studies on the effects of agonists and antagonists on platelet shape change and platelet aggregation in whole blood. Blood Coagul. Fibrinolysis 1996, 7, 245–248. [Google Scholar] [CrossRef]
- Cho, M.J.; Liu, J.; Pestina, T.I.; Steward, S.A.; Jackson, C.W.; Gartner, T.K. αIIbβ3-mediated outside-in signal-ing induced by the agonist peptide LSARLAF utilizes ADP and thromboxane A2 receptors to cause α-granule secretion by platelets. J. Thromb. Haemost. 2003, 1, 363–373. [Google Scholar] [CrossRef]
- Brahmer, A.; Neuberger, E.; Esch-Heisser, L.; Haller, N.; Jørgensen, M.M.; Baek, R.; Möbius, W.; Simon, P.; Krämer-Albers, E.-M. Platelets, endothelial cells and leukocytes contribute to the exercise-triggered release of extracellular vesicles into the circulation. J. Extracell. Vesicles 2019, 8, 1615820. [Google Scholar] [CrossRef]
- Nederveen, J.P.; Warnier, G.; Di Carlo, A.; Nilsson, M.I.; Tarnopolsky, M.A. Extracellular Vesicles and Exo-somes: Insights from Exercise Science. Front. Physiol. 2021, 11, 604274. [Google Scholar] [CrossRef]
- Elzanowska, J.; Semira, C.; Costa-Silva, B. DNA in extracellular vesicles: Biological and clinical aspects. Mol. Oncol. 2020, 15, 1701–1714. [Google Scholar] [CrossRef]
- Sinauridze, E.I.; Kireev, D.A.; Popenko, N.Y.; Pichugin, A.; Panteleev, M.; Krymskaya, O.V.; Ataullakhanov, F.I. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb. Haemost. 2007, 97, 425–434. [Google Scholar] [CrossRef]
- Subrahmanian, S.; Borczuk, A.; Salvatore, S.; Fung, K.M.; Merrill, J.T.; Laurence, J.; Ahamed, J. Tissue factor upregula-tion is associated with SARS-CoV-2 in the lungs of COVID-19 patients. J. Thromb. Haemost. 2021, 19, 2268–2274. [Google Scholar] [CrossRef]
- Busch, M.; Timmermans, S.A.; Nagy, M.; Visser, M.; Huckriede, J.; Aendekerk, J.P.; De Vries, F.; Potjewijd, J.; Jallah, B.; Ysermans, R.; et al. Neutrophils and Contact Activation of Coagulation as Potential Drivers of COVID-19. Circulation 2020, 142, 1787–1790. [Google Scholar] [CrossRef]
- Gorog, D.A.; Storey, R.F.; Gurbel, P.A.; Tantry, U.S.; Berger, J.S.; Chan, M.Y.; Duerschmied, D.; Smyth, S.S.; Parker, W.A.E.; Ajjan, R.A.; et al. Current and novel biomarkers of thrombotic risk in COVID-19: A Consensus Statement from the International COVID-19 Thrombosis Biomarkers Colloquium. Nat. Rev. Cardiol. 2022, 19, 475–495. [Google Scholar] [CrossRef]
- Bevers, E.M.; Comfurius, P.; Van Rijn, J.L.M.L.; Hemker, H.C. Generation of Prothrombin-Converting Activity and the Exposure of Phosphatidylserine at the Outer Surface of Platelets. Eur. J. Biochem. 1982, 122, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Lhermusier, T.; Chap, H.; Payrastre, B. Platelet membrane phospholipid asymmetry: From the characterization of a scramblase activity to the identification of an essential protein mutated in Scott syndrome. J. Thromb. Haemost. 2011, 9, 1883–1891. [Google Scholar] [CrossRef]
- Ofosu, F.A.; Dewar, L.; Craven, S.J.; Song, Y.; Cedrone, A.; Freedman, J.; Fenton, J.W. Coordinate Activation of Human Platelet Protease-activated Receptor-1 and -4 in Response to Subnanomolar α-Thrombin. J. Biol. Chem. 2008, 283, 26886–26893. [Google Scholar] [CrossRef] [PubMed]
- Duvernay, M.T.; Temple, K.J.; Maeng, J.G.; Blobaum, A.L.; Stauffer, S.R.; Lindsley, C.W.; Hamm, H.E. Contributions of Protease-Activated Receptors PAR1 and PAR4 to Thrombin-Induced GPIIbIIIa Activation in Human Platelets. Mol. Pharmacol. 2016, 91, 39–47. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Pultar, J.; Weikert, C.; Eichelberger, B.; Panzer, B.; Huber, K.; Lang, I.M.; Koppensteiner, R.; Panzer, S.; Gremmel, T. Protease-activated receptor-mediated platelet aggregation in acute coronary syndrome patients on potent P2Y12 inhibitors. Res. Pract. Thromb. Haemost. 2019, 3, 383–390. [Google Scholar] [CrossRef]
- Gremmel, T.; Michelson, A.D.; Wadowski, P.P.; Pultar, J.; Weikert, C.; Tscharre, M.; Lee, S.; Panzer, S.; Frelinger, A.L. Sex-specific platelet activation through protease-activated receptor-1 in patients undergoing cardiac catheterization. Atherosclerosis 2021, 339, 12–19. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Pultar, J.; Weikert, C.; Eichelberger, B.; Tscharre, M.; Koppensteiner, R.; Panzer, S.; Gremmel, T. Platelet-to-Lymphocyte Ratio as Marker of Platelet Activation in Patients on Potent P2Y12 Inhibitors. J. Cardiovasc. Pharmacol. Ther. 2022, 27, 10742484221096524. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.-D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Weikert, C.; Pultar, J.; Lee, S.; Eichelberger, B.; Koppensteiner, R.; Lang, I.M.; Panzer, S.; Gremmel, T. Ticagrelor Inhibits Toll-Like and Protease-Activated Receptor Mediated Platelet Activation in Acute Coronary Syndromes. Cardiovasc. Drugs Ther. 2020, 34, 53–63. [Google Scholar] [CrossRef]
- Biswas, S.; Zimman, A.; Gao, D.; Byzova, T.V.; Podrez, E.A. TLR2 Plays a Key Role in Platelet Hyperreactivity and Accelerated Thrombosis Associated with Hyperlipidemia. Circ. Res. 2017, 121, 951–962. [Google Scholar] [CrossRef]
- Into, T.; Kanno, Y.; Dohkan, J.-I.; Nakashima, M.; Inomata, M.; Shibata, K.-I.; Lowenstein, C.J.; Matsushita, K. Pathogen Recognition by Toll-like Receptor 2 Activates Weibel-Palade Body Exocytosis in Human Aortic Endothelial Cells. J. Biol. Chem. 2007, 282, 8134–8141. [Google Scholar] [CrossRef]
- Singh, B.; Biswas, I.; Bhagat, S.; Surya Kumari, S.; Khan, G.A. HMGB1 facilitates hypoxia-induced vWF upregu-lation through TLR2-MYD88-SP1 pathway. Eur. J. Immunol. 2016, 46, 2388–2400. [Google Scholar] [CrossRef]
- Carestia, A.; Kaufman, T.; Rivadeneyra, L.; Landoni, V.I.; Pozner, R.G.; Negrotto, S.; D’Atri, L.P.; Gómez, R.M.; Schattner, M. Mediators and molec-ular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J. Leukoc. Biol. 2016, 99, 153–162. [Google Scholar] [CrossRef]
- Fujimura, Y.; Holland, L.Z. COVID-19 microthrombosis: Unusually large VWF multimers are a platform for activation of the alternative complement pathway under cytokine storm. Int. J. Hematol. 2022, 115, 457–469. [Google Scholar] [CrossRef]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.W.; Savani, R.C.; Zaki, H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway. Elife 2021, 10, e68563. [Google Scholar] [CrossRef]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef]
- Fard, M.B.; Fard, S.B.; Ramazi, S.; Atashi, A.; Eslamifar, Z. Thrombosis in COVID-19 infection: Role of platelet activation-mediated immunity. Thromb. J. 2021, 19, 59. [Google Scholar] [CrossRef]
- Teixeira, P.C.; Dorneles, G.P.; Filho, P.C.S.; da Silva, I.M.; Schipper, L.L.; Postiga, I.A.; Neves, C.A.M.; Junior, L.C.R.; Peres, A.; de Souto, J.T.; et al. Increased LPS levels coexist with systemic inflammation and result in monocyte activation in severe COVID-19 patients. Int. Immunopharmacol. 2021, 100, 108125. [Google Scholar] [CrossRef]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Eichelberger, B.; Kopp, C.W.; Pultar, J.; Seidinger, D.; Koppensteiner, R.; Lang, I.M.; Panzer, S.; Gremmel, T. Disaggregation Following Agonist-Induced Platelet Activation in Patients on Dual Antiplatelet Therapy. J. Cardiovasc. Transl. Res. 2017, 10, 359–367. [Google Scholar] [CrossRef]
- Bohula, E.A.; Berg, D.D.; Lopes, M.S.; Connors, J.M.; Babar, I.; Barnett, C.F.; Chaudhry, S.-P.; Chopra, A.; Ginete, W.; Ieong, M.H.; et al. Anticoagulation and Antiplatelet Therapy for Prevention of Venous and Arterial Thrombotic Events in Critically Ill Patients with COVID-19: COVID-PACT. Circulation 2022, 146, 1344–1356. [Google Scholar] [CrossRef]
- Abani, O.; Abbas, A.; Abbas, F.; Abbas, M.; Abbasi, S.; Abbass, H.; Abbott, A.; Abdallah, N.; Abdelaziz, A.; Abdelfattah, M.; et al. Aspirin in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2022, 399, 143–151. [Google Scholar] [CrossRef]
- Berger, J.S.; Kornblith, L.Z.; Gong, M.N.; Reynolds, H.R.; Cushman, M.; Cheng, Y.; McVerry, B.J.; Kim, K.S.; Lopes, R.D.; Atassi, B.; et al. Effect of P2Y12 Inhibitors on Survival Free of Organ Support among Non-Critically Ill Hospitalized Patients with COVID-19. JAMA 2022, 327, 227. [Google Scholar] [CrossRef]
- Lawler, P.R.; Goligher, E.C.; Berger, J.S.; Neal, M.D.; McVerry, B.J.; Nicolau, J.C. Therapeutic Anticoagulation with Heparin in Noncritically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 385, 790–802. [Google Scholar] [CrossRef]
- Goligher, E.C.; Bradbury, C.A.; McVerry, B.J.; Lawler, P.R.; Berger, J.S.; Gong, M.N. Therapeutic Anticoagulation with Heparin in Critically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 385, 777–789. [Google Scholar] [CrossRef]
- Ionescu, F.; Jaiyesimi, I.; Petrescu, I.; Lawler, P.R.; Castillo, E.; Munoz-Maldonado, Y.; Imam, Z.; Narasimhan, M.; Abbas, A.E.; Konde, A.; et al. Association of anti-coagulation dose and survival in hospitalized COVID-19 patients: A retrospective propensity score-weighted analysis. Eur. J. Haematol. 2021, 106, 165–174. [Google Scholar] [CrossRef]
- Bakchoul, T.; Hammer, S.; Lang, P.; Rosenberger, P. Fibrinolysis shut down in COVID-19 patients: Report on two severe cases with potential diagnostic and clinical relevance. Thromb. Updat. 2020, 1, 100008. [Google Scholar] [CrossRef]
- Wygrecka, M.; Birnhuber, A.; Seeliger, B.; Michalick, L.; Pak, O.; Schultz, A.-S.; Schramm, F.; Zacharias, M.; Gorkiewicz, G.; David, S.; et al. Altered fibrin clot structure and dysregulated fibrinolysis contribute to thrombosis risk in severe COVID-19. Blood Adv. 2022, 6, 1074–1087. [Google Scholar] [CrossRef]
- Ji, H.-L.; Dai, Y.; Zhao, R. Fibrinolytic therapy for COVID-19: A review of case series. Acta Pharmacol. Sin. 2021, 43, 2168–2170. [Google Scholar] [CrossRef]
- Zuo, Y.; Warnock, M.; Harbaugh, A.; Yalavarthi, S.; Gockman, K.; Zuo, M.; Madison, J.A.; Knight, J.S.; Kanthi, Y.; Lawrence, D.A. Plasma tissue plasminogen activator and plasminogen activator inhibitor-1 in hospitalized COVID-19 patients. Sci. Rep. 2021, 11, 1580. [Google Scholar] [CrossRef]
- Rovai, E.S.; Alves, T.; Holzhausen, M. Protease-activated receptor 1 as a potential therapeutic target for COVID-19. Exp. Biol. Med. 2020, 246, 688–694. [Google Scholar] [CrossRef]
- Pultar, J.; Wadowski, P.P.; Panzer, S.; Gremmel, T. Oral antiplatelet agents in cardiovascular disease. Vasa 2019, 48, 291–302. [Google Scholar] [CrossRef]
- Katsoularis, I.; Fonseca-Rodríguez, O.; Farrington, P.; Jerndal, H.; Lundevaller, E.H.; Sund, M.; Lindmark, K.; Connolly, A.-M.F. Risks of deep vein thrombosis, pulmonary embolism, and bleeding after COVID-19: Nationwide self-controlled cases series and matched cohort study. BMJ 2022, 377, e069590. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W.; Litvinov, R.I. Fibrin Formation, Structure and Properties. In Fibrous Proteins: Structures and Mechanisms; Springer International Publishing: Cham, Switzerland, 2017; pp. 405–456. [Google Scholar]
- Kaplan, Z.S.; Zarpellon, A.; Alwis, I.; Yuan, Y.; McFadyen, J.; Ghasemzadeh, M.; Schoenwaelder, S.M.; Ruggeri, Z.M.; Jackson, S.P. Thrombin-dependent intravascular leukocyte trafficking regulated by fibrin and the platelet receptors GPIb and PAR4. Nat. Commun. 2015, 6, 7835. [Google Scholar] [CrossRef]
- Nguyen, D.; Jeon, H.-M.; Lee, J. Tissue factor links inflammation, thrombosis, and senescence in COVID-19. Sci. Rep. 2022, 12, 19842. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N.; Sachettor, A.T.A. Tissue factor and COVID-19: An update. Curr. Drug Targets 2022, 23, 1573–1577. [Google Scholar] [CrossRef]
- Rosell, A.; Havervall, S.; Von Meijenfeldt, F.; Hisada, Y.; Aguilera, K.; Grover, S.P.; Lisman, T.; Mackman, N.; Thålin, C. Patients With COVID-19 Have Elevated Levels of Circulating Extracellular Vesicle Tissue Factor Activity That Is Associated with Severity and Mortality—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 878–882. [Google Scholar] [CrossRef] [PubMed]
- Guervilly, C.; Bonifay, A.; Burtey, S.; Sabatier, F.; Cauchois, R.; Abdili, E.; Arnaud, L.; Lano, G.; Pietri, L.; Robert, T.; et al. Dissemination of extreme levels of extracellular vesicles: Tissue factor activity in patients with severe COVID-19. Blood Adv. 2021, 5, 628–634. [Google Scholar] [CrossRef]
- Claus, R.A.; Bockmeyer, C.L.; Sossdorf, M.; Lösche, W. The balance between von-Willebrand factor and its cleaving protease ADAMTS13: Biomarker in systemic inflammation and development of organ failure? Curr. Mol. Med. 2010, 10, 236–248. [Google Scholar] [CrossRef]
- Fernández-Pérez, M.P.; Águila, S.; Reguilón-Gallego, L.; de los Reyes-García, A.M.; Miñano, A.; Bravo-Pérez, C.; María, E.; Corral, J.; García-Barberá, N.; Gómez-Verdú, J.M.; et al. Neutrophil extracellular traps and von Willebrand factor are allies that negatively influence COVID-19 outcomes. Clin. Transl. Med. 2021, 11, e268. [Google Scholar] [CrossRef] [PubMed]
- Menter, T.; Haslbauer, J.D.; Nienhold, R.; Savic, S.; Hopfer, H.; Deigendesch, N.; Frank, S.; Turek, D.; Willi, N.; Pargger, H.; et al. Post-mortem examination of COVID19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings of lungs and other organs suggesting vascular dysfunction. Histopathology 2020, 77, 198–209. [Google Scholar] [CrossRef]
- Yang, J.; Wu, Z.; Long, Q.; Huang, J.; Hong, T.; Liu, W.; Lin, J. Insights into Immunothrombosis: The Interplay among Neutrophil Extracellular Trap, von Willebrand Factor, and ADAMTS13. Front. Immunol. 2020, 11, 610696. [Google Scholar] [CrossRef]
- Furlan, M. Von Willebrand factor: Molecular size and functional activity. Ann. Hematol. 1996, 72, 341–348. [Google Scholar] [CrossRef]
- Singh, B.; Biswas, I.; Garg, I.; Sugadev, R.; Singh, A.K.; Dey, S.; Khan, G.A. Von Willebrand Factor Antagonizes Nitric Oxide Synthase to Promote Insulin Resistance during Hypoxia. Biochemistry 2013, 53, 115–126. [Google Scholar] [CrossRef]
- Grässle, S.; Huck, V.; Pappelbaum, K.I.; Gorzelanny, C.; Aponte-Santamaría, C.; Baldauf, C.; Gräter, F.; Schneppenheim, R.; Obser, T.; Schneider, S.W. Von Willebrand Factor Directly Interacts with DNA From Neutrophil Extracellular Traps. Arter. Thromb. Vasc. Biol. 2014, 34, 1382–1389. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Jenne, C.N.; Surewaard, B.G.J.; Thanabalasuriar, A.; Lee, W.-Y.; Sanz, M.-J.; Mowen, K.; Opdenakker, G.; Kubes, P. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat. Commun. 2015, 6, 6673. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef]
- Freeman, C.G.; Parish, C.; Knox, K.J.; Blackmore, J.L.; Lobov, S.A.; King, D.W.; Senden, T.; Stephens, R.W. The accumulation of circulating histones on heparan sulphate in the capillary glycocalyx of the lungs. Biomaterials 2013, 34, 5670–5676. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Bhandari, A.A.; Wagner, D.D. Histones induce rapid and profound thrombocytopenia in mice. Blood 2011, 118, 3708–3714. [Google Scholar] [CrossRef]
- Moore, B.J.B.; June, C.H. Cytokine release syndrome in severe COVID-19. Science 2020, 368, 473–474. [Google Scholar] [CrossRef]
- Gordon, A.C.; Mouncey, P.R.; Al-Beidh, F.; Rowan, K.M.; Nichol, A.D.; Arabi, Y.M.; Annane, D.; Beane, A.; Van Bentum-Puijk, W.; Berry, L.R.; et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 384, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Thiam, H.R.; Wong, S.L.; Wagner, D.D.; Waterman, C.M. Cellular Mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 2020, 36, 191–218. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Chen, X.; Liu, X. NETosis and Neutrophil Extracellular Traps in COVID-19: Immunothrombosis and Beyond. Front. Immunol. 2022, 13, 838011. [Google Scholar] [CrossRef]
- Hepprich, M.; Mudry, J.M.; Gregoriano, C.; Jornayvaz, F.R.; Carballo, S.; Wojtusciszyn, A.; Bart, P.A.; Chiche, J.D.; Fischli, S.; Baumgartner, T.; et al. Canakinumab in patients with COVID-19 and type 2 diabetes—A multicentre, randomised, double-blind, placebo-controlled trial. EClinicalMedicine 2022, 53, 101649. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240. [Google Scholar] [CrossRef]
- Falk, E.; Shah, P.K.; Fuster, V. Coronary Plaque Disruption. Circulation 1995, 92, 657–671. [Google Scholar] [CrossRef]
- Nazy, I.; Elliott, T.D.; Arnold, D.M. Platelet factor 4 inhibits ADAMTS13 activity and regulates the multimeric distribution of von Willebrand factor. Br. J. Haematol. 2020, 190, 594–598. [Google Scholar] [CrossRef]
- Wang, A.; Liu, F.; Dong, N.; Ma, Z.; Zhang, J.; Su, J.; Zhao, Y.; Ruan, C. Thrombospondin-1 and ADAMTS13 competitively bind to VWF A2 and A3 domains in vitro. Thromb. Res. 2010, 126, e260–e265. [Google Scholar] [CrossRef]
- Zuo, Y.; Estes, S.K.; Ali, R.A.; Gandhi, A.A.; Yalavarthi, S.; Shi, H.; Sule, G.; Gockman, K.; Madison, J.A.; Zuo, M.; et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci. Transl. Med. 2020, 12, eabd3876. [Google Scholar] [CrossRef]
- Ehrenfeld, M.; Tincani, A.; Andreoli, L.; Cattalini, M.; Greenbaum, A.; Kanduc, D.; Alijotas-Reig, J.; Zinserling, V.; Semenova, N.; Amital, H.; et al. Covid-19 and autoimmunity. Autoimmun. Rev. 2020, 19, 102597. [Google Scholar] [CrossRef]
- George, P.M.; Reed, A.; Desai, S.R.; Devaraj, A.; Faiez, T.S.; Laverty, S.; Kanwal, A.; Esneau, C.; Liu, M.K.; Kamal, F.; et al. A persistent neutrophil-associated immune signature characterizes post–COVID-19 pulmonary sequelae. Sci. Transl. Med. 2022, 14, eabo5795. [Google Scholar] [CrossRef]
- Mantovani, A.; Morrone, M.C.; Patrono, C.; Santoro, M.G.; Schiaffino, S.; Remuzzi, G.; Bussolati, G.; Cappuccinelli, P.; Fitzgerald, G.; Bacci, M.L.; et al. Long Covid: Where we stand and challenges ahead. Cell Death Differ. 2022, 29, 1891–1900. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.R.; Roch, B. Neutrophil Extracellular Traps and By-Products Play a Key Role in COVID-19: Pathogenesis, Risk Factors, and Therapy. J. Clin. Med. 2020, 9, 2942. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Ahamed, J.; Laurence, J. Long COVID endotheliopathy: Hypothesized mechanisms and potential therapeutic approaches. J. Clin. Investig. 2022, 132, e161167. [Google Scholar] [CrossRef]
- Wang, C.; Yu, C.; Jing, H.; Wu, X.; Novakovic, V.A.; Xie, R.; Shi, J. Long COVID: The Nature of Thrombotic Se-quelae Determines the Necessity of Early Anticoagulation. Front. Cell. Infect. Microbiol. 2022, 12, 861703. [Google Scholar] [CrossRef]
- Xie, Y.; Choi, T.; Al-Aly, Z. Nirmatrelvir and the Risk of Post-Acute Sequelae of COVID-19. medRxiv 2022. [Google Scholar] [CrossRef]
- Ali, R.A.; Gandhi, A.A.; Meng, H.; Yalavarthi, S.; Vreede, A.P.; Estes, S.K.; Palmer, O.R.; Bockenstedt, P.L.; Pinsky, D.J.; Greve, J.M.; et al. Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat. Commun. 2019, 10, 1916. [Google Scholar] [CrossRef]
- Mitsios, A.; Chrysanthopoulou, A.; Arampatzioglou, A.; Angelidou, I.; Vidali, V.; Ritis, K.; Skendros, P.; Stakos, D. Ticagrelor Exerts Immune-Modulatory Effect by Attenuating Neutrophil Extracellular Traps. Int. J. Mol. Sci. 2020, 21, 3625. [Google Scholar] [CrossRef]
- Wolach, O.; Sellar, R.S.; Martinod, K.; Cherpokova, D.; McConkey, M.; Chappell, R.J.; Silver, A.J.; Adams, D.; Castellano, C.A.; Schneider, R.K.; et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, eaan8292. [Google Scholar] [CrossRef]
- Annane, D.; Heming, N.; Grimaldi-Bensouda, L.; Frémeaux-Bacchi, V.; Vigan, M.; Roux, A.-L.; Marchal, A.; Michelon, H.; Rottman, M.; Moine, P. Eculizumab as an emergency treatment for adult patients with severe COVID-19 in the intensive care unit: A proof-of-concept study. Eclinicalmedicine 2020, 28, 100590. [Google Scholar] [CrossRef] [PubMed]
- Diurno, F.; Numis, F.G.; Porta, G.; Cirillo, F.; Maddaluno, S.; Ragozzino, A.; De Negri, P.; Di Gennaro, C.; Pagano, A.; Allegorico, E.; et al. Eculizumab treatment in patients with COVID-19: Preliminary results from real life ASL Napoli 2 Nord experience. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4040–4047. [Google Scholar]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.M.J.; Oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflug. Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef]
- Schierke, F.; Wyrwoll, M.J.; Wisdorf, M.; Niedzielski, L.; Maase, M.; Ruck, T.; Meuth, S.G.; Kusche-Vihrog, K. Nanomechanics of the endothelial glycocalyx contribute to Na+-induced vascular inflammation. Sci. Rep. 2017, 7, srep46476. [Google Scholar] [CrossRef]
- Min, B.; Fairchild, R.L. Over-salting ruins the balance of the immune menu. J. Clin. Investig. 2015, 125, 4002–4004. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Adamson, R.H.; Curry, F.-R.E.; Tarbell, J.M. Sphingosine-1-phosphate protects endothelial glycocalyx by inhibiting syndecan-1 shedding. Am. J. Physiol. Circ. Physiol. 2014, 306, H363–H372. [Google Scholar] [CrossRef]
- Becker, B.F.; Chappell, D.; Bruegger, D.; Annecke, T.; Jacob, M. Therapeutic strategies targeting the endothelial glycocalyx: Acute deficits, but great potential. Cardiovasc. Res. 2010, 87, 300–310. [Google Scholar] [CrossRef]
- Broekhuizen, L.N.; Lemkes, B.A.; Mooij, H.L.; Meuwese, M.C.; Verberne, H.; Holleman, F.; Schlingemann, R.O.; Nieuwdorp, M.; Stroes, E.S.G.; Vink, H. Effect of sulodexide on endothelial glycocalyx and vascular permeability in patients with type 2 diabetes mellitus. Diabetologia 2010, 53, 2646–2655. [Google Scholar] [CrossRef]
- Uchimido, R.; Schmidt, E.P.; Shapiro, N.I. The glycocalyx: A novel diagnostic and therapeutic target in sepsis. Crit. Care 2019, 23, 16. [Google Scholar] [CrossRef] [PubMed]
- Boels, M.G.; Avramut, M.C.; Koudijs, A.; Dane, M.J.; Lee, D.H.; van der Vlag, J.; Koster, A.J.; van Zonneveld, A.J.; van Faassen, E.; Gröne, H.-J.; et al. Atrasentan Reduces Albuminuria by Restoring the Glomerular Endothelial Glycocalyx Barrier in Diabetic Nephropathy. Diabetes 2016, 65, 2429–2439. [Google Scholar] [CrossRef] [PubMed]
- Chappell, D.; Jacob, M.; Hofmann-Kiefer, K.; Bruegger, D.; Rehm, M.; Conzen, P.; Welsch, U.; Becker, B.F. Hydrocortisone Preserves the Vascular Barrier by Protecting the Endothelial Glycocalyx. Anesthesiology 2007, 107, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Brettner, F.; Chappell, D.; Nebelsiek, T.; Hauer, D.; Schelling, G.; Becker, B.F.; Rehm, M.; Weis, F. Preinterventional hydrocortisone sustains the endothelial glycocalyx in cardiac surgery. Clin. Hemorheol. Microcirc. 2019, 71, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Chappell, D.; Dörfler, N.; Jacob, M.; Rehm, M.; Welsch, U.; Conzen, P.; Becker, B.F. Glycocalyx protection reduces leukocyte adhesion after ischemia/reperfusion. Shock 2010, 34, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Meuwese, M.C.; Mooij, H.L.; Nieuwdorp, M.; van Lith, B.; Marck, R.; Vink, H.; Kastelein, J.J.P.; Stroes, E.S.G. Partial recovery of the endothelial glycocalyx upon rosuvastatin therapy in patients with heterozygous familial hypercholesterolemia. J. Lipid Res. 2009, 50, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.; Huber, J.; Ziehl, A. Complementary Sets of Autoantibodies Induced by SARS-CoV-2, Adenovirus and Bacterial Antigens Cross-React with Human Blood Protein Antigens in COVID-19 Coagulopathies. Int. J. Mol. Sci. 2022, 23, 11500. [Google Scholar] [CrossRef]
- Xu, S.-W.; Ilyas, I.; Weng, J.-P. Endothelial dysfunction in COVID-19: An overview of evidence, biomarkers, mechanisms and potential therapies. Acta Pharmacol. Sin. 2022, 1–15, (Epub ahead of print). [Google Scholar] [CrossRef]
- Caillon, A.; Trimaille, A.; Favre, J.; Jesel, L.; Morel, O.; Kauffenstein, G. Role of neutrophils, platelets, and ex-tracellular vesicles and their interactions in COVID-19-associated thrombopathy. J. Thromb. Haemost. 2022, 20, 17–31. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wadowski, P.P.; Panzer, B.; Józkowicz, A.; Kopp, C.W.; Gremmel, T.; Panzer, S.; Koppensteiner, R. Microvascular Thrombosis as a Critical Factor in Severe COVID-19. Int. J. Mol. Sci. 2023, 24, 2492. https://doi.org/10.3390/ijms24032492
Wadowski PP, Panzer B, Józkowicz A, Kopp CW, Gremmel T, Panzer S, Koppensteiner R. Microvascular Thrombosis as a Critical Factor in Severe COVID-19. International Journal of Molecular Sciences. 2023; 24(3):2492. https://doi.org/10.3390/ijms24032492
Chicago/Turabian StyleWadowski, Patricia P., Benjamin Panzer, Alicja Józkowicz, Christoph W. Kopp, Thomas Gremmel, Simon Panzer, and Renate Koppensteiner. 2023. "Microvascular Thrombosis as a Critical Factor in Severe COVID-19" International Journal of Molecular Sciences 24, no. 3: 2492. https://doi.org/10.3390/ijms24032492
APA StyleWadowski, P. P., Panzer, B., Józkowicz, A., Kopp, C. W., Gremmel, T., Panzer, S., & Koppensteiner, R. (2023). Microvascular Thrombosis as a Critical Factor in Severe COVID-19. International Journal of Molecular Sciences, 24(3), 2492. https://doi.org/10.3390/ijms24032492