

Molecular Dynamics Approach to the Physical Mixture of In2O3 and ZrO2: Defect Formation and Ionic Diffusion
 ,
,
Abstract
1. Introduction
2. Methodology
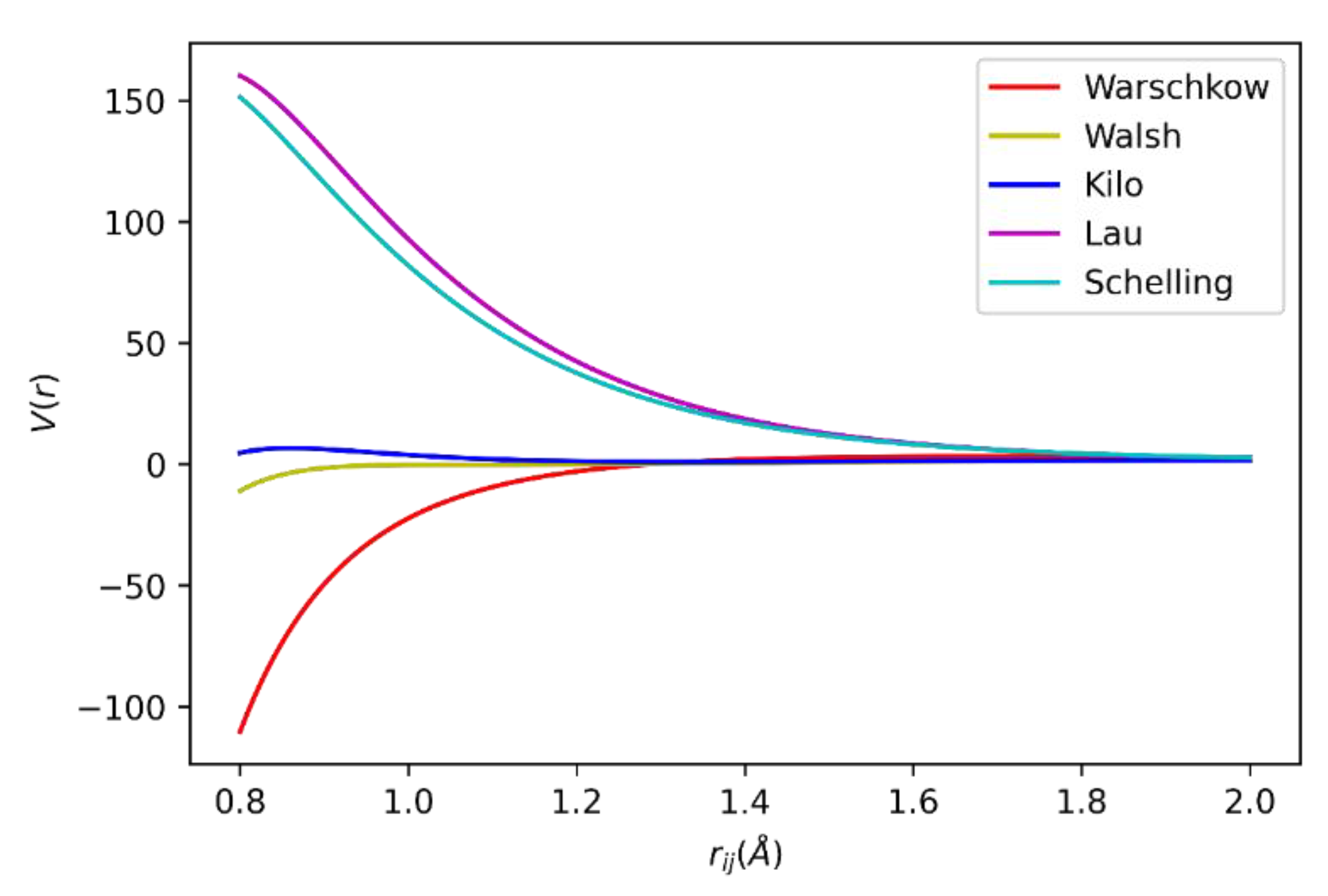
2.1. In2O3 Model Verification and Selection
2.2. Defect Energy
2.3. ZrO2 Model Selection
2.4. Defective In2O3 and ZrO2
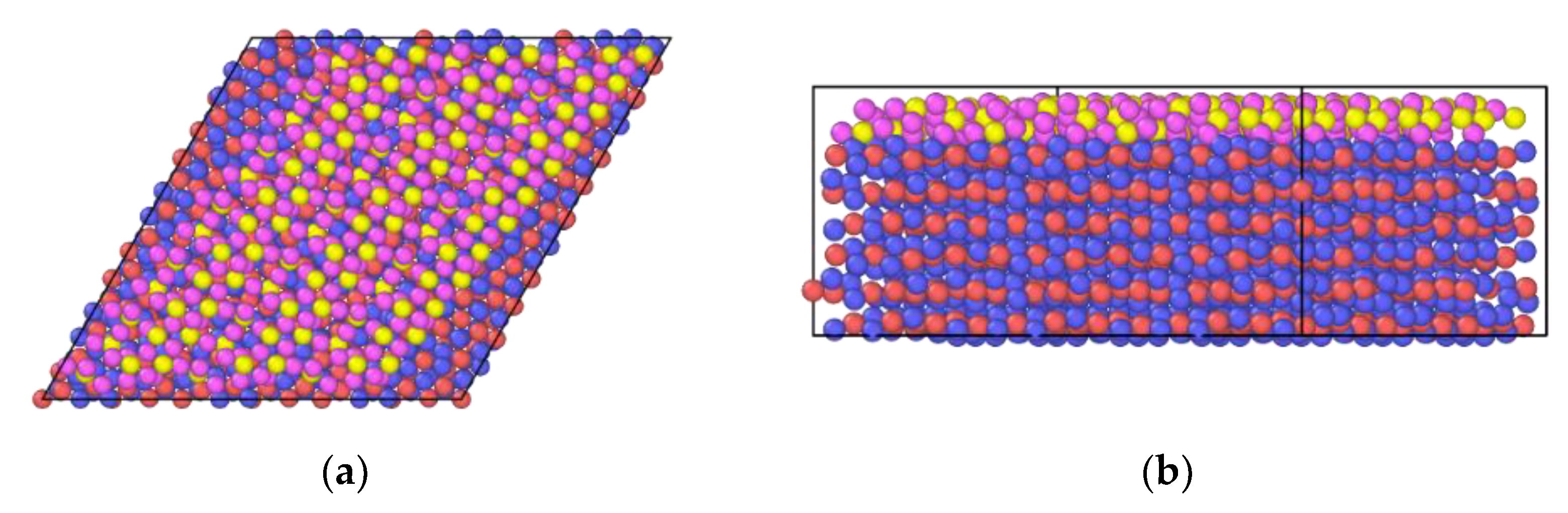
2.5. In2O3-ZrO2 Physical Mixture
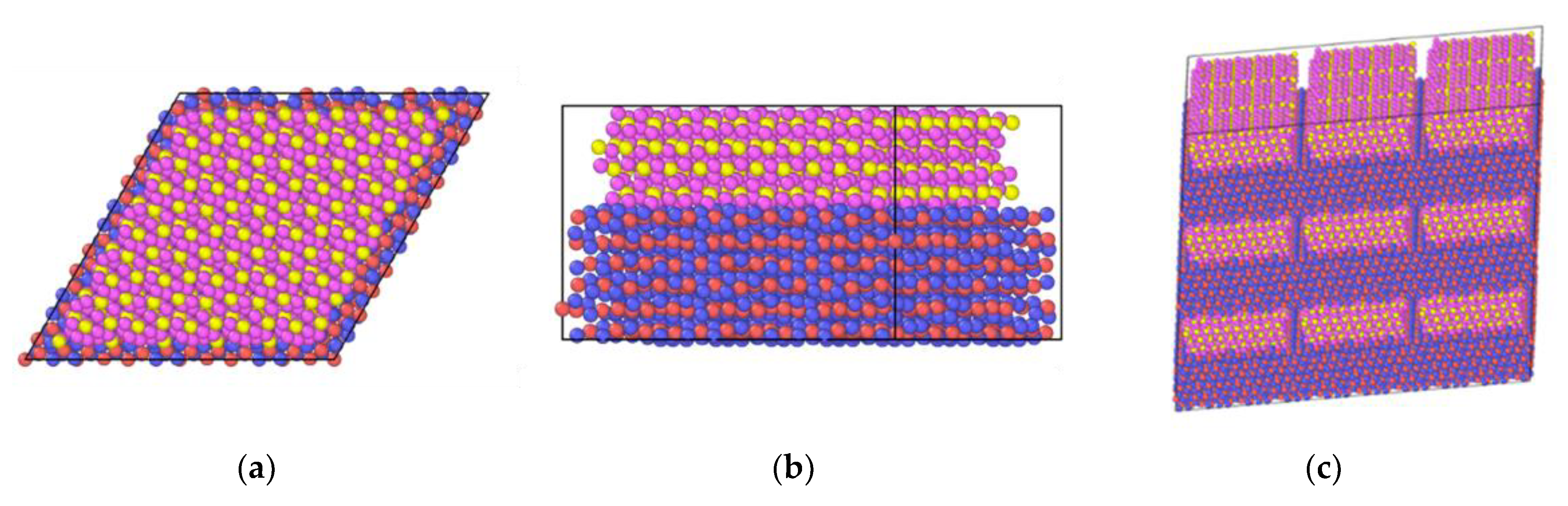
2.6. In2O3-ZrO2 Interface
2.7. In2O3-ZrO2 Physical Mixture—ZrO2 Monolayer
2.8. MSD and Diffusion
2.9. Defect Formation
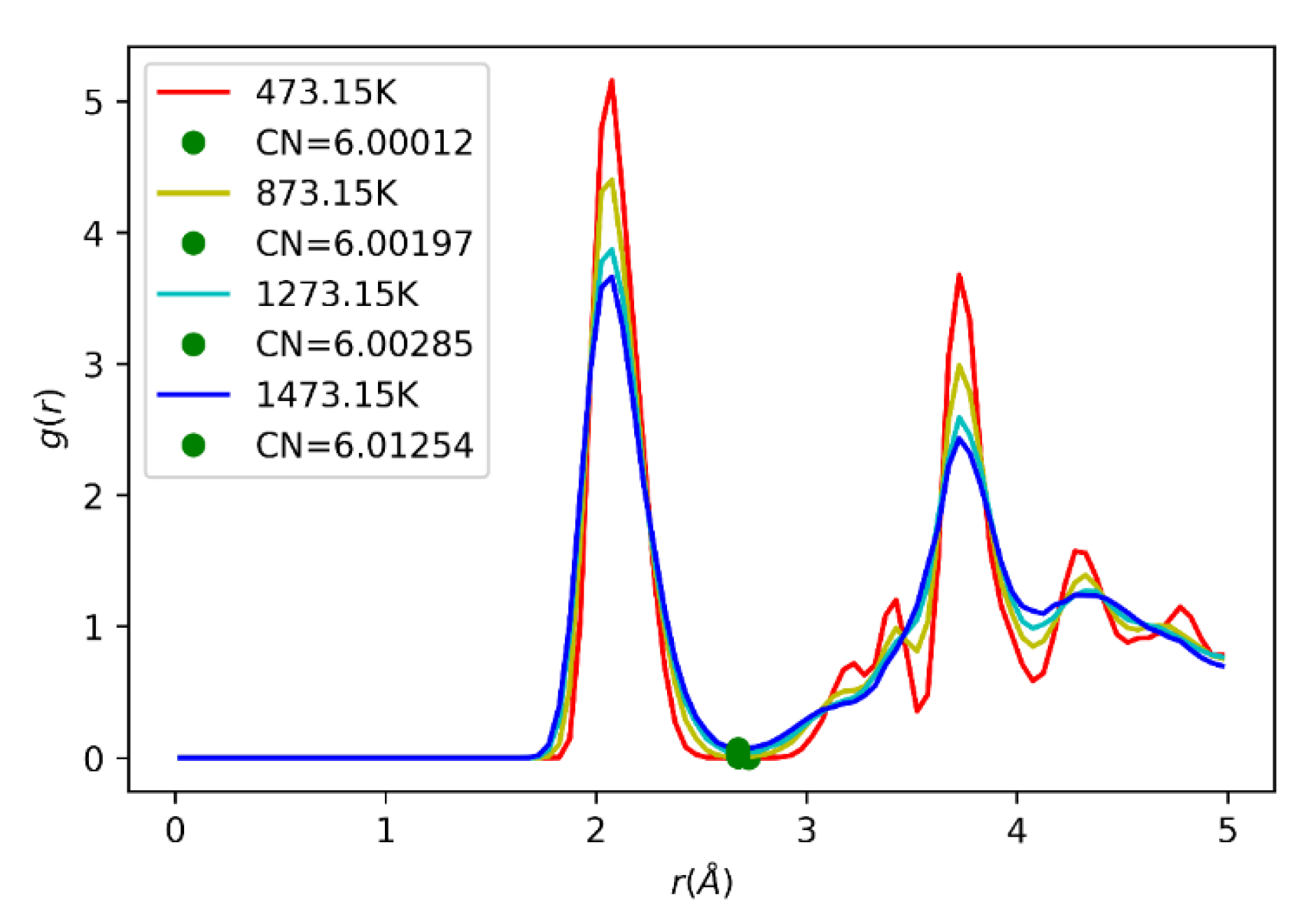
2.10. Radial Distribution Function (RDF)
3. Results and Discussion
3.1. In2O3 Model Verification and Selection
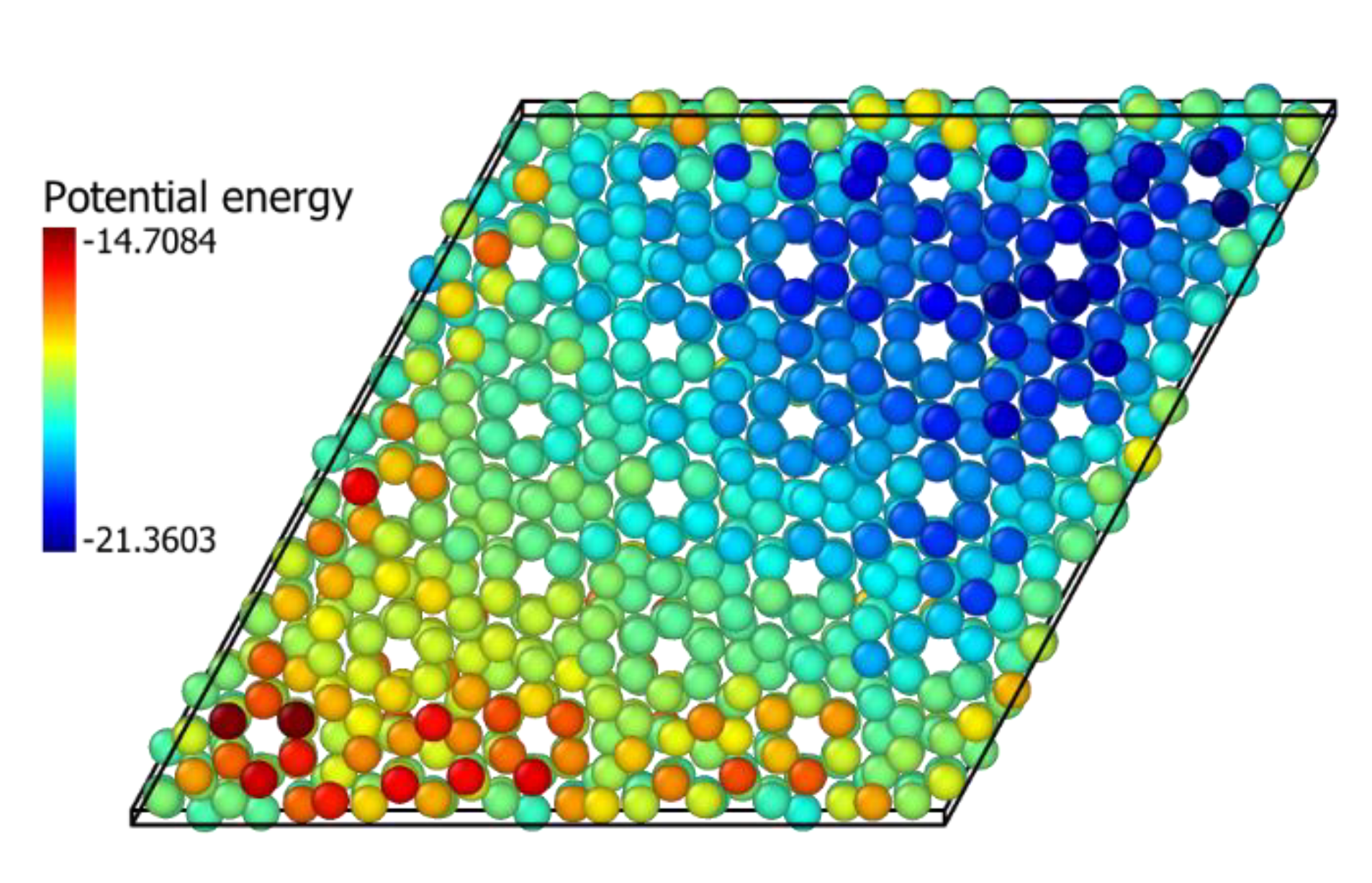
3.2. In2O3 Defect Energy
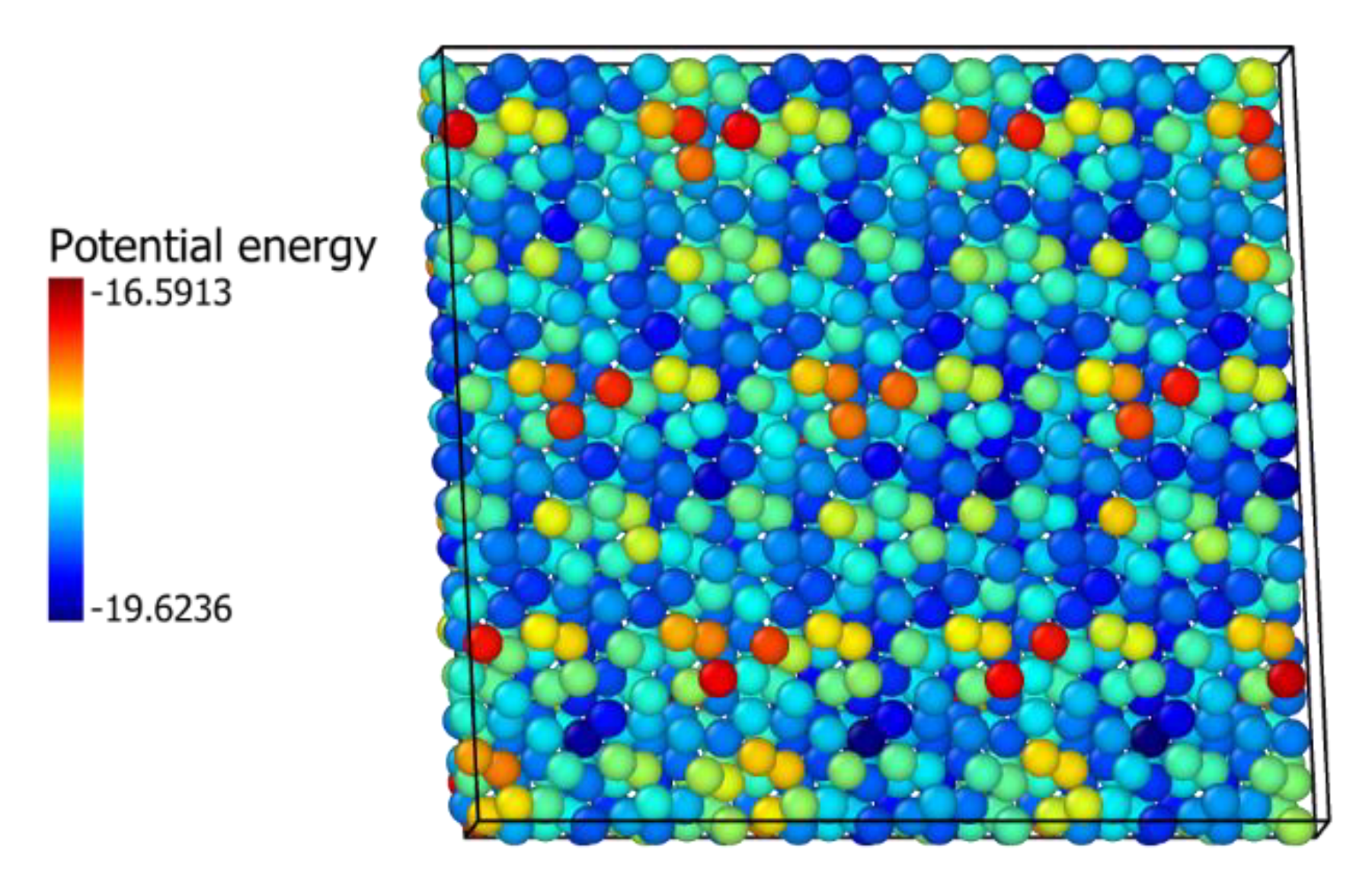
3.3. ZrO2 Model Selection
3.4. Defective In2O3 and ZrO2
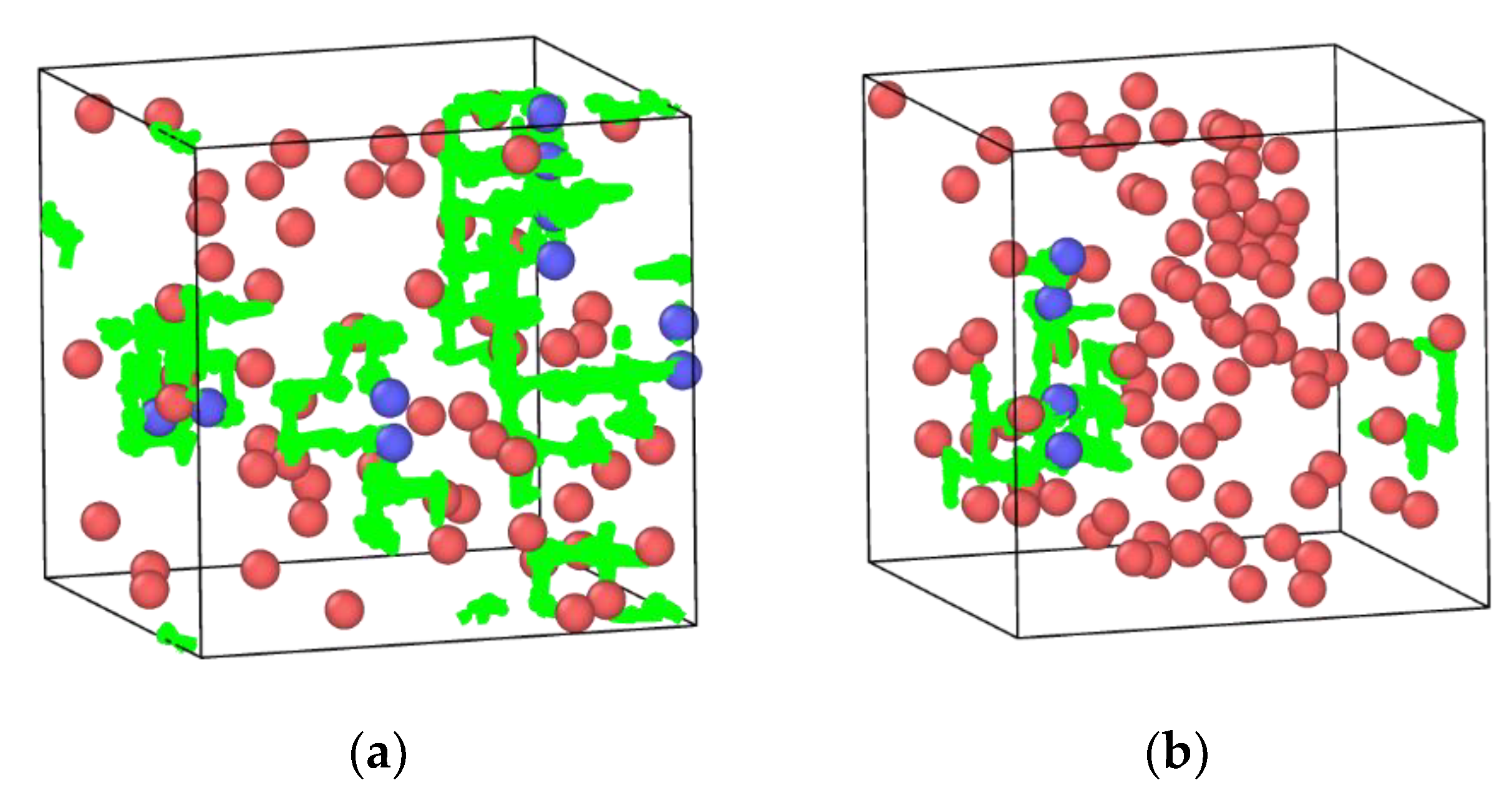
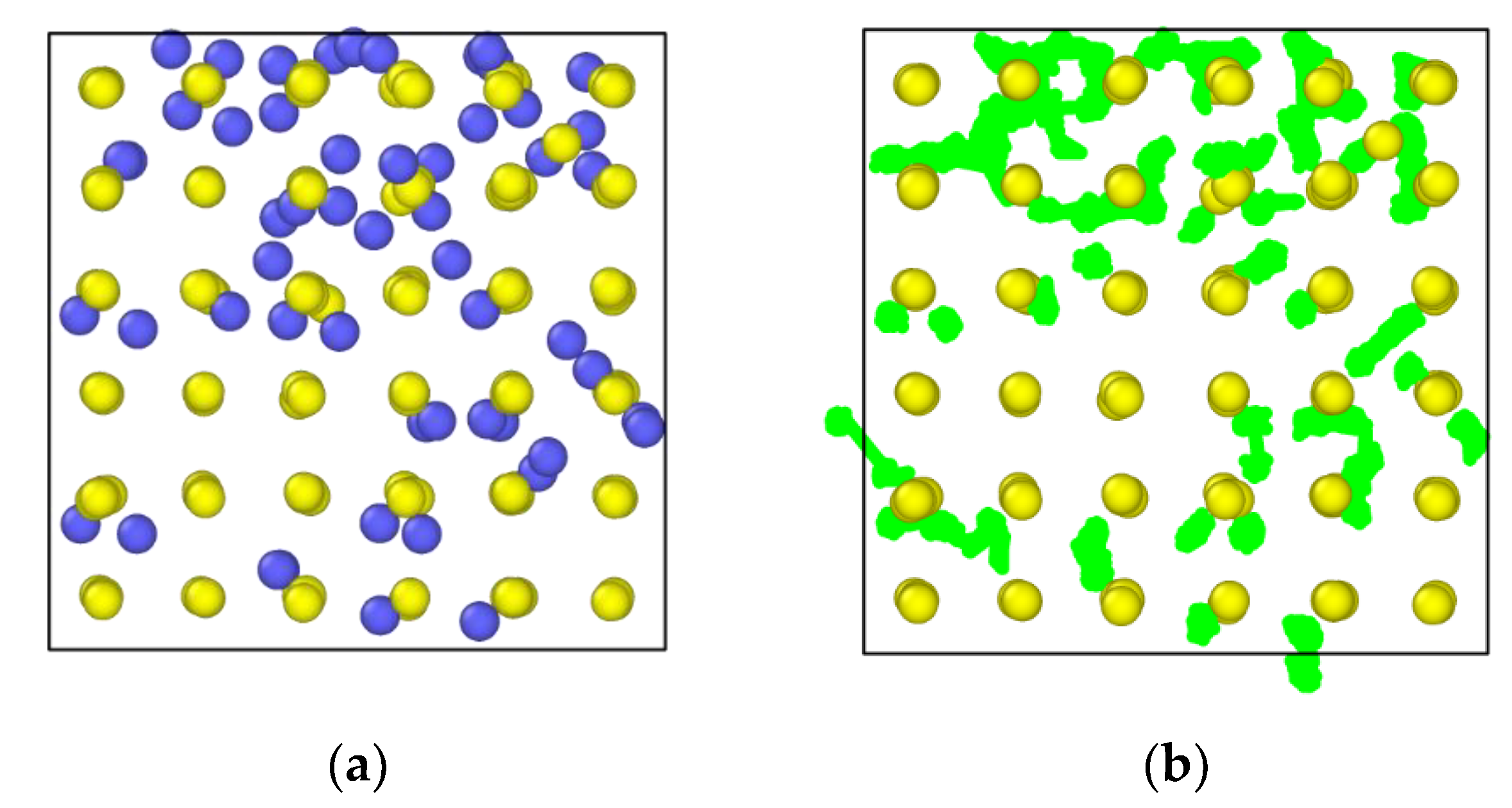
3.5. In2O3-ZrO2 Physical Mixture
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Walsh, A.; Catlow, C.R.A.; Sokol, A.A.; Woodley, S.M. Physical Properties, Intrinsic Defects, and Phase Stability of Indium Sesquioxide. Chem. Mater. 2009, 21, 4962–4969. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, G.; Zhu, J.; Zhang, X.; Ding, F.; Zhang, A.; Guo, X.; Song, C. CO2 Hydrogenation to Methanol over In2O3-Based Catalysts: From Mechanism to Catalyst Development. ACS Catal. 2021, 11, 1406–1423. [Google Scholar] [CrossRef]
- Wang, J.; Liu, C.Y.; Senftle, T.P.; Zhu, J.; Zhang, G.; Guo, X.; Song, C. Variation in the In2O3 Crystal Phase Alters Catalytic Performance toward the Reverse Water Gas Shift Reaction. ACS Catal. 2020, 10, 3264–3273. [Google Scholar] [CrossRef]
- Martin, O.; Martín, A.J.; Mondelli, C.; Mitchell, S.; Segawa, T.F.; Hauert, R.; Drouilly, C.; Curulla-Ferré, D.; Pérez-Ramírez, J. Indium Oxide as a Superior Catalyst for Methanol Synthesis by CO2 Hydrogenation. Angew. Chemie Int. Ed. 2016, 55, 6261–6265. [Google Scholar] [CrossRef] [PubMed]
- Zonetti, P.C.; Bridi, V.L.; Gonzalez, G.G.; Moreira, C.R.; Alves, O.C.; de Avillez, R.R.; Appel, L.G. Isobutene from Ethanol: Describing the Synergy between In2O3 and m-ZrO2. ChemCatChem 2019, 11, 4011–4020. [Google Scholar] [CrossRef]
- Bronsato, B.J.d.S.; Zonetti, P.C.; Moreira, C.R.; Mendoza, C.D.; Maia da Costa, M.E.H.; Alves, O.C.; de Avillez, R.R.; Appel, L.G. How the Interaction between In2O3-ZrO2 Promotes the Isobutene Synthesis from Ethanol? Catal. Today 2020, 381, 224–233. [Google Scholar] [CrossRef]
- Frei, M.S.; Mondelli, C.; Cesarini, A.; Krumeich, F.; Hauert, R.; Stewart, J.A.; Curulla Ferré, D.; Pérez-Ramírez, J. Role of Zirconia in Indium Oxide-Catalyzed CO2 Hydrogenation to Methanol. ACS Catal. 2020, 10, 1133–1145. [Google Scholar] [CrossRef]
- Chen, Y.; Zhai, Z.; Liu, J.; Zhang, J.; Geng, Z.; Lyu, H. The Synergistic Effects of Cu Clusters and In2O3 on Ethanol Synthesis from Acetic Acid Hydrogenation. Phys. Chem. Chem. Phys. 2019, 21, 23906–23915. [Google Scholar] [CrossRef]
- Chen, Y.; Li, G.; Zhang, M.; Jiang, H.; Lyu, H.; Liu, J. Effect of Surface Oxygen Vacancy Sites on Ethanol Synthesis from Acetic Acid Hydrogenation on a Defective In2O3 (110) Surface. Phys. Chem. Chem. Phys. 2018, 20, 7156–7166. [Google Scholar]
- Doornkamp, C.; Ponec, V. The Universal Character of the Mars and Van Krevelen Mechanism. J. Mol. Catal. A Chem. 2000, 162, 19–32. [Google Scholar] [CrossRef]
- Chen, B.W.J.; Xu, L.; Mavrikakis, M. Computational Methods in Heterogeneous Catalysis. Chem. Rev. 2021, 121, 1007–1048. [Google Scholar] [CrossRef]
- Dou, M.; Zhang, M.; Chen, Y.; Yu, Y. Mechanistic Insight into the Modification of the Surface Stability of In2O3 Catalyst Through Metal Oxide Doping. Catal. Lett. 2018, 148, 3723–3731. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, W.; Chen, Y. Insight of DFT and Ab Initio Atomistic Thermodynamics on the Surface Stability and Morphology of In2O3. Appl. Surf. Sci. 2018, 434, 1344–1352. [Google Scholar] [CrossRef]
- Walsh, A.; Catlow, C.R.A. Structure, Stability and Work Functions of the Low Index Surfaces of Pure Indium Oxide and Sn-Doped Indium Oxide (ITO) from Density Functional Theory. J. Mater. Chem. 2010, 20, 10438–10444. [Google Scholar] [CrossRef]
- Ziemba, M.; Schumacher, L.; Hess, C. Reduction Behavior of Cubic In2O3 Nanoparticles by Combined Multiple in Situ Spectroscopy and DFT. J. Phys. Chem. Lett. 2021, 12, 3749–3754. [Google Scholar] [CrossRef]
- Walsh, A.; Sokol, A.A.; Catlow, C.R.A. Free Energy of Defect Formation: Thermodynamics of Anion Frenkel Pairs in Indium Oxide. Phys. Rev. B-Condens. Matter Mater. Phys. 2011, 83, 224105. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Cao, A.; Wang, Z.; Li, H. Relations between Surface Oxygen Vacancies and Activity of Methanol Formation from CO2 Hydrogenation over In2O3 Surfaces. ACS Catal. 2021, 11, 1780–1786. [Google Scholar] [CrossRef]
- Warschkow, O.; Ellis, D.E.; González, G.B.; Mason, T.O. Defect Structures of Tin-Doped Indium Oxide. J. Am. Ceram. Soc. 2003, 86, 1700–1706. [Google Scholar] [CrossRef]
- Hou, Q.; Buckeridge, J.; Lazauskas, T.; Mora-Fonz, D.; Sokol, A.A.; Woodley, S.M.; Catlow, C.R.A. Defect Formation in In2O3 and SnO2: A New Atomistic Approach Based on Accurate Lattice Energies. J. Mater. Chem. C 2018, 6, 12386–12395. [Google Scholar] [CrossRef]
- Walsh, A.; Woodley, S.M.; Catlow, C.R.A.; Sokol, A.A. Potential Energy Landscapes for Anion Frenkel-Pair Formation in Ceria and India. Solid State Ion. 2011, 184, 52–56. [Google Scholar] [CrossRef]
- Bush, T.S.; Gale, J.D.; Catlow, C.R.A.; Battle, P.D. Self-Consistent Interatomic Potentials for the Simulation of Binary and Ternary Oxides. J. Mater. Chem. 1994, 4, 831–837. [Google Scholar] [CrossRef]
- Schelling, P.K.; Phillpot, S.R. Mechanism of Thermal Transport in Zirconia and Yttria-Stabilized Zirconia by Molecular-Dynamics Simulation. J. Am. Ceram. Soc. 2001, 84, 2997–3007. [Google Scholar] [CrossRef]
- Kilo, M.; Argirusis, C.; Borchardt, G.; Jackson, R.A. Oxygen Diffusion in Yttria Stabilised Zirconia—Experimental Results and Molecular Dynamics Calculations. Phys. Chem. Chem. Phys. 2003, 5, 2219–2224. [Google Scholar] [CrossRef]
- Lau, K.C.; Dunlap, B.I. Molecular Dynamics Simulation of Yttria-Stabilized Zirconia (YSZ) Crystalline and Amorphous Solids. J. Phys. Condens. Matter 2011, 23, 035401. [Google Scholar] [CrossRef]
- Van Duin, A.C.T.; Merinov, B.V.; Han, S.S.; Dorso, C.O.; Goddard, W.A. ReaxFF Reactive Force Field for the Y-Doped BaZrO3 Proton Conductor with Applications to Diffusion Rates for Multigranular Systems. J. Phys. Chem. A 2008, 112, 11414–11422. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, R.A. The Classical Equation of State of Gaseous Helium, Neon and Argon. Proc. R. Soc. London Ser. A. Math. Phys. Sci. 1938, 168, 264–283. [Google Scholar]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; in’t Veld Pieter, J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS- a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Larsen, A.H.; Mortensen, J.J.; Blomqvist, J.; Castelli, I.E.; Christensen, R.; Dułak, M.; Friis, J.; Groves, M.N.; Hammer, B.; Hargus, C.; et al. The Atomic Simulation Environment—A Python Library for Working with Atoms. J. Phys. Energy 2017, 29, 273002. [Google Scholar]
- Daoud, S.; Loucif, K.; Bioud, N.; Lebgaa, N. First-Principles Study of Structural, Elastic and Mechanical Properties of Zinc-Blende Boron Nitride (B3-BN). Acta Phys. Pol. A 2012, 122, 109–115. [Google Scholar] [CrossRef]
- Murnaghan, F.D. The Compressibility of Media Under Extreme Pressures. Proc. Natl. Acad. Sci. USA 1944, 30, 244–247. [Google Scholar] [CrossRef]
- Aroyo, M.I.; Perez-Mato, J.M.; Capillas, C.; Kroumova, E.; Ivantchev, S.; Madariaga, G.; Kirov, A.; Wondratschek, H. Bilbao Crystallographic Server: I. Databases and Crystallographic Computing Programs. Zeitschrift fur Krist. 2006, 221, 15–27. [Google Scholar] [CrossRef]
- Wyckoff Positions. Available online: https://www.cryst.ehu.es/cryst/get_wp.html (accessed on 3 December 2021).
- Bandura, A.V.; Lukyanov, S.I.; Evarestov, R.A. Atom–Atom Force Field for Simulation of Zirconia Bulk, Nanosheets and Nanotubes. Mol. Simul. 2017, 43, 886–899. [Google Scholar] [CrossRef]
- Lewis, G.V.; Catlow, C.R.A. Potential models for ionic oxides. J. Phys. C Solid State Phys. 1985, 18, 1149–1161. [Google Scholar] [CrossRef]
- Lee, J.G. Computational Materials Science: An Introduction, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2017; p. 43. [Google Scholar]
- Stukowski, A. Visualization and Analysis of Atomistic Simulation Data with OVITO-the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Marezio, M. Refinement of the Crystal Structure of In2O3 at Two Wavelengths. Acta Crystallogr. 1966, 20, 723–728. [Google Scholar] [CrossRef]
- Liu, D.; Lei, W.W.; Zou, B.; Yu, S.D.; Hao, J.; Wang, K.; Liu, B.B.; Cui, Q.L.; Zou, G.T. High-Pressure x-Ray Diffraction and Raman Spectra Study of Indium Oxide. J. Appl. Phys. 2008, 104, 083506. [Google Scholar] [CrossRef]
- Weiher, R.L.; Ley, R.P. Thermal Expansion of Indium Oxide. J. Appl. Phys. 1963, 34, 1833–1834. [Google Scholar] [CrossRef]
- Kundra, K.D.; S. Z. Ali. Thermal Expansion of In2O3. J. Appl. Cryst. 1970, 543–545. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Morishita, K.; Iwakiri, H.; Kaneta, Y. Stress Dependence of Oxygen Diffusion in ZrO2 Film. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2013, 303, 42–45. [Google Scholar] [CrossRef]
- Liu, C.; Sun, J.; Smith, C.; Wang, Y. A Study of ZnxZryOz Mixed Oxides for Direct Conversion of Ethanol to Isobutene. Appl. Catal. A Gen. 2013, 467, 91–97. [Google Scholar] [CrossRef]
- Sun, K.; Shen, C.; Zou, R.; Liu, C.J. Highly active Pt/In2O3-ZrO2 catalyst for CO2 hydrogenation to methanol with enhanced CO tolerance: The effects of ZrO2. Appl. Catal. B: Environ. 2023, 320, 122018. [Google Scholar] [CrossRef]
- Silva-Calpa, L.D.; Zonetti, P.C.; de Oliveira, D.C.; de Avillez, R.R.; Appel, L.G. Acetone from Ethanol Employing ZnxZr1−xO2−y. Catal. Today 2017, 289, 264–272. [Google Scholar] [CrossRef]
- Sun, J.; Baylon, R.A.L.; Liu, C.; Mei, D.; Martin, K.J.; Venkitasubramanian, P.; Wang, Y. Key Roles of Lewis Acid-Base Pairs on ZnxZryOz in Direct Ethanol/Acetone to Isobutene Conversion. J. Am. Chem. Soc. 2016, 138, 507–517. [Google Scholar] [CrossRef] [PubMed]
- McFarland, E.W.; Metiu, H. Catalysis by Doped Oxides. Chem. Rev. 2013, 113, 4391–4427. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
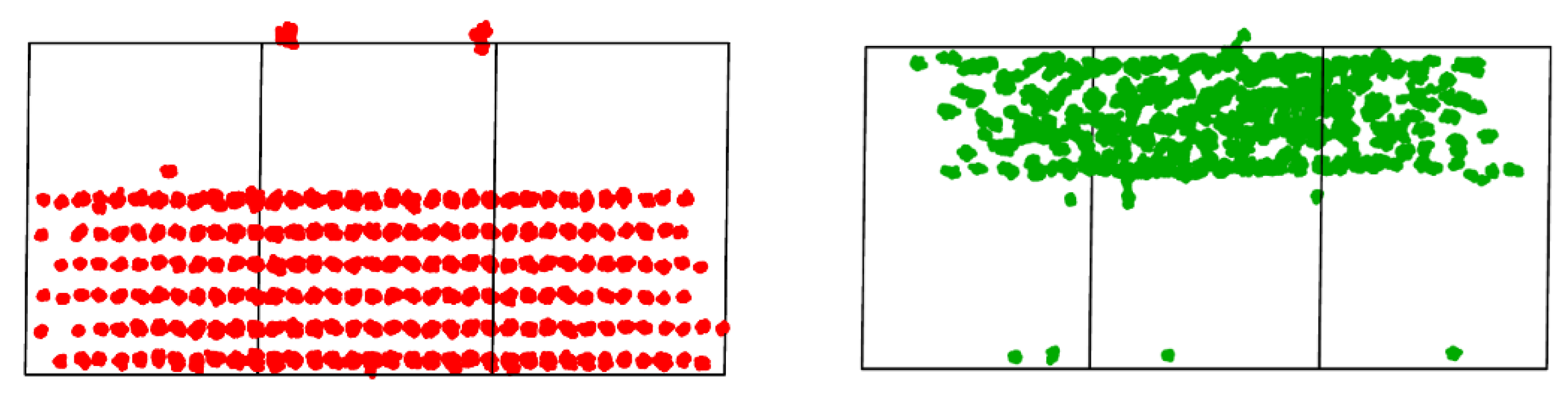{kind=link}
| A (eV) | ρ (Å) | C (eV Å6) | Reference | |
|---|---|---|---|---|
| In+3—O−2 | 1498.651 | 0.3405 | 33.2 | [1] |
| Zr+4—O−2 | 1024.600 | 0.3760 | 0.0 | [23] |
| O−2—O−2 | 22,764.000 | 0.1490 | 32.0 | [1] |
| Walsh’s Potential | Warschkow’s Potential | ||||||
|---|---|---|---|---|---|---|---|
| Properties | Experiment | Reference [1] | Core-Shell Model Results | Rigid Ion Model Results | Reference | Core-Shell Model Results | Rigid Ion Model Results |
| Lattice parameter (Å) | [37] 10.117 | 10.1210 | 10.0502 | 10.1188 | 10.1200 | 10.4186 | 10.1182 |
| Bulk Modulus (GPa) | [38] 194.24 | 193.7700 | 204.9747 | 196.8721 | 222.7900 | 179.8271 | 226.5402 |
| Linear expansion coefficient, α (°C−1) | [39] 6.15 × 10−6 | NA | 5.25 × 10−6 | 6.14 × 10−6 | NA | 1.25 × 10−5 | 7.22 × 10−6 |
| [40] 7.5 × 10−6 | |||||||
| A (eV) | ρ (Å) | C (eV Å6) | Reference | |
|---|---|---|---|---|
| In2O3 crystal | ||||
| In+3—O−2 | 1498.651 | 0.340500 | 33.2000 | [1] |
| O−2—O−2 | 22,764.000 | 0.149000 | 32.0000 | [1] |
| In+3—O−2 | 2719.770 | 0.291700 | 0.0000 | [18] |
| O−2—O−2 | 25.410 | 0.693700 | 32.3200 | [18] |
| ZrO2 crystal | ||||
| Zr+4—O−2 | 1024.600 | 0.376000 | 0.0000 | [23] |
| O−2—O−2 | 22,764.000 | 0.149000 | 27.8900 | [23] |
| Zr+4—O−2 | 1292.850 | 0.358388 | 19.3646 | [24] |
| O−2—O−2 | 13,098.900 | 0.219670 | 49.2998 | [24] |
| Zr+4—O−2 | 1502.110 | 0.3450 | 5.1000 | [22] |
| O−2—O−2 | 9547.960 | 0.2240 | 32.0000 | [22] |
| 3 × 3 × 3 Supercell | 4 × 4 × 4 Supercell | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Reference | Core-Shell Model | Rigid Ion Model | Core-Shell Model | Rigid Ion Model | |||||
| Defect Type | Walsh et al., 2009 [1] | Simulated Values | Difference from Reference | Simulated Values | Difference from Reference | Simulated Values | Difference from Reference | Simulated Values | Difference from Reference |
| Lattice energy | −140.60 | −141.40 | 0.6% | −140.57 | 0.0% | −141.40 | 0.6% | −140.57 | 0.0% |
| In vacancy (8b) | 49.92 | 49.82 | 0.2% | 50.08 | 0.3% | 50.21 | 0.6% | 50.31 | 0.8% |
| In vacancy (24d) | 50.05 | 50.42 | 0.7% | 50.99 | 1.9% | 50.72 | 1.3% | 51.22 | 2.3% |
| O vacancy | 20.99 | 21.71 | 3.4% | 21.13 | 0.7% | 21.89 | 4.3% | 21.23 | 1.1% |
| In intersticial (8a) | −33.57 | −34.71 | 3.4% | −33.99 | 1.3% | −34.38 | 2.4% | −33.76 | 0.6% |
| In intersticial (16c) | −36.21 | −35.75 | 1.3% | −35.48 | 2.0% | −35.41 | 2.2% | −35.27 | 2.6% |
| In intersticial (24d) | −34.89 | −34.09 | 2.3% | −33.41 | 4.2% | −33.74 | 3.3% | −33.08 | 5.2% |
| O intersticial (8a) | −13.29 | −14.20 | 6.8% | −13.13 | 1.2% | −14.03 | 5.6% | −13.03 | 2.0% |
| O intersticial (16c) | −14.61 | −15.43 | 5.6% | −14.54 | 0.5% | −15.25 | 4.4% | −14.44 | 1.2% |
| O intersticial (24d) | −12.08 | −13.03 | 7.9% | −11.70 | 3.1% | −12.84 | 6.3% | −11.60 | 4.0% |
| cation Frenkel pair | 6.85 | 7.04 | 2.8% | 7.40 | 8.0% | 7.40 | 8.0% | 6.48 | 5.4% |
| anion Frenkel pair | 3.19 | 3.14 | 1.6% | 3.30 | 3.4% | 3.32 | 4.1% | 3.43 | 7.5% |
| Schottky defect. | 4.44 | 4.67 | 5.2% | 4.58 | 3.2% | 4.94 | 11.3% | 4.71 | 6.1% |
| Differences mean | 3.2% | 2.3% | 4.2% | 3.0% | |||||
| Schelling | Kilo | Lewis-Catlow | Lau-Dunlap | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Properties | Experimental Values for monoclinic ZrO2 | Simulated Values | Difference from Experiment | Simulated Values | Difference from Experiment | Simulated Values | Difference from Experiment | Simulated Values | Difference from Experiment |
| a (Å) | 5.149 | 5.234 | 1.7% | 5.291 | 2.8% | 4.954 | 3.8% | 5.201 | 1.0% |
| b (Å) | 5.208 | 4.904 | 5.8% | 4.973 | 4.5% | 4.954 | 4.9% | 4.871 | 6.5% |
| c (Å) | 5.316 | 5.574 | 4.9% | 5.624 | 5.8% | 4.954 | 6.8% | 5.564 | 4.7% |
| β (°) | 99.23 | 90.00 | 9.3% | 89.99 | 9.3% | 90.00 | 9.3% | 90.00 | 9.3% |
| Bulk modulus (Gpa) | 201 | 235 | 16.9% | 196 | 2.5% | 327 | 62.7% | 236 | 17.4% |
| Mean difference | 7.7% | 5.0% | 17.5% | 7.8% | |||||
| Defects | Pseudo-Monoclinic ZrO2 | Cubic In2O3 |
|---|---|---|
| 3% Schottky | 1.0466 | 0.3626 |
| Frenkel | 1.5041 * | 0.4065 |
| Oxygen vacancies | 2.4801 | 0.5114 |
| 12% substitutional In | 0.3093 | |
| 18% substitutional In | 0.4974 | |
| 9% substitutional Zr | 1.3753 | |
| 22% substitutional Zr | 1.2548 |
| Defects | Pseudo-Monoclinic ZrO2 | Cubic In2O3 |
|---|---|---|
| 3% Schottky | 0.87% | 0.00% |
| Frenkel | 0.42% | 0.51% |
| Oxygen vacancies | 1.19% | 0.00% |
| 12% substitutional In | 0.54% | |
| 18% substitutional In | 0.45% | |
| 9% substitutional Zr | 0.67% | |
| 22% substitutional Zr | 1.90% |
| In2O3 Oxygen | ZrO2 Oxygen | |||
|---|---|---|---|---|
| Physical Mixture | No Defects | 3% Schottky | No Defects | 3% Schottky |
| Pseudo-monoclinic ZrO2(111) over cubic In2O3(111) | 2.0224 | 0.4331 | 0.5101 | 0.8796 |
| Pseudo-monoclinic ZrO2(102) over cubic In2O3(111) | 0.9851 | 0.4600 | 0.6161 | 0.7661 |
| ZrO2(211) monolayer over cubic In2O3(111) | 1.0464 | 0.9555 | 0.7971 | 0.6970 |
| Cubic In2O3(111) over pseudo-monoclinic ZrO2(111) | 0.4262 | 0.8195 | 0.8433 | 0.8307 |
| In2O3 Oxygen | ZrO2 Oxygen | |||
|---|---|---|---|---|
| Physical Mixture | No Defects | 3% Schottky | No Defects | 3% Schottky |
| Pseudo-monoclinic ZrO2(111) over cubic In2O3(111) | 1.02% | 1.61% | 11.55% | 13.17% |
| Pseudo-monoclinic ZrO2(102) over cubic In2O3(111) | 1.49% | 2.08% | 12.27% | 14.87% |
| ZrO2(211) monolayer over cubic In2O3(111) | 1.86% | 2.46% | 20.72% | 22.73% |
| Cubic In2O3(111) over pseudo-monoclinic ZrO2(111) | 5.89% | 7.52% | 3.19% | 4.12% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fornasari, L.E.; Bronsato, B.J.d.S.; Appel, L.G.; de Avillez, R.R. Molecular Dynamics Approach to the Physical Mixture of In2O3 and ZrO2: Defect Formation and Ionic Diffusion. Int. J. Mol. Sci. 2023, 24, 2426. https://doi.org/10.3390/ijms24032426
Fornasari LE, Bronsato BJdS, Appel LG, de Avillez RR. Molecular Dynamics Approach to the Physical Mixture of In2O3 and ZrO2: Defect Formation and Ionic Diffusion. International Journal of Molecular Sciences. 2023; 24(3):2426. https://doi.org/10.3390/ijms24032426
Chicago/Turabian StyleFornasari, Lorenzo E., Bruna J. da S. Bronsato, Lucia G. Appel, and Roberto R. de Avillez. 2023. "Molecular Dynamics Approach to the Physical Mixture of In2O3 and ZrO2: Defect Formation and Ionic Diffusion" International Journal of Molecular Sciences 24, no. 3: 2426. https://doi.org/10.3390/ijms24032426
APA StyleFornasari, L. E., Bronsato, B. J. d. S., Appel, L. G., & de Avillez, R. R. (2023). Molecular Dynamics Approach to the Physical Mixture of In2O3 and ZrO2: Defect Formation and Ionic Diffusion. International Journal of Molecular Sciences, 24(3), 2426. https://doi.org/10.3390/ijms24032426