
Characterization of the WRKY Gene Family Related to Anthocyanin Biosynthesis and the Regulation Mechanism under Drought Stress and Methyl Jasmonate Treatment in Lycoris radiata

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Identification and Characterization of LrWRKY Proteins in L. radiata
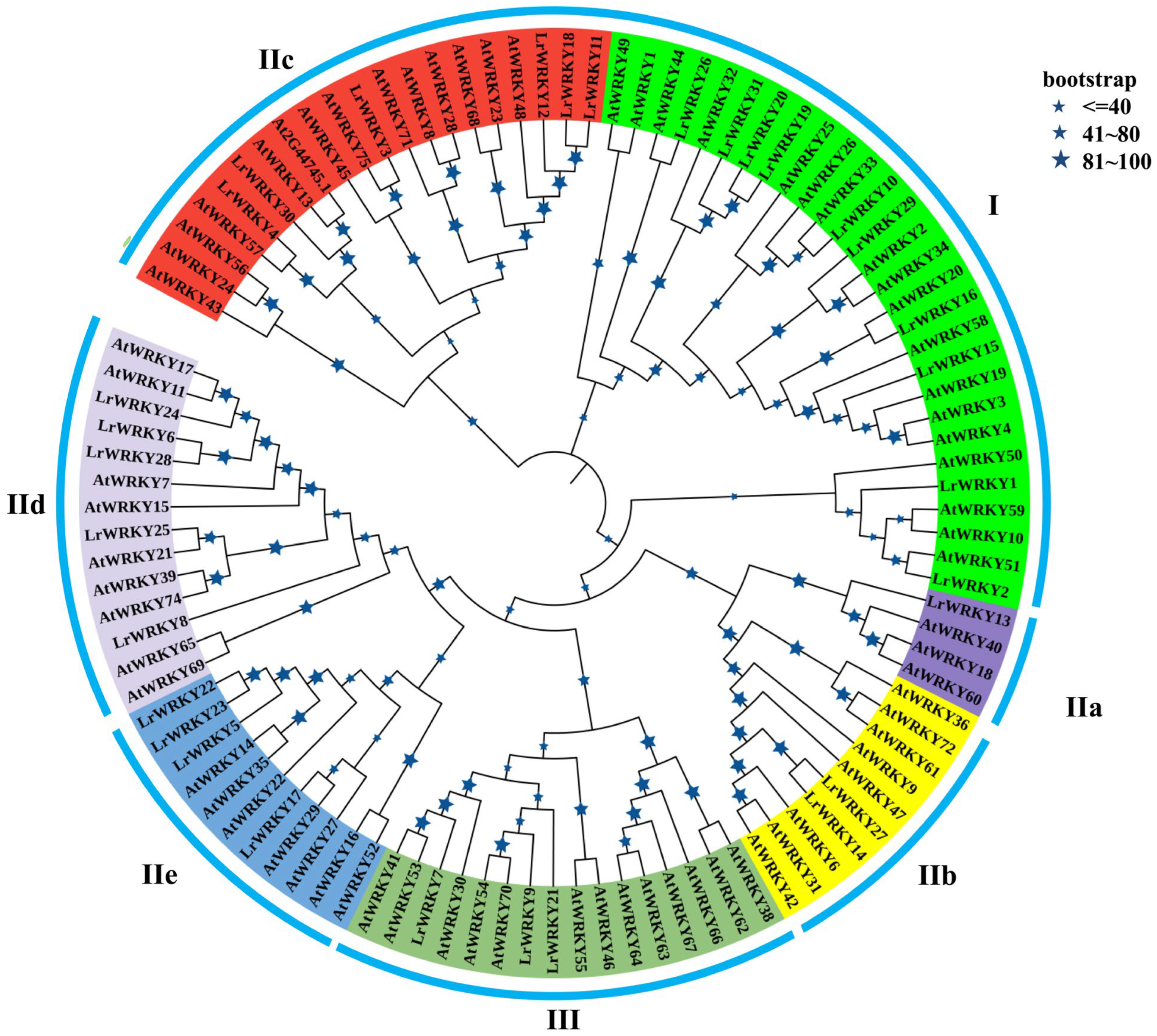
2.2. Phylogenetic Analysis and Classification of LrWRKY Proteins
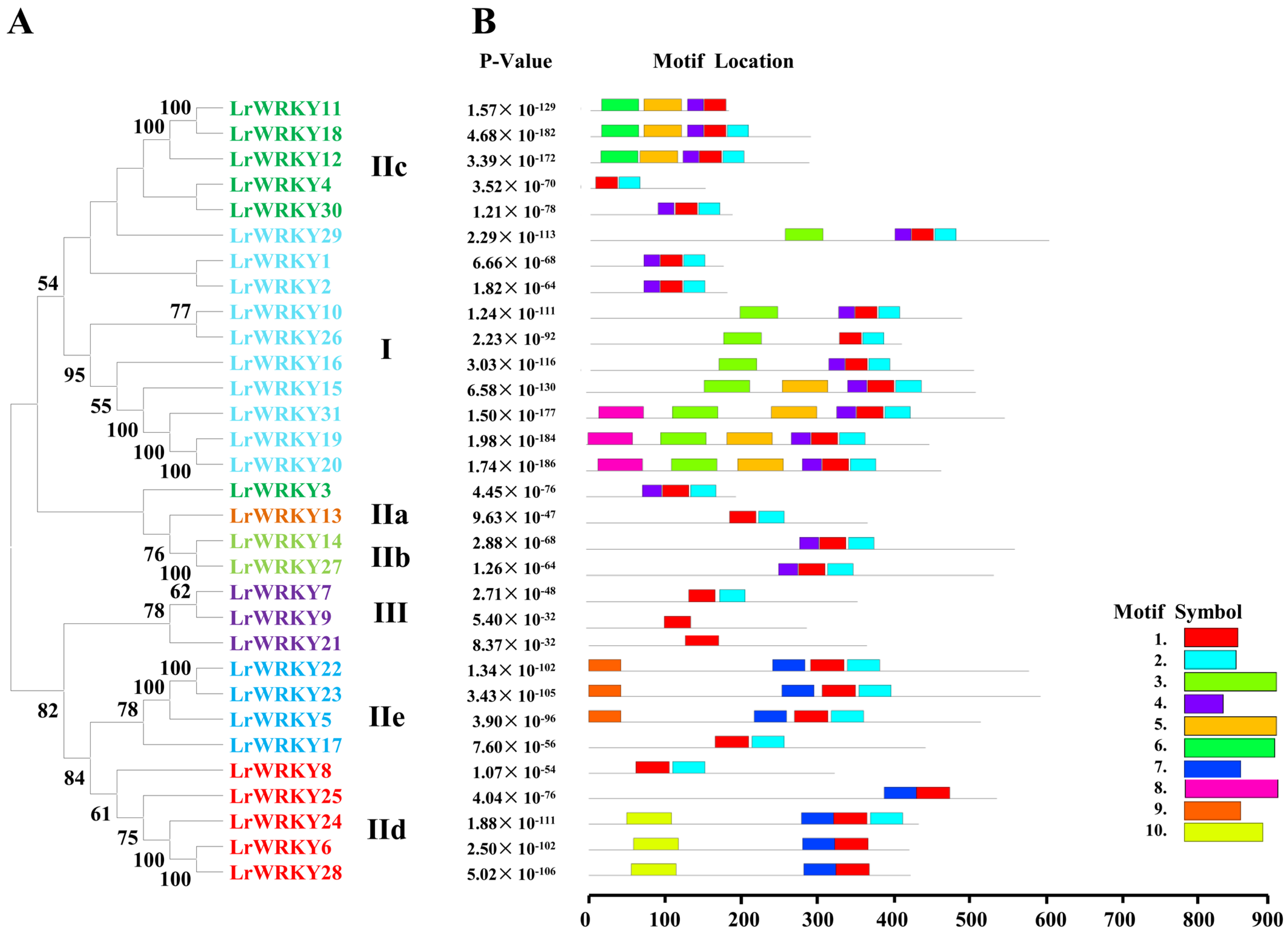
2.3. Multiple Sequence Alignment, Conserved Motifs, and WRKY Domains of LrWRKY Proteins
2.4. Multiple Sequence Alignment, Conserved Motifs, and WRKY Domains of LrWRKY Proteins
2.5. Multiple Sequence Alignment, Conserved Motifs, and WRKY Domains of LrWRKY Proteins
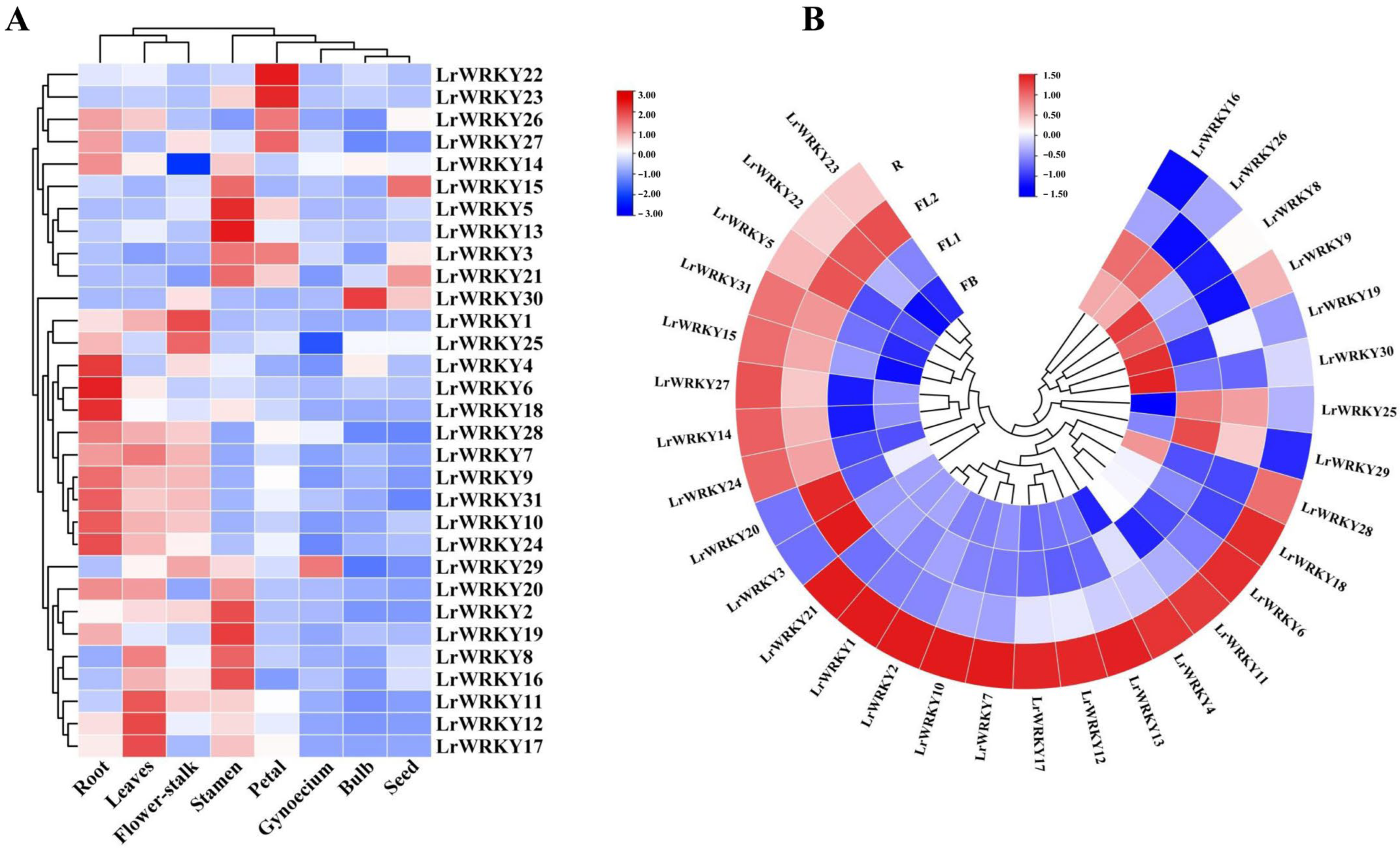
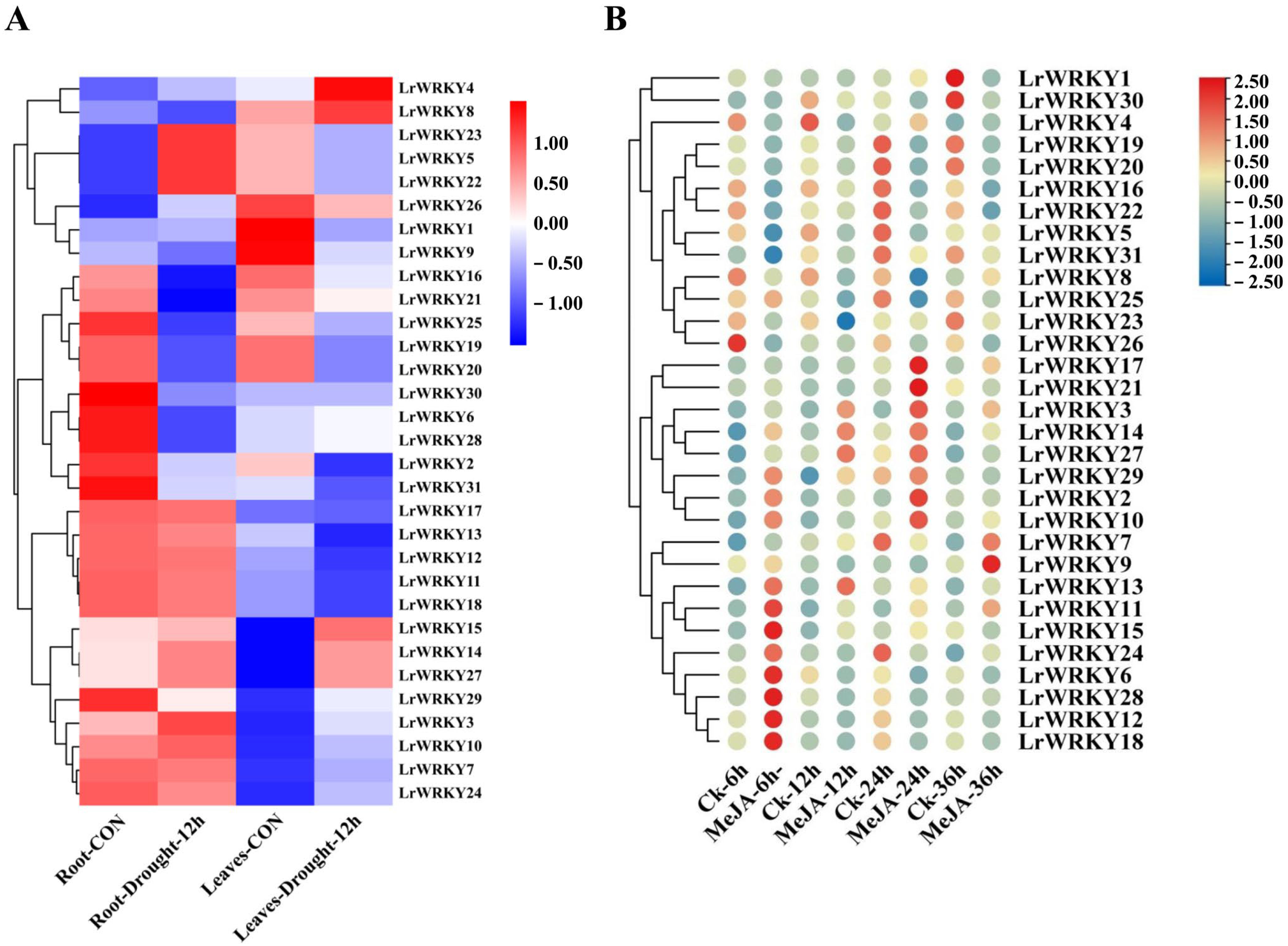
2.6. Expression Patterns of LrWRKYs in Response to Drought Stress and MeJA Treatment
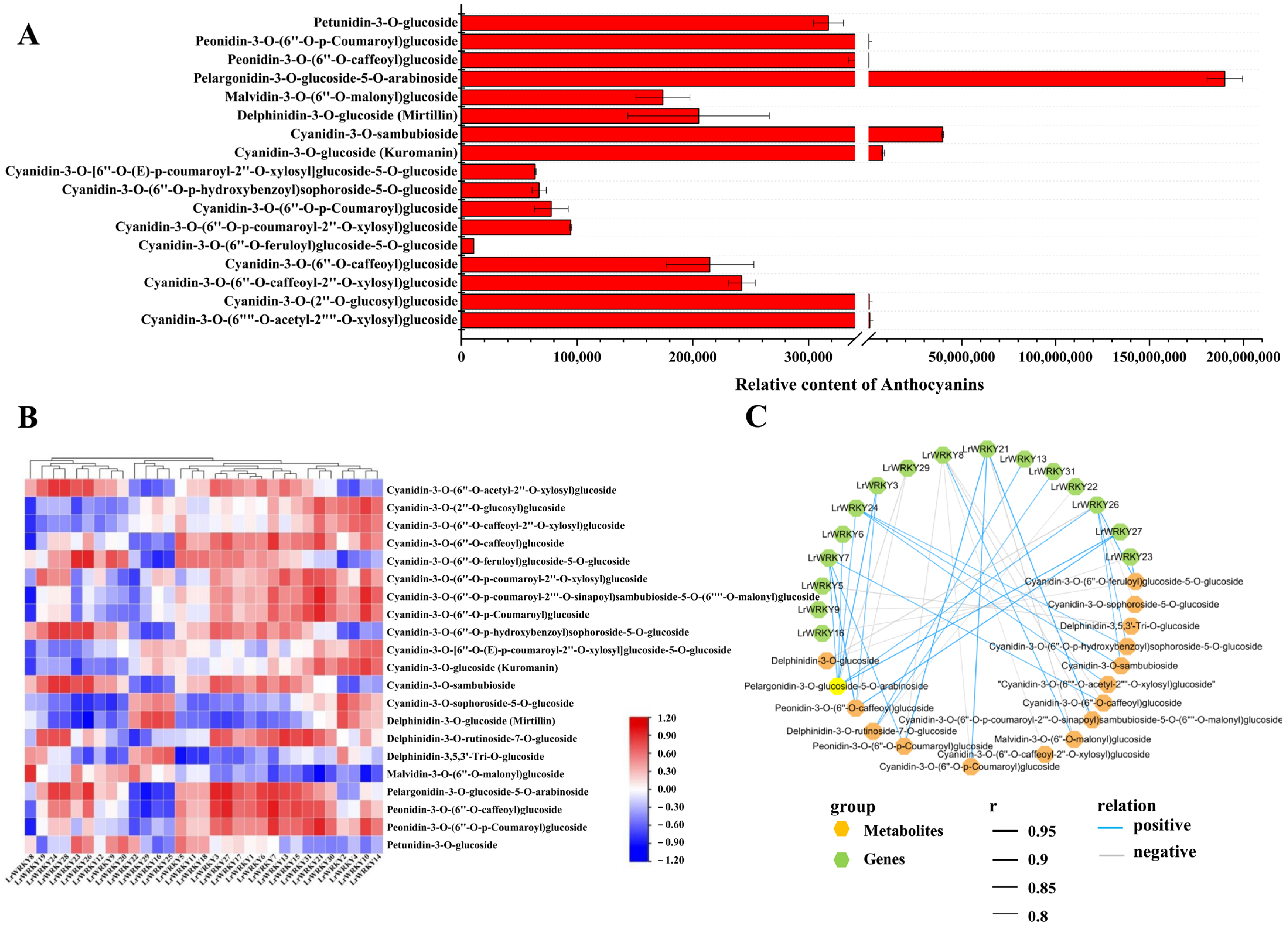
2.7. Identification of the Main Anthocyanin Pigments in L. radiata Petals
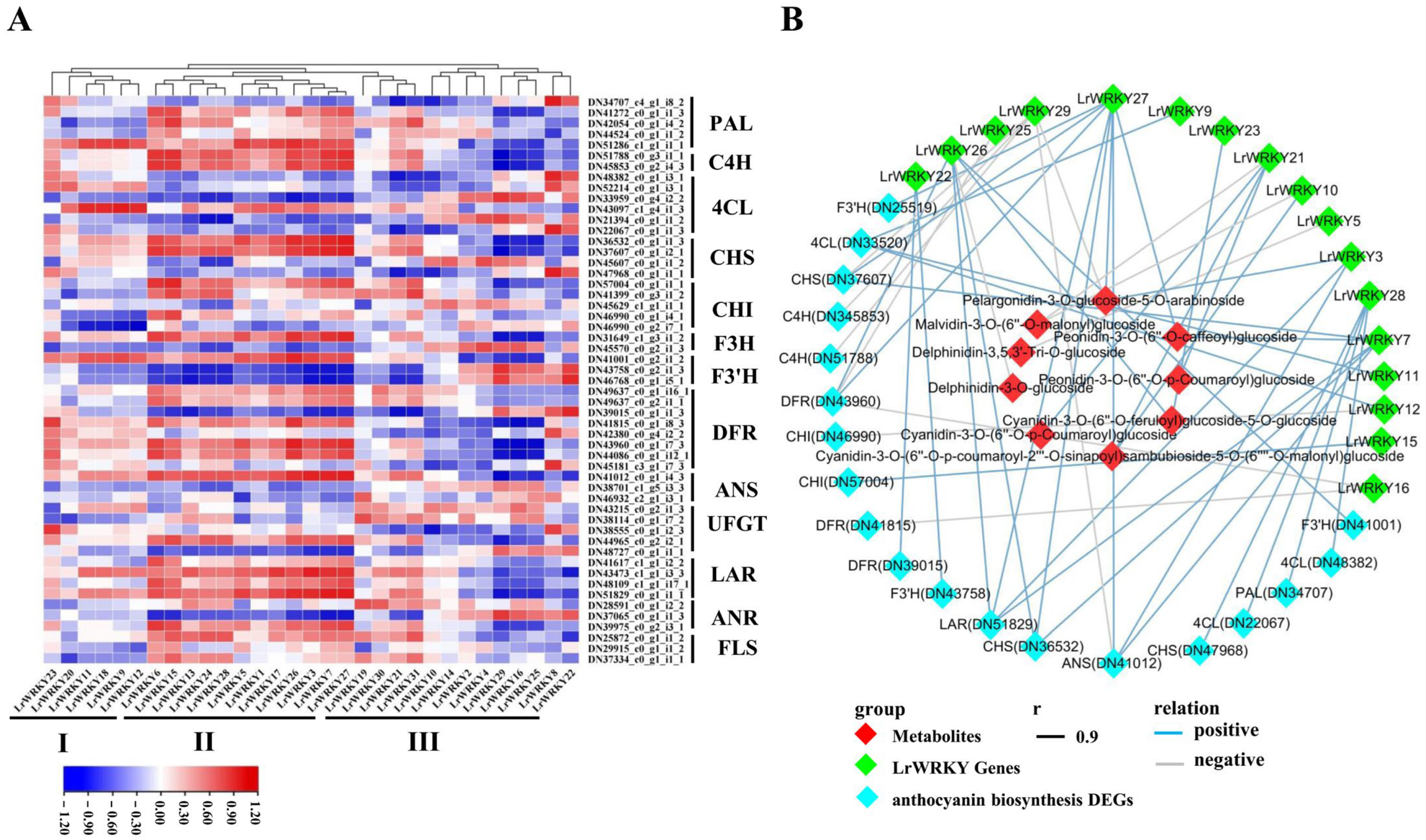
2.8. Integrating Related LrWRKY Genes and Metabolites in Anthocyanin Biosynthesis Pathway
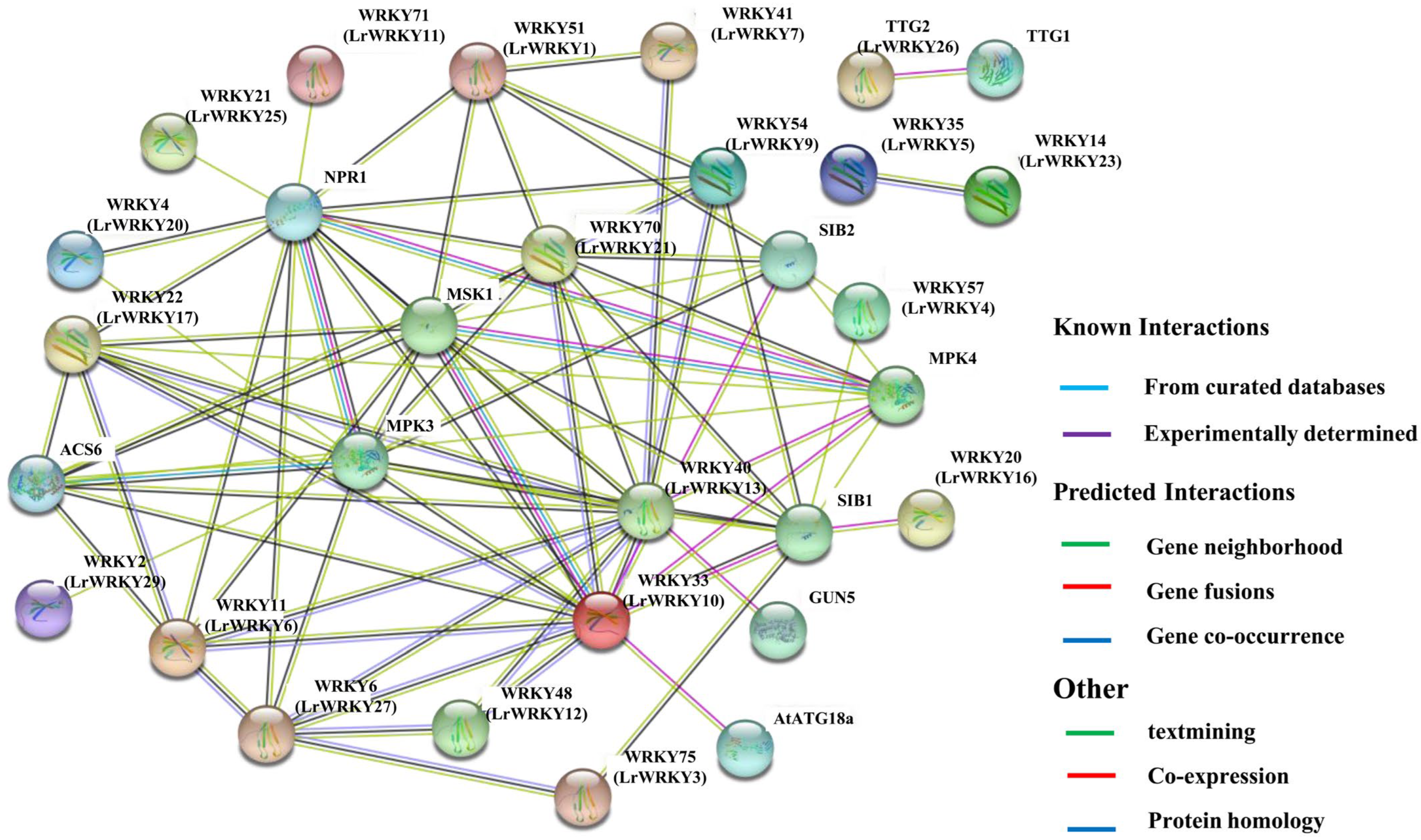
2.9. PPI Networks of LrWRKY Proteins
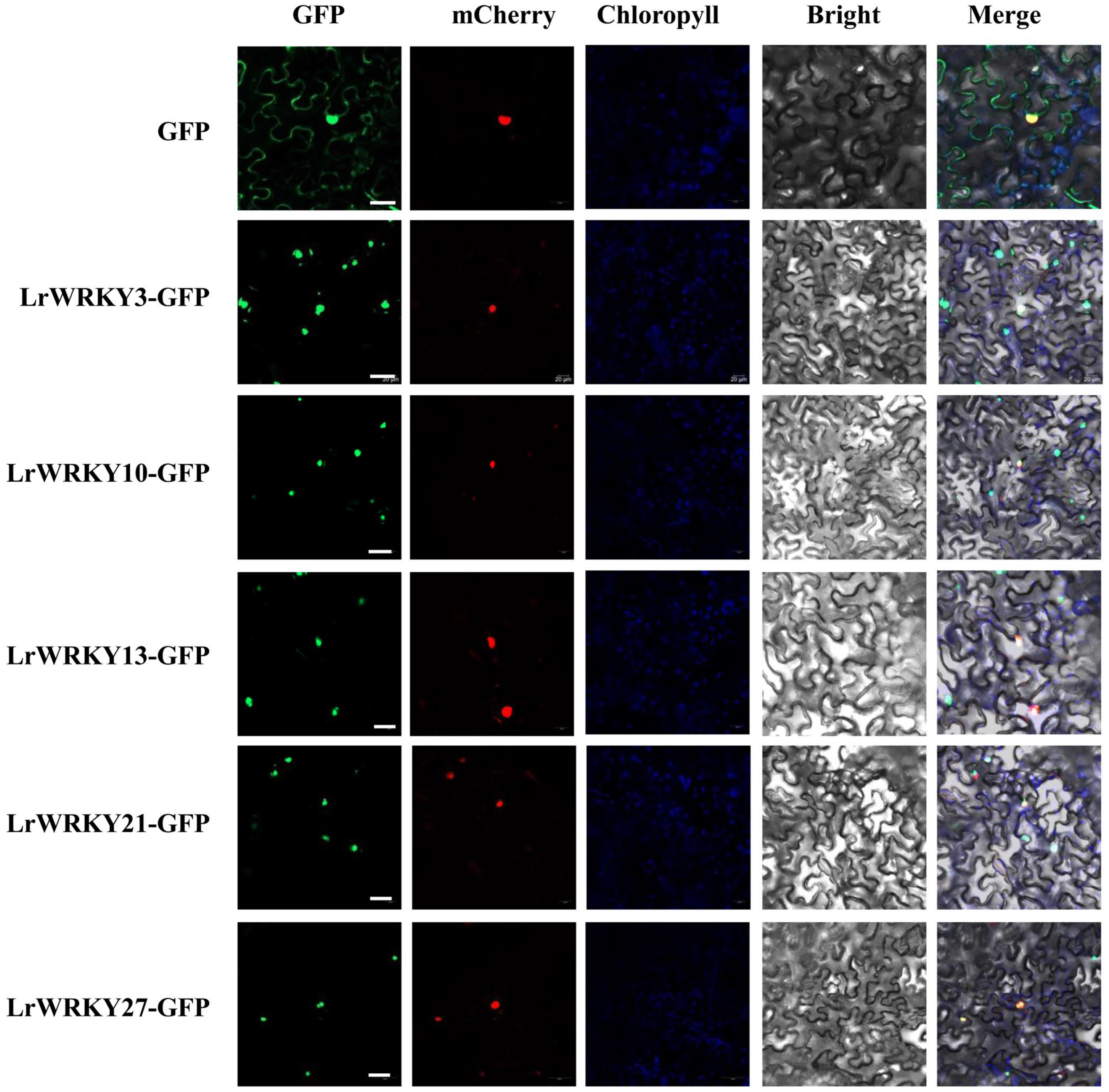
2.10. Subcellular Localization of LrWRKY Proteins
3. Discussion
3.1. Characterization of the WRKY Gene Family in L. radiata
3.2. The WRKY Gene Expression Patterns Facilitate Their Functional Analysis
3.3. LrWRKYs Are Related to L. radiata Petal Color Change
4. Materials and Methods
4.1. Plant Materials and Plant Treatments
4.2. Transcriptome-Wide Identification and Expression Profiling of LrWRKY Genes
4.3. Phylogenetic Tree and Protein Motif Analyses of LrWRKY Proteins
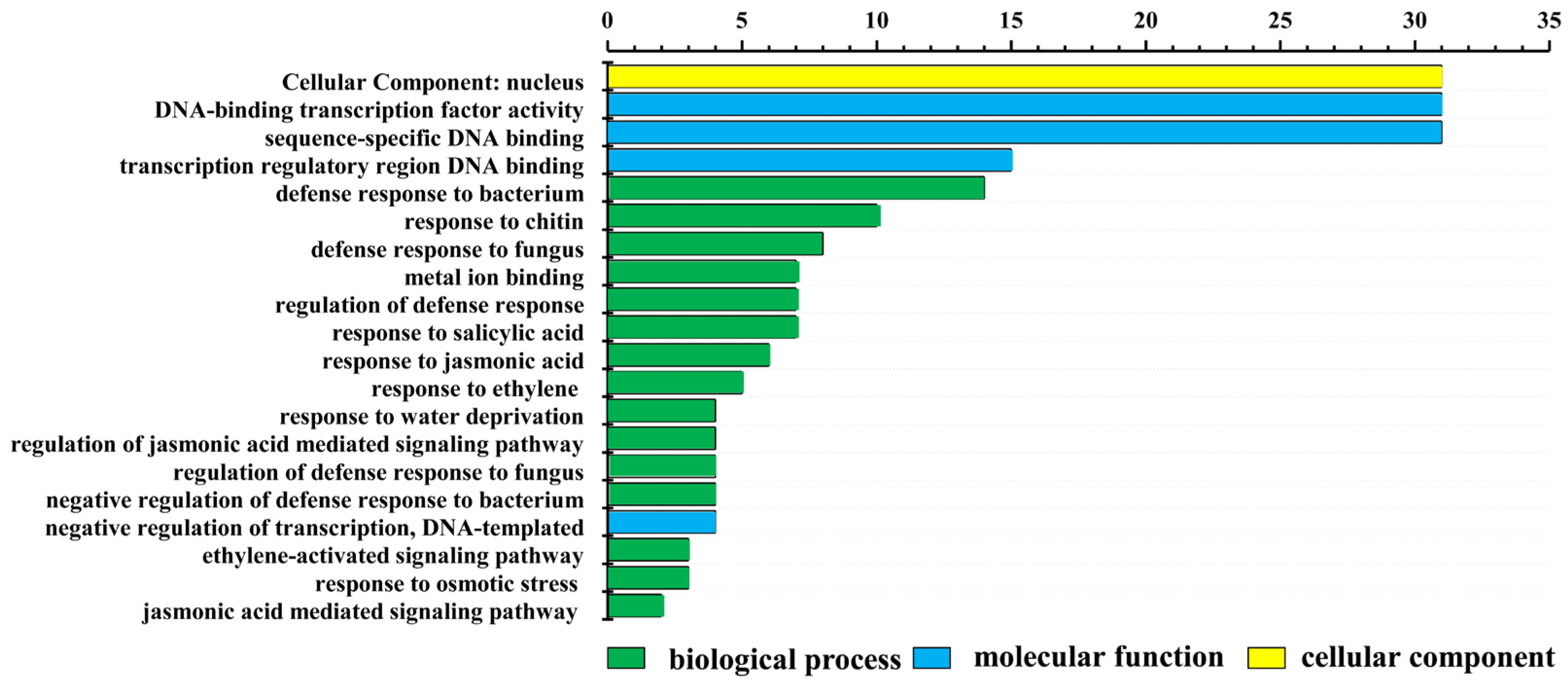
4.4. Gene Ontology Annotation of LrWRKYs
4.5. Expression Profiles Analysis and Real-Time Quantitative PCR Analysis of LrWRKYs Genes
4.6. GenesGene Cloning and Construction of Expression Vectors
4.7. Anthocyanin Quantification and Data Analysis of L. radiata
4.8. Subcellular Localization Analysis of LrWRKY Proteins
4.9. PPI Network Prediction of LrWRKY Proteins
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Javed, T.; Shabbir, R.; Ali, A.; Afzal, I.; Zaheer, U.; Gao, S.J. Transcription factors in plant stress responses: Challenges and potential for Sugarcane improvement. Plants 2020, 9, 491. [Google Scholar] [CrossRef] [PubMed]
- Romani, F.; Moreno, J.E. Molecular mechanisms involved in functional macroevolution of plant transcription factors. New Phytol. 2021, 230, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Wani, S.H.; Anand, S.; Singh, B.; Bohra, A.; Joshi, R. WRKY transcription factors and plant defense responses: Latest discoveries and future prospects. Plant Cell Rep. 2021, 40, 1071–1085. [Google Scholar] [CrossRef] [PubMed]
- Javed, T.; Zhou, J.R.; Li, J.; Hu, Z.T.; Wang, Q.N.; Gao, S.J. Identification and expression profiling of WRKY family genes in sugarcane in response to bacterial pathogen infection and nitrogen implantation dosage. Front. Plant Sci. 2022, 13, 917953. [Google Scholar] [CrossRef]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.J. WRKY transcription factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Jiang, Y.; Guo, Y.; Huang, J.; Zhou, M.; Tang, Y.; Sui, J.; Wang, J.; Qiao, L. A novel salt inducible WRKY transcription factor gene, AhWRKY75, confers salt tolerance in transgenic peanut. Plant Physiol. Biochem. 2021, 160, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hua, X.; Zhong, W.; Yuan, Y.; Wang, Y.; Wang, Z.; Ming, R.; Zhang, J. Genome-wide identification and expression profile analysis of WRKY family genes in the autopolyploid Saccharum spontaneum. Plant Cell Physiol. 2020, 61, 616–630. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L. The WRKY transcription factor superfamily: Its origin in eukaryotes and expansion in plants. BMC Evol. Biol. 2005, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, R.; Wang, Y.; Wu, C.; Huang, J. Genome-wide identification of WRKY transcription factors in Chinese jujube (Ziziphus jujuba Mill.) and their involvement in fruit developing, ripening, and abiotic stress. Genes 2019, 10, 360. [Google Scholar] [CrossRef]
- Eulgem, T.; Rushton, P.J.; Robatzek, S.; Somssich, I.E. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000, 5, 199–206. [Google Scholar] [CrossRef]
- Dong, J.; Chen, C.; Chen, Z. Expression profiles of the Arabidopsis WRKY gene superfamily during plant defense response. Plant Mol. Biol. 2003, 51, 21–37. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Mao, S.; Gao, Y.; Zhu, L.; Wu, D.; Cui, Y.; Li, J.; Qian, W. Genome-wide identification and expression analysis of WRKY transcription factors under multiple stresses in Brassica napus. PLoS ONE 2016, 11, e0157558. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, J.; Wang, L.; Wang, S. Genome-wide investigation of WRKY transcription factors involved in terminal drought stress response in common bean. Front. Plant Sci. 2017, 8, 380. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Chen, C.; Li, C.; Liu, J.; Liu, C.; He, Y. Genome-wide investigation of WRKY gene family in pineapple: Evolution and expression profiles during development and stress. BMC Genom. 2018, 19, 490. [Google Scholar] [CrossRef] [PubMed]
- Baillo, E.H.; Hanif, M.S.; Guo, Y.; Zhang, Z.; Xu, P.; Algam, S.A. Genome-wide identification of WRKY transcription factor family members in sorghum (Sorghum bicolor (L.) moench). PLoS ONE 2020, 15, e0236651. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhu, S.; Xu, L.; Zhu, L.; Wang, D.; Liu, Y.; Liu, S.; Hao, Z.; Lu, Y.; Yang, L.; et al. Genome-wide identification of the Liriodendron chinense WRKY gene family and its diverse roles in response to multiple abiotic stress. BMC Plant Biol. 2022, 22, 25. [Google Scholar] [CrossRef]
- Ayoub Khan, M.; Dongru, K.; Yifei, W.; Ying, W.; Penghui, A.; Zicheng, W. Characterization of WRKY gene family in whole-genome and exploration of flowering improvement genes in Chrysanthemum lavandulifolium. Front. Plant Sci. 2022, 13, 861193. [Google Scholar] [CrossRef]
- Yan, L.; Jin, H.; Raza, A.; Huang, Y.; Gu, D.; Zou, X. WRKY genes provide novel insights into their role against Ralstonia solanacearum infection in cultivated peanut (Arachis hypogaea L.). Front. Plant Sci. 2022, 13, 986673. [Google Scholar] [CrossRef]
- Phukan, U.J.; Jeena, G.S.; Shukla, R.K. WRKY transcription factors: Molecular regulation and stress responses in plants. Front. Plant Sci. 2016, 7, 760. [Google Scholar] [CrossRef]
- Jiang, J.; Ma, S.; Ye, N.; Jiang, M.; Cao, J.; Zhang, J. WRKY transcription factors in plant responses to stresses. J. Integr. Plant Biol. 2017, 59, 86–101. [Google Scholar] [CrossRef]
- Li, W.; Pang, S.; Lu, Z.; Jin, B. Function and mechanism of WRKY transcription factors in abiotic stress responses of plants. Plants 2020, 9, 1515. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, L.; Yan, Y.; Zhang, S.; Li, H.; Gao, Z.; Wang, C.; Guo, X. GhWRKY21 regulates ABA-mediated drought tolerance by fine-tuning the expression of GhHAB in cotton. Plant Cell Rep. 2021, 40, 2135–2150. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Wang, Y.; Xu, P.; Zhang, Z. Overexpression of a WRKY transcription factor TaWRKY2 enhances drought stress tolerance in transgenic wheat. Front. Plant Sci. 2018, 9, 997. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Chen, Z.; Liu, Y.; Zhang, H.; Zhang, M.; Liu, Q.; Hong, X.; Zhu, J.K.; Gong, Z. ABO3, a WRKY transcription factor, mediates plant responses to abscisic acid and drought tolerance in Arabidopsis. Plant J. 2010, 63, 417–429. [Google Scholar] [CrossRef]
- El-Esawi, M.A.; Al-Ghamdi, A.A.; Ali, H.M.; Ahmad, M. Overexpression of AtWRKY30 transcription factor enhances heat and drought stress tolerance in wheat (Triticum aestivum L.). Genes 2019, 10, 163. [Google Scholar] [CrossRef]
- Ali, M.A.; Azeem, F.; Nawaz, M.A.; Acet, T.; Abbas, A.; Imran, Q.M.; Shah, K.H.; Rehman, H.M.; Chung, G.; Yang, S.H.; et al. Transcription factors WRKY11 and WRKY17 are involved in abiotic stress responses in Arabidopsis. J. Plant Physiol. 2018, 226, 12–21. [Google Scholar] [CrossRef]
- Babitha, K.C.; Ramu, S.V.; Pruthvi, V.; Mahesh, P.; Nataraja, K.N.; Udayakumar, M. Co-expression of AtbHLH17 and AtWRKY28 confers resistance to abiotic stress in Arabidopsis. Transg. Res. 2013, 22, 327–341. [Google Scholar] [CrossRef]
- Chen, J.N.; Nolan, T.M.; Ye, H.X.; Zhang, M.C.; Tong, H.N.; Xin, P.Y.; Chu, J.F.; Chu, C.C.; Li, Z.H.; Yin, Y.H. Arabidopsis WRKY46, WRKY54, and WRKY70 transcription factors are involved in brassinosteroid-regulated plant growth and drought responses. Plant Cell 2017, 29, 1425–1439. [Google Scholar] [CrossRef]
- Ma, J.; Gao, X.; Liu, Q.; Shao, Y.; Zhang, D.; Jiang, L.; Li, C. Overexpression of TaWRKY146 Increases drought tolerance through inducing stomatal closure in Arabidopsis thaliana. Front. Plant Sci. 2017, 8, 2036. [Google Scholar] [CrossRef]
- Wang, C.T.; Ru, J.N.; Liu, Y.W.; Li, M.; Zhao, D.; Yang, J.F.; Fu, J.D.; Xu, Z.S. Maize WRKY transcription factor ZmWRKY106 confers drought and heat tolerance in transgenic plants. Int. J. Mol. Sci. 2018, 19, 3046. [Google Scholar] [CrossRef]
- Hu, Q.; Ao, C.; Wang, X.; Wu, Y.; Du, X. GhWRKY1-like, a WRKY transcription factor, mediates drought tolerance in Arabidopsis via modulating ABA biosynthesis. BMC Plant Biol. 2021, 21, 458. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Qiao, L.Y.; Guo, H.Y.; Guo, L.P.; Ren, F.; Bai, J.F.; Wang, Y.K. Genome-wide identification of wheat WRKY gene family reveals that TaWRKY75-A is referred to drought and salt resistances. Front. Plant Sci. 2021, 12, 812. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Luo, W.; Ge, M.; Guan, Y.; Tang, Y.; Chen, W.; Lv, J. A group I WRKY Gene, TaWRKY133, negatively regulates drought resistance in transgenic plants. Int. J. Mol. Sci. 2022, 23, 12026. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, D.; Zhao, X.; Zhang, M.; Wang, Q.; Hou, X.; Di, D.; Su, B.; Wang, S.; Sun, P. Drought-responsive WRKY transcription factor genes IgWRKY50 and IgWRKY32 from Iris germanica enhance drought resistance in transgenic Arabidopsis. Front. Plant Sci. 2022, 13, 983600. [Google Scholar] [CrossRef]
- Huang, Z.; Song, L.; Xiao, Y.; Zhong, X.; Wang, J.; Xu, W.; Jiang, C.Z. Overexpression of Myrothamnus flabellifolia MfWRKY41 confers drought and salinity tolerance by enhancing root system and antioxidation ability in Arabidopsis. Front. Plant Sci. 2022, 13, 967352. [Google Scholar] [CrossRef]
- Song, G.; Son, S.; Lee, K.S.; Park, Y.J.; Suh, E.J.; Lee, S.I.; Park, S.R. OsWRKY114 negatively regulates drought tolerance by restricting stomatal closure in rice. Plants 2022, 11, 1938. [Google Scholar] [CrossRef]
- Fan, C.; Yao, H.; Qiu, Z.; Ma, H.; Zeng, B. Genome-wide analysis of Eucalyptus grandis WRKY genes family and their expression profiling in response to hormone and abiotic stress treatment. Gene 2018, 678, 38–48. [Google Scholar] [CrossRef]
- Jiang, Y.; Duan, Y.; Yin, J.; Ye, S.; Zhu, J.; Zhang, F.; Lu, W.; Fan, D.; Luo, K. Genome-wide identification and characterization of the Populus WRKY transcription factor family and analysis of their expression in response to biotic and abiotic stresses. J. Exp. Bot. 2014, 65, 6629–6644. [Google Scholar] [CrossRef]
- Wang, L.; Liu, F.; Zhang, X.; Wang, W.; Sun, T.; Chen, Y.; Dai., M.; Yu, S.; Xu, L.; Su, Y.; et al. Expression characteristics and functional analysis of the ScWRKY3 gene from sugarcane. Int. J. Mol. Sci. 2018, 19, 4059. [Google Scholar] [CrossRef]
- Wang, D.; Wang, L.; Su, W.; Ren, Y.; You, C.; Zhang, C.; Que, Y.; Su, Y. A class III WRKY transcription factor in sugarcane was involved in biotic and abiotic stress responses. Sci. Rep. 2020, 10, 20964. [Google Scholar] [CrossRef]
- Kang, G.; Yan, D.; Chen, X.; Yang, L.; Zeng, R. HbWRKY82, a novel IIc WRKY transcription factor from Hevea brasiliensis associated with abiotic stress tolerance and leaf senescence in Arabidopsis. Physiol. Plant. 2021, 171, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, Y.; Yu, L.; Jiang, H.; Guo, Z.; Xu, H.; Jiang, S.; Fang, H.; Zhang, J.; Su, M.; et al. MdWRKY11 participates in anthocyanin accumulation in red-fleshed apples by affecting MYB transcription factors and the photoresponse factor MdHY5. J. Agric. Food Chem. 2019, 67, 8783–8793. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wu, J.; Hu, K.D.; Wei, S.W.; Sun, H.Y.; Hu, L.Y.; Han, Z.; Yao, G.F.; Zhang, H. PyWRKY26 and PybHLH3 cotargeted the PyMYB114 promoter to regulate anthocyanin biosynthesis and transport in red-skinned pears. Hortic. Res. 2020, 7, 37. [Google Scholar] [CrossRef]
- Cong, L.; Qu, Y.; Sha, G.; Zhang, S.; Ma, Y.; Chen, M.; Zhai, R.; Yang, C.; Xu, L.; Wang, Z. PbWRKY75 promotes anthocyanin synthesis by activating PbDFR, PbUFGT, and PbMYB10b in pear. Physiol. Plant. 2021, 173, 1841–1849. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, Z.; Zhao, Y.; Guo, D.; Zhao, X.; Gao, W.; Zhang, J.; Song, B. StWRKY13 promotes anthocyanin biosynthesis in potato (Solanum tuberosum) tubers. Funct. Plant Biol. 2021, 49, 102–114. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Mao, Z.; Liu, W.; Ding, L.; Zhang, X.; Yang, Y.; Wu, S.; Chen, X.; Wang, Y. Transcription factor McWRKY71 induced by ozone stress regulates anthocyanin and proanthocyanidin biosynthesis in Malus crabapple. Ecotoxicol. Environ. Saf. 2022, 232, 113274. [Google Scholar] [CrossRef]
- Su, M.; Zuo, W.; Wang, Y.; Liu, W.; Zhang, Z.; Wang, N.; Chen, X. The WKRY transcription factor MdWRKY75 regulates anthocyanins accumulation in apples (Malus domestica). Funct. Plant Biol. 2022, 49, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Cahlíková, L.; Breiterová, K.; Opletal, L. Chemistry and biological activity of alkaloids from the genus Lycoris (Amaryllidaceae). Molecules 2020, 5, 4797. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z. Amaryllidaceae and sceletium alkaloids. Nat. Prod. Rep. 2011, 28, 1126–1142. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Wang, Y.Y.; Su, J.; Li, Y.; Cai, X.H.; Luo, X.D. Amaryllidaceae alkaloids from Lycoris radiata. Helv. Chim. Acta 2011, 94, 178–183. [Google Scholar] [CrossRef]
- Chen, G.L.; Tian, Y.Q.; Wu, J.L.; Li, N.; Guo, M.Q. Antiproliferative activities of Amaryllidaceae alkaloids from Lycoris radiata targeting DNA topoisomerase I. Sci. Rep. 2016, 6, 38284. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.Y.; Xu, X.L.; Yang, L.J.; Jiang, J.G. Identification of narciclasine from Lycoris radiata (L’Her.) Herb. and its inhibitory effect on LPS-induced inflammatory responses in macrophages. Food Chem. Toxicol. 2019, 125, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Shu, X.; Zhang, F.; Zhuang, W.; Wang, T.; Wang, Z. Comparative transcriptome analysis identifies key regulatory genes involved in anthocyanin metabolism during flower development in Lycoris radiata. Front. Plant Sci. 2021, 12, 761862. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Xu, S.; Wang, N.; Xia, B.; Jiang, Y.; Wang, R. Transcriptome analysis of secondary metabolism pathway, transcription factors, and transporters in response to methyl jasmonate in Lycoris aurea. Front. Plant Sci. 2017, 7, 1971. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Yeo, H.J.; Park, Y.E.; Baek, S.A.; Kim, J.K.; Park, S.U. Transcriptome analysis and metabolic profiling of Lycoris radiata. Biology 2019, 8, 63. [Google Scholar] [CrossRef]
- Yue, Y.; Liu, J.; Shi, T.; Chen, M.; Li, Y.; Du, J.; Jiang, H.; Yang, X.; Hu, H.; Wang, L. Integrating transcriptomic and GC-MS metabolomic analysis to characterize color and aroma formation during tepal development in Lycoris longituba. Plants 2019, 8, 53. [Google Scholar] [CrossRef]
- Li, Q.; Xu, J.; Yang, L.; Zhou, X.; Cai, Y.; Zhang, Y. Transcriptome analysis of different tissues reveals key genes associated with galanthamine biosynthesis in Lycoris longituba. Front. Plant Sci. 2020, 11, 519752. [Google Scholar] [CrossRef]
- Ren, Z.M.; Zhang, D.; Jiao, C.; Li, D.Q.; Wu, Y.; Wang, X.Y.; Gao, C.; Lin, Y.F.; Ruan, Y.L.; Xia, Y.P. Comparative transcriptome and metabolome analyses identified the mode of sucrose degradation as a metabolic marker for early vegetative propagation in bulbs of Lycoris. Plant J. 2022, 112, 115–134. [Google Scholar] [CrossRef]
- Bakshi, M.; Oelmüller, R. WRKY transcription factors: Jack of many trades in plants. Plant Signal. Behav. 2014, 9, e27700. [Google Scholar] [CrossRef]
- Zhao, N.; He, M.; Li, L.; Cui, S.; Hou, M.; Wang, L.; Mu, G.; Liu, L.; Yang, X. Identification and expression analysis of WRKY gene family under drought stress in peanut (Arachis hypogaea L.). PLoS ONE 2020, 15, e0231396. [Google Scholar] [CrossRef]
- Cheng, W.; Jiang, Y.; Peng, J.; Guo, J.; Lin, M.; Jin, C.; Huang, J.; Tang, W.; Guan, D.; He, S. The transcriptional reprograming and functional identification of WRKY family members in pepper’s response to Phytophthora capsici infection. BMC Plant Biol. 2020, 20, 256. [Google Scholar] [CrossRef]
- Abdullah-Zawawi, M.R.; Ahmad-Nizammuddin, N.F.; Govender, N.; Harun, S.; Mohd-Assaad, N.; Mohamed-Hussein, Z.A. Comparative genome-wide analysis of WRKY, MADS-box and MYB transcription factor families in Arabidopsis and rice. Sci. Rep. 2021, 11, 19678. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Yu, X.; Ma, W.; Sun, X.; Ge, Y.; Yue, X.; Han, J.; Zhao, J.; Lu, Y.; Liu, M. Genome-wide identification and expression analysis of WRKY family genes under soft rot in Chinese cabbage. Front. Genet. 2022, 13, 958769. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhang, D.; Zhao, T.; Yang, H.; Jiang, J.; Li, J.; Zhang, H.; Xu, X. Genome-wide analysis of the WRKY gene family unveil evolutionary history and expression characteristics in tomato and its wild relatives. Front. Genet. 2022, 13, 962975. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, W.; Wang, D.; Hu, S.; Zhang, Q.; Wang, Z.; Cui, L. Genome-wide identification and characterization of the WRKY gene family in scutellaria baicalensis georgi under diverse abiotic stress. Int. J. Mol. Sci. 2022, 23, 4225. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Cui, H.; Shi, Y.; Xue, J.; Ji, C.; Zhang, C.; Yuan, L.; Li, R. Genome-wide identification and functional characterization of the Camelina sativa WRKY gene family in response to abiotic stress. BMC Genom. 2020, 21, 786. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; White, M.J.; MacRae, T.H. Transcription factors and their genes in higher plants functional domains, evolution and regulation. Eur. J. Biochem. 1999, 262, 247–257. [Google Scholar] [CrossRef]
- Wei, Y.; Shi, H.; Xia, Z.; Tie, W.; Ding, Z.; Yan, Y.; Wang, W.; Hu, W.; Li, K. Genome-wide identification and expression analysis of the WRKY gene family in cassava. Front. Plant Sci. 2016, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.H.; Sun, P.W.; Tang, X.L.; Gao, Z.H.; Zhang, Z.; Wei, J.H. Genome-wide analysis of WRKY transcription factors in Aquilaria sinensis (Lour.) Gilg. Sci. Rep. 2020, 10, 3018. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.; Choi, J.P.; Park, I.; Yang, K.; Kim, M.K.; Lee, Y.H.; Nou, I.S.; Kim, D.S.; Min, S.R.; et al. Functional innovations of three chronological mesohexaploid Brassica rapa genomes. BMC Genom. 2014, 15, 606. [Google Scholar] [CrossRef]
- Li, J.; Brader, G.; Palva, E.T. The WRKY70 transcription factor: A node of convergence for jasmonate-mediated and salicylate-mediated signals in plant defense. Plant Cell 2004, 16, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Devaiah, B.N.; Karthikeyan, A.S.; Raghothama, K.G. WRKY75 transcription factor is a modulator of phosphate acquisition and root development in Arabidopsis. Plant Physiol. 2007, 143, 1789–1801. [Google Scholar] [CrossRef]
- Lei, R.; Li, X.; Ma, Z.; Lv, Y.; Hu, Y.; Yu, D. Arabidopsis WRKY2 and WRKY34 transcription factors interact with VQ20 protein to modulate pollen development and function. Plant J. 2017, 91, 962–976. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, L.; Wu, S.; Chen, Y.; Yu, D.; Chen, L. AtWRKY75 positively regulates age-triggered leaf senescence through gibberellin pathway. Plant Divers. 2020, 43, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Li, Z.; Huang, P.; Li, B.; Fang, S.; Chu, J.; Guo, H. A Tripartite amplification loop involving the transcription factor WRKY75, salicylic acid, and reactive oxygen species accelerates feaf senescence. Plant Cell 2017, 29, 2854–2870. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, X.; Nvsvrot, T.; Huang, L.; Cai, G.; Ding, Y.; Ren, W.; Wang, N. The transcription factor WRKY75 regulates the development of adventitious roots, lateral buds and callus by modulating hydrogen peroxide content in poplar. J. Exp. Bot. 2022, 73, 1483–1498. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, L.; Wang, H.; Zhang, L.; Wang, F.; Yu, D. Arabidopsis transcription factor WRKY8 functions antagonistically with its interacting partner VQ9 to modulate salinity stress tolerance. Plant J. 2013, 74, 730–745. [Google Scholar] [CrossRef]
- Chen, H.; Lai, Z.; Shi, J.; Xiao, Y.; Chen, Z.; Xu, X. Roles of Arabidopsis WRKY18, WRKY40 and WRKY60 transcription factors in plant responses to abscisic acid and abiotic stress. BMC Plant Biol. 2010, 10, 281. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, J.; Zheng, Z.; Fan, B.; Yu, J.Q.; Chen, Z. Characterization of the promoter and extended C-terminal domain of Arabidopsis WRKY33 and functional analysis of tomato WRKY33 homologues in plant stress responses. J. Exp. Bot. 2015, 66, 4567–4583. [Google Scholar] [CrossRef]
- Wasternack, C. Jasmonates: An update on biosynthesis, signal trans-duction and action in plant stress response, growth and development. Ann. Bot. 2007, 100, 681–697. [Google Scholar] [CrossRef]
- Pauwels, L.; Inzé, D.; Goossens, A. Jasmonate-inducible gene: What does it mean? Trends Plant Sci. 2009, 14, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Browse, J.; Pauwels, L.; Inzé, D.; Goossens, A. Jasmonate passes muster: A receptor and targets for the defense hormone. Annu. Rev. Plant Biol. 2009, 60, 183–205. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Wu, S.; Tian, L. Methyl jasmonate elicits distinctive hydrolyzable tannin, flavonoid, and phyto-oxylipin responses in pomegranate (Punica granatum L.) leaves. Planta 2021, 254, 89. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Zhou, H.; Wang, L.; Zhao, J.; Ma, L.; Ling, J.; Li, G.; Huang, W.; Li, P.; Zhang, Y. Post-harvest application of methyl jasmonate or prohydrojasmon affects color development and anthocyanins biosynthesis in peach by regulation of sucrose metabolism. Front. Nutr. 2022, 9, 871467. [Google Scholar] [CrossRef]
- Zhou, W.; Shi, M.; Deng, C.; Lu, S.; Huang, F.; Wang, Y.; Kai, G. The methyl jasmonate-responsive transcription factor SmMYB1 promotes phenolic acid biosynthesis in Salvia miltiorrhiza. Hortic. Res. 2021, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Journot-catalino, N.; Somssich, I.E.; Roby, D.; Kroj, T. The transcription factors WRKY11 and WRKY17 act as negative regulators of basal resistance in Arabidopsis thaliana. Plant Cell 2006, 18, 3289–3302. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, L.; Xiang, S.; Chen, Y.; Zhang, H.; Yu, D. The transcription factor WRKY75 positively regulates jasmonate-mediated plant defense to necrotrophic fungal pathogens. J. Exp. Bot. 2021, 72, 1473–1489. [Google Scholar] [CrossRef] [PubMed]
- Ji, N.; Li, Y.; Wang, J.; Zuo, X.; Li, M.; Jin, P.; Zheng, Y. Interaction of PpWRKY46 and PpWRKY53 regulates energy metabolism in MeJA primed disease resistance of peach fruit. Plant Physiol. Biochem. 2022, 171, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gao, M.; Li, Y.; Zhang, J.; Su, H.; Cao, M.; Liu, Z.; Zhang, X.; Zhao, B.; Guo, Y.D.; et al. The transcription factor SlWRKY37 positively regulates jasmonic acid- and dark-induced leaf senescence in tomato. J. Exp. Bot. 2022, 73, 6207–6225. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, S.; Peng, D.; Wu, Q.; Liao, X.; Xiang, K.; Wang, Z.; Tembrock, L.R.; Bendahmane, M.; Bao, M.; et al. Integrated multi-omic data and analyses reveal the pathways underlying key ornamental traits in carnation flowers. Plant Biotechnol. J. 2022, 20, 1182–1196. [Google Scholar] [CrossRef]
- Shao, D.; Liang, Q.; Wang, X.; Zhu, Q.H.; Liu, F.; Li, Y.; Zhang, X.; Yang, Y.; Sun, J.; Xue, F. Comparative metabolome and transcriptome analysis of anthocyanin biosynthesis in white and pink petals of cotton (Gossypium hirsutum L.). Int. J. Mol. Sci. 2022, 23, 10137. [Google Scholar] [CrossRef] [PubMed]
- Pal, L.; Dwivedi, V.; Gupta, S.K.; Saxena, S.; Pandey, A.; Chattopadhyay, D. Biochemical analysis of anthocyanin and proanthocyanidin and their regulation in determining chickpea flower and seed coat colours. J. Exp. Bot. 2022, 7, erac392. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Li, Z.; Jiang, X.; Qin, Y. Comparison of metabolome and transcriptome of flavonoid biosynthesis in two colors of coreopsis tinctoria Nutt. Front. Plant Sci. 2022, 13, 810422. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gao, R.; Zhang, J.; Wang, Y.; Kong, P.; Lu, K.; Adnan Liu, M.; Ao, F.; Zhao, C.; Wang, L.; et al. The biochemical and molecular investigation of flower color and scent sheds lights on further genetic modification of ornamental traits in Clivia miniata. Hortic. Res. 2022, 9, uhac114. [Google Scholar] [CrossRef]
- Xing, A.; Wang, X.; Nazir, M.F.; Zhang, X.; Wang, X.; Yang, R.; Chen, B.; Fu, G.; Wang, J.; Ge, H.; et al. Transcriptomic and metabolomic profiling of flavonoid biosynthesis provides novel insights into petals coloration in Asian cotton (Gossypium arboreum L.). BMC Plant Biol. 2022, 22, 416. [Google Scholar] [CrossRef]
- Wang, X.; Li, L.; Liu, C.; Zhang, M.; Wen, Y. An integrated metabolome and transcriptome analysis of the Hibiscus syriacus L. petals reveal the molecular mechanisms of anthocyanin accumulation. Front. Genet. 2022, 13, 995748. [Google Scholar] [CrossRef]
- Sun, X.; He, L.; Guo, Z.; Xiao, Z.; Su, J.; Liu, X.; Zhou, H.; Li, C.; Gao, H. Comparative transcriptome analyses reveal genes related to pigmentation in the petals of a flower color variation cultivar of Rhododendron obtusum. Mol. Biol. Rep. 2022, 49, 2641–2653. [Google Scholar] [CrossRef]
- Verweij, W.; Spelt, C.E.; Bliek, M.; de Vries, M.; Wit, N.; Faraco, M.; Koes, R.; Quattrocchio, F.M. Functionally similar WRKY proteins regulate vacuolar acidification in petunia and hair development in Arabidopsis. Plant Cell 2016, 28, 786–803. [Google Scholar] [CrossRef]
- Lloyd, A.; Brockman, A.; Aguirre, L.; Campbell, A.; Bean, A.; Cantero, A.; Gonzalez, A. Advances in the MYB-bHLH-WD repeat (MBW) pigment regulatory model: Addition of a WRKY factor and co-option of an anthocyanin MYB for betalain regulation. Plant Cell Physiol. 2017, 58, 1431–1441. [Google Scholar] [CrossRef]
- Yang, F.S.; Nie, S.; Liu, H.; Shi, T.L.; Tian, X.C.; Zhou, S.S.; Bao, Y.T.; Jia, K.H.; Guo, J.F.; Zhao, W.; et al. Chromosome-level genome assembly of a parent species of widely cultivated azaleas. Nat. Commun. 2020, 11, 5269. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent updates, new developments and status in 2020. Nucleic Acids Res. 2021, 49, D458–D460. [Google Scholar] [CrossRef] [PubMed]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; De Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Yang, X.; Zheng, J.; Wang, L.; Yang, X.; Tu, Y.; Ye, J.; Zhang, W.; Liao, Y.; Cheng, S.; et al. Unraveling the regulatory mechanism of color diversity in camellia japonica petals by integrative transcriptome and metabolome analysis. Front. Plant Sci. 2021, 12, 685136. [Google Scholar] [CrossRef]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The human metabolome database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef]
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2022, 12, gkac1000. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, N.; Song, G.; Zhang, F.; Shu, X.; Cheng, G.; Zhuang, W.; Wang, T.; Li, Y.; Wang, Z. Characterization of the WRKY Gene Family Related to Anthocyanin Biosynthesis and the Regulation Mechanism under Drought Stress and Methyl Jasmonate Treatment in Lycoris radiata. Int. J. Mol. Sci. 2023, 24, 2423. https://doi.org/10.3390/ijms24032423
Wang N, Song G, Zhang F, Shu X, Cheng G, Zhuang W, Wang T, Li Y, Wang Z. Characterization of the WRKY Gene Family Related to Anthocyanin Biosynthesis and the Regulation Mechanism under Drought Stress and Methyl Jasmonate Treatment in Lycoris radiata. International Journal of Molecular Sciences. 2023; 24(3):2423. https://doi.org/10.3390/ijms24032423
Chicago/Turabian StyleWang, Ning, Guowei Song, Fengjiao Zhang, Xiaochun Shu, Guanghao Cheng, Weibing Zhuang, Tao Wang, Yuhang Li, and Zhong Wang. 2023. "Characterization of the WRKY Gene Family Related to Anthocyanin Biosynthesis and the Regulation Mechanism under Drought Stress and Methyl Jasmonate Treatment in Lycoris radiata" International Journal of Molecular Sciences 24, no. 3: 2423. https://doi.org/10.3390/ijms24032423
APA StyleWang, N., Song, G., Zhang, F., Shu, X., Cheng, G., Zhuang, W., Wang, T., Li, Y., & Wang, Z. (2023). Characterization of the WRKY Gene Family Related to Anthocyanin Biosynthesis and the Regulation Mechanism under Drought Stress and Methyl Jasmonate Treatment in Lycoris radiata. International Journal of Molecular Sciences, 24(3), 2423. https://doi.org/10.3390/ijms24032423