

Glutamine Metabolism in Cancer Stem Cells: A Complex Liaison in the Tumor Microenvironment


Abstract
1. Glutamine Metabolism
2. Glutamine Metabolism in CSC
2.1. Glutamine and Redox Homeostasis in CSC
2.2. Glutamine, TCA Cycle, Anaplerotic and Cataplerotic Fluxes in CSC
2.3. Glutamine and Epigenetic Modifications in CSC
2.4. Stem Marker Proteins and Glutamine Metabolism: Direct Interplay
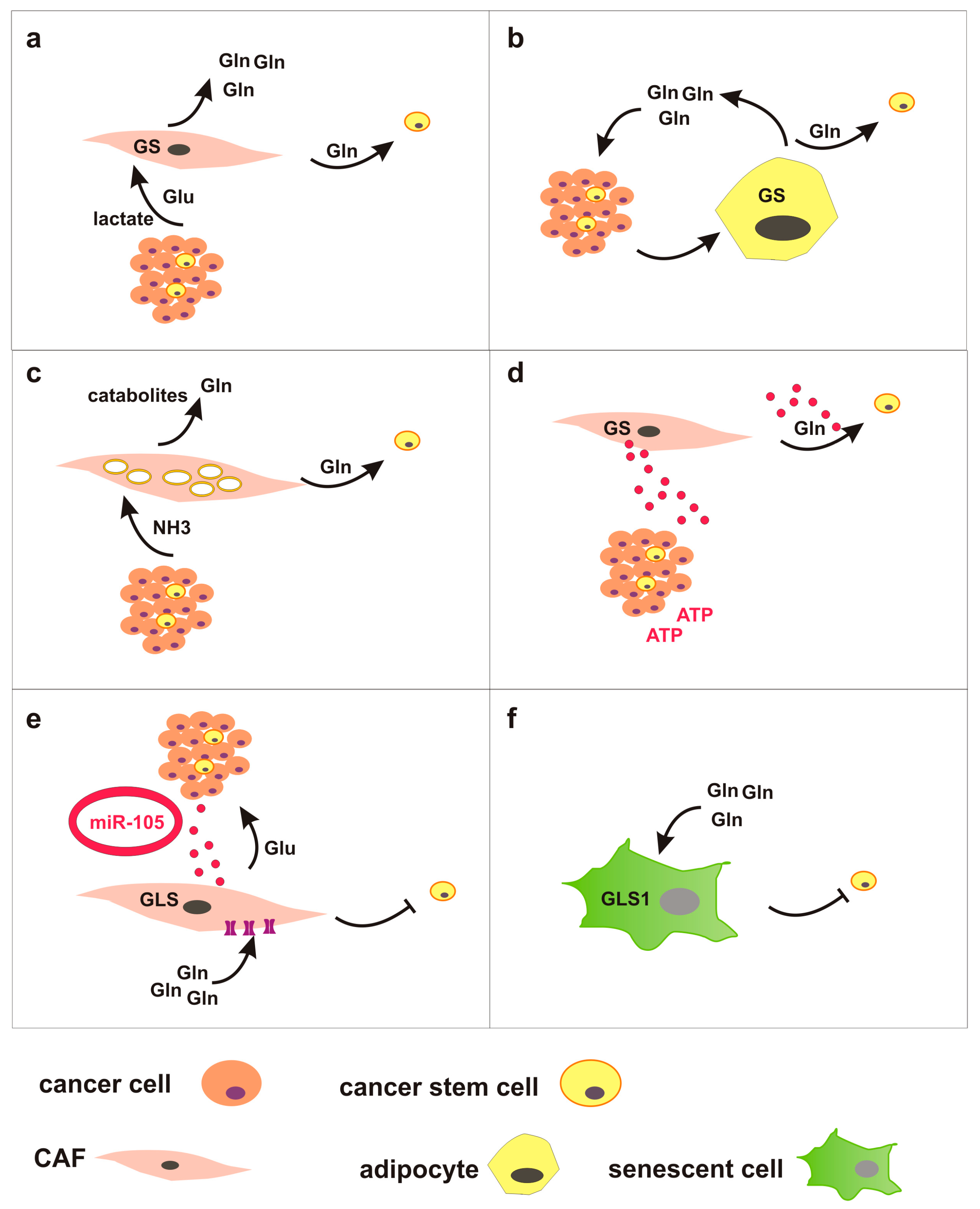
3. Glutamine Metabolism in Tumor Microenvironment: Tumor-Stroma Crosstalk Might Regulate CSC
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pan, M.; Reid, M.A.; Lowman, X.H.; Kulkarni, R.P.; Tran, T.Q.; Liu, X.; Yang, Y.; Hernandez-Davies, J.E.; Rosales, K.K.; Li, H.; et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat. Cell. Biol. 2016, 18, 1090–1101. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Ploessl, K.; Zhou, R.; Mankoff, D.; Kung, H.F. Metabolic Imaging of Glutamine in Cancer. J. Nucl. Med. 2017, 58, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Pochini, L.; Scalise, M.; Galluccio, M.; Indiveri, C. Membrane transporters for the special amino acid glutamine: Structure/function relationships and relevance to human health. Front. Chem. 2014, 2, 61. [Google Scholar] [CrossRef] [PubMed]
- Bröer, A.; Rahimi, F.; Bröer, S. Deletion of Amino Acid Transporter ASCT2 (SLC1A5) Reveals an Essential Role for Transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to Sustain Glutaminolysis in Cancer Cells. J. Biol. Chem. 2016, 291, 13194–13205. [Google Scholar] [CrossRef]
- Cassago, A.; Ferreira, A.P.; Ferreira, I.M.; Fornezari, C.; Gomes, E.R.; Greene, K.S.; Pereira, H.M.; Garratt, R.C.; Dias, S.M.; Ambrosio, A.L. Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab. 2020, 31, 267–283.e12. [Google Scholar] [CrossRef]
- Masisi, B.K.; El Ansari, R.; Alfarsi, L.; Rakha, E.A.; Green, A.R.; Craze, M.L. The role of glutaminase in cancer. Histopathology 2020, 76, 498–508. [Google Scholar] [CrossRef]
- Saha, S.K.; Islam, S.M.R.; Abdullah-Al-Wadud, M.; Islam, S.; Ali, F.; Park, K.S. Multiomics Analysis Reveals that GLS and GLS2 Differentially Modulate the Clinical Outcomes of Cancer. J. Clin. Med. 2019, 8, 355. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Ko, B.; Hensley, C.T.; Jiang, L.; Wasti, A.T.; Kim, J.; Sudderth, J.; Calvaruso, M.A.; Lumata, L.; Mitsche, M.; et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol. Cell. 2014, 56, 414–424. [Google Scholar] [CrossRef]
- Coloff, J.L.; Murphy, J.P.; Braun, C.R.; Harris, I.S.; Shelton, L.M.; Kami, K.; Gygi, S.P.; Selfors, L.M.; Brugge, J.S. Differential Glutamate Metabolism in Proliferating and Quiescent Mammary Epithelial Cells. Cell. Metab. 2016, 23, 867–880. [Google Scholar] [CrossRef]
- Fan, J.; Kamphorst, J.J.; Mathew, R.; Chung, M.K.; White, E.; Shlomi, T.; Rabinowitz, J.D. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 2013, 9, 712. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Arismendi-Morillo, G.; Mukherjee, P.; Chinopoulos, C. On the Origin of ATP Synthesis in Cancer. iScience 2020, 23, 101761. [Google Scholar] [CrossRef] [PubMed]
- Chinopoulos, C.; Seyfried, T.N. Mitochondrial Substrate-Level Phosphorylation as Energy Source for Glioblastoma: Review and Hypothesis. ASN Neuro. 2018, 10, 1759091418818261. [Google Scholar] [CrossRef]
- Taylor, E.B. Functional Properties of the Mitochondrial Carrier System. Trends Cell Biol. 2017, 27, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Frigerio, F.; Casimir, M.; Carobbio, S.; Maechler, P. Tissue specificity of mitochondrial glutamate pathways and the control of metabolic homeostasis. Biochim. Biophys. Acta 2008, 1777, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Jyotsana, N.; Ta, K.T.; DelGiorno, K.E. The Role of Cystine/Glutamate Antiporter SLC7A11/xCT in the Pathophysiology of Cancer. Front. Oncol. 2022, 12, 858462. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.H.; Coloff, J.L. The Diverse Functions of Non-Essential Amino Acids in Cancer. Cancers 2019, 11, 675. [Google Scholar] [CrossRef]
- Wang, T.; Liu, L.; Chen, X.; Shen, Y.; Lian, G.; Shah, N.; Davidoff, A.M.; Yang, J.; Wang, R. MYCN drives glutaminolysis in neuroblastoma and confers sensitivity to an ROS augmenting agent. Cell Death Dis. 2018, 9, 220. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marcos, P.J.; Nóbrega-Pereira, S. NADPH: New oxygen for the ROS theory of aging. Oncotarget 2016, 7, 50814–50815. [Google Scholar] [CrossRef]
- Alzial, G.; Renoult, O.; Paris, F.; Gratas, C.; Clavreul, A.; Pecqueur, C. Wild-type isocitrate dehydrogenase under the spotlight in glioblastoma. Oncogene 2022, 41, 613–621. [Google Scholar] [CrossRef]
- Kim, G.W.; Lee, D.H.; Jeon, Y.H.; Yoo, J.; Kim, S.Y.; Lee, S.W.; Cho, H.Y.; Kwon, S.H. Glutamine Synthetase as a Therapeutic Target for Cancer Treatment. Int. J. Mol. Sci. 2021, 22, 1701. [Google Scholar] [CrossRef]
- Issaq, S.H.; Mendoza, A.; Fox, S.D.; Helman, L.J. Glutamine synthetase is necessary for sarcoma adaptation to glutamine deprivation and tumor growth. Oncogenesis 2019, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Bott, A.J.; Peng, I.C.; Fan, Y.; Faubert, B.; Zhao, L.; Li, J.; Neidler, S.; Sun, Y.; Jaber, N.; Krokowski, D.; et al. Oncogenic Myc Induces Expression of Glutamine Synthetase through Promoter Demethylation. Cell Metab. 2015, 22, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Kung, H.N.; Marks, J.R.; Chi, J.T. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet. 2011, 7, e1002229. [Google Scholar] [CrossRef]
- Hakvoort, T.B.; He, Y.; Kulik, W.; Vermeulen, J.L.; Duijst, S.; Ruijter, J.M.; Runge, J.H.; Deutz, N.E.; Koehler, S.E.; Lamers, W.H. Pivotal role of glutamine synthetase in ammonia detoxification. Hepatology 2017, 65, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Eid, T.; Hassel, B.; Danbolt, N.C. Novel aspects of glutamine synthetase in ammonia homeostasis. Neurochem. Int. 2020, 140, 104809. [Google Scholar] [CrossRef]
- Bisht, S.; Nigam, M.; Kunjwal, S.S.; Sergey, P.; Mishra, A.P.; Sharifi-Rad, J. Cancer Stem Cells: From an Insight into the Basics to Recent Advances and Therapeutic Targeting. Stem Cells Int. 2022, 2022, 9653244. [Google Scholar] [CrossRef]
- Beck, B.; Blanpain, C. Unravelling cancer stem cell potential. Nat. Rev. Cancer. 2013, 13, 727–738. [Google Scholar] [CrossRef]
- Liu, H.G.; Chen, C.; Yang, H.; Pan, Y.F.; Zhang, X.H. J Cancer stem cell subsets and their relationships. Transl. Med. 2011, 9, 50. [Google Scholar] [CrossRef]
- Akrap, N.; Andersson, D.; Bom, E.; Gregersson, P.; Stahlberg, A.; Landberg, G. Identification of Distinct Breast Cancer Stem Cell Populations Based on Single-Cell Analyses of Functionally Enriched Stem and Progenitor Pools. Stem Cell Rep. 2016, 6, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Song, X.; Xu, D.; Tiek, D.; Goenka, A.; Wu, B.; Sastry, N.; Hu, B.; Cheng, S.Y. Stem cell programs in cancer initiation, progression, and therapy resistance. Theranostics 2020, 10, 8721–8743. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Yadav, U.P.; Singh, T.; Kumar, P.; Sharma, P.; Kaur, H.; Sharma, S.; Singh, S.; Kumar, S.; Mehta, K. Metabolic Adaptations in Cancer Stem Cells. Front. Oncol. 2020, 10, 1010. [Google Scholar] [CrossRef]
- Sharif, T.; Dai, C.; Martell, E.; Ghassemi-Rad, M.S.; Hanes, M.R.; Murphy, P.J.; Kennedy, B.E.; Venugopal, C.; Subapanditha, M.; Giacomantonio, C.A.; et al. TAp73 Modifies Metabolism and Positively Regulates Growth of Cancer Stem-Like Cells in a Redox-Sensitive Manner. Clin. Cancer Res. 2019, 25, 2001–2017. [Google Scholar] [CrossRef]
- Jaggupilli, A.; Ly, S.; Nguyen, K.; Anand, V.; Yuan, B.; El-Dana, F.; Yan, Y.; Arvanitis, Z.; Piyarathna, D.W.B.; Putluri, N.; et al. Metabolic stress induces GD2+ cancer stem cell-like phenotype in triple-negative breast cancer. Br. J. Cancer 2022, 126, 615–627. [Google Scholar] [CrossRef]
- Mukha, A.; Kahya, U.; Linge, A.; Chen, O.; Löck, S.; Lukiyanchuk, V.; Richter, S.; Alves, T.C.; Peitzsch, M.; Telychko, V.; et al. GLS-driven glutamine catabolism contributes to prostate cancer radiosensitivity by regulating the redox state, stemness and ATG5-mediated autophagy. Theranostics 2021, 11, 7844–7868. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Tsuchihashi, K.; Ishimoto, T.; Yae, T.; Motohara, T.; Sugihara, E.; Onishi, N.; Masuko, T.; Yoshizawa, K.; Kawashiri, S.; et al. xCT inhibition depletes CD44v-expressing tumor cells that are resistant to EGFR-targeted therapy in head and neck squamous cell carcinoma. Cancer Res. 2013, 73, 1855–1866. [Google Scholar] [CrossRef]
- Li, D.; Fu, Z.; Chen, R.; Zhao, X.; Zhou, Y.; Zeng, B.; Yu, M.; Zhou, Q.; Lin, Q.; Gao, W.; et al. Inhibition of glutamine metabolism counteracts pancreatic cancer stem cell features and sensitizes cells to radiotherapy. Oncotarget 2015, 6, 31151–31163. [Google Scholar] [CrossRef]
- Marsboom, G.; Zhang, G.F.; Pohl-Avila, N.; Zhang, Y.; Yuan, Y.; Kang, H.; Hao, B.; Brunengraber, H.; Malik, A.B.; Rehman, J. Glutamine Metabolism Regulates the Pluripotency Transcription Factor OCT4. Cell Rep. 2016, 16, 323–332. [Google Scholar] [CrossRef]
- Battula, V.L.; Shi, Y.; Evans, K.W.; Wang, R.Y.; Spaeth, E.L.; Jacamo, R.O.; Guerra, R.; Sahin, A.A.; Marini, F.C.; Hortobagyi, G.; et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J. Clin. Investig. 2012, 122, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, S.; Umene, K.; Yamasaki, J.; Suina, K.; Otsuki, Y.; Yoshikawa, M.; Minami, Y.; Masuko, T.; Kawaguchi, S.; Nakayama, H.; et al. Glutaminolysis-related genes determine sensitivity to xCT-targeted therapy in head and neck squamous cell carcinoma. Cancer Sci. 2019, 110, 3453–3463. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016, 532, 255–258. [Google Scholar] [CrossRef]
- Zhao, C.; Setrerrahmane, S.; Xu, H. Enrichment and characterization of cancer stem cells from a human non-small cell lung cancer cell line. Oncol. Rep. 2015, 34, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Gheytanchi, E.; Naseri, M.; Karimi-Busheri, F.; Atyabi, F.; Mirsharif, E.S.; Bozorgmehr, M.; Ghods, R.; Madjd, Z. Morphological and molecular characteristics of spheroid formation in HT-29 and Caco-2 colorectal cancer cell lines. Cancer Cell Int. 2021, 21, 204. [Google Scholar] [CrossRef]
- Herreros-Pomares, A.; de-Maya-Girones, J.D.; Calabuig-Fariñas, S.; Lucas, R.; Martínez, A.; Pardo-Sánchez, J.M.; Alonso, S.; Blasco, A.; Guijarro, R.; Martorell, M.; et al. Lung tumorspheres reveal cancer stem cell-like properties and a score with prognostic impact in resected non-small-cell lung cancer. Cell Death Dis. 2019, 10, 660. [Google Scholar] [CrossRef]
- Calvert, A.E.; Chalastanis, A.; Wu, Y.; Hurley, L.A.; Kouri, F.M.; Bi, Y.; Kachman, M.; May, J.L.; Bartom, E.; Hua, Y.; et al. Cancer-Associated IDH1 Promotes Growth and Resistance to Targeted Therapies in the Absence of Mutation. Cell Rep. 2017, 19, 1858–1873. [Google Scholar] [CrossRef]
- Verma, N.; Vinik, Y.; Saroha, A.; Nair, N.U.; Ruppin, E.; Mills, G.; Karn, T.; Dubey, V.; Khera, L.; Raj, H.; et al. Synthetic lethal combination targeting BET uncovered intrinsic susceptibility of TNBC to ferroptosis. Sci. Adv. 2020, 6, eaba8968. [Google Scholar] [CrossRef]
- Liao, J.; Liu, P.P.; Hou, G.; Shao, J.; Yang, J.; Liu, K.; Lu, W.; Wen, S.; Hu, Y.; Huang, P. Regulation of stem-like cancer cells by glutamine through β-catenin pathway mediated by redox signaling. Mol. Cancer 2017, 16, 51. [Google Scholar] [CrossRef]
- Li, B.; Cao, Y.; Meng, G.; Qian, L.; Xu, T.; Yan, C.; Luo, O.; Wang, S.; Wei, J.; Ding, Y.; et al. Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBioMedicine 2019, 39, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Prasad, P.; Ghosh, S.; Roy, S.S. Glutamine deficiency promotes stemness and chemoresistance in tumor cells through DRP1-induced mitochondrial fragmentation. Cell Mol. Life Sci. 2021, 78, 4821–4845. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.K.; Lau, P.M.; Kwan, Y.W.; Kong, S.K. Mitochondrial Fuel Dependence on Glutamine Drives Chemo-Resistance in the Cancer Stem Cells of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 3315. [Google Scholar] [CrossRef] [PubMed]
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino acids in cancer. J. Exp. Mol. Med. 2020, 52, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Kawana, K.; Adachi, K.; Fujimoto, A.; Yoshida, M.; Nakamura, H.; Nishida, H.; Inoue, T.; Taguchi, A.; Takahashi, J.; et al. Spheroid cancer stem cells display reprogrammed metabolism and obtain energy by actively running the tricarboxylic acid (TCA) cycle. Oncotarget 2016, 7, 33297–33305. [Google Scholar] [CrossRef]
- Koch, K.; Hartmann, R.; Tsiampali, J.; Uhlmann, C.; Nickel, A.C.; He, X.; Kamp, M.A.; Sabel, M.; Barker, R.A.; Steiger, H.J.; et al. A comparative pharmaco-metabolomic study of glutaminase inhibitors in glioma stem-like cells confirms biological effectiveness but reveals differences in target-specificity. Cell Death Discov. 2020, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Restall, I.J.; Cseh, O.; Richards, L.M.; Pugh, T.J.; Luchman, H.A.; Weiss, S. Brain Tumor Stem Cell Dependence on Glutaminase Reveals a Metabolic Vulnerability through the Amino Acid Deprivation Response Pathway. Cancer Res. 2020, 80, 5478–5490. [Google Scholar] [CrossRef]
- Bridges, R.J.; Stanley, M.S.; Anderson, M.W.; Cotman, C.W.; Chamberlin, A.R. Conformationally defined neurotransmitter analogues. Selective inhibition of glutamate uptake by one pyrrolidine-2,4-dicarboxylate diastereomer. J. Med. Chem. 1991, 34, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Chang, L.C.; Takahashi, K.; Liu, Q.; Schulte, D.A.; Lai, L.; Ibabao, B.; Lin, Y.; Stouffer, N.; Das Mukhopadhyay, C.; et al. Small-molecule activator of glutamate transporter EAAT2 translation provides neuroprotection. J. Clin. Investig. 2014, 124, 1255–1267. [Google Scholar] [CrossRef]
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers 2019, 11, 292. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Cheng, M.; Koch, K.; Marchionni, L.; Fan, X.; Raabe, E.H.; Maciaczyk, J.; Glunde, K.; Eberhart, C.G. Alterations in cellular metabolome after pharmacological inhibition of Notch in glioblastoma cells. Int. J. Cancer 2016, 138, 1246–1255. [Google Scholar] [CrossRef]
- Obara-Michlewska, M.; Szeliga, M. Targeting Glutamine Addiction in Gliomas. Cancers 2020, 12, 310. [Google Scholar] [CrossRef]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.Ø.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef]
- Vande Voorde, J.; Ackermann, T.; Pfetzer, N.; Sumpton, D.; Mackay, G.; Kalna, G.; Nixon, C.; Blyth, K.; Gottlieb, E.; Tardito, S. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci. Adv. 2019, 5, eaau7314. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, K.; Xie, Q.; Wu, Q.; Mack, S.C.; Shi, Y.; Kim, L.J.Y.; Prager, B.C.; Flavahan, W.A.; Liu, X.; et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat. Neurosci. 2017, 5, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Chen, P.; Yan, H.X.; Fu, G.B.; Luo, F.F.; Zhang, J.; Zhao, S.M.; Zhai, B.; Yu, J.H.; Chen, L.; et al. Targeting mTORC2/HDAC3 Inhibits Stemness of Liver Cancer Cells Against Glutamine Starvation. Adv. Sci. 2022, 9, e2103887. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.Y.; Lee, M.S.; Jadhav, U.; Naqvi, I.; Madha, S.; Adler, A.; Mistry, M.; Naumenko, S.; Lewis, C.A.; Hitchcock, D.S.; et al. Adaptation of pancreatic cancer cells to nutrient deprivation is reversible and requires glutamine synthetase stabilization by mTORC1. Proc. Natl. Acad. Sci. USA 2021, 118, e2003014118. [Google Scholar] [CrossRef] [PubMed]
- Pacifico, F.; Badolati, N.; Mellone, S.; Stornaiuolo, M.; Leonardi, A.; Crescenzi, E. Glutamine promotes escape from therapy-induced senescence in tumor cells. Aging 2021, 13, 20962–20991. [Google Scholar] [CrossRef] [PubMed]
- Karimi-Busheri, F.; Rasouli-Nia, A.; Mackey, J.R.; Weinfeld, M. Senescence evasion by MCF-7 human breast tumor-initiating cells. Breast Cancer Res. 2010, 12, R31. [Google Scholar] [CrossRef]
- Achuthan, S.; Santhoshkumar, T.R.; Prabhakar, J.; Nair, S.A.; Pillai, M.R. Drug-induced senescence generates chemoresistant stemlike cells with low reactive oxygen species. J. Biol. Chem. 2011, 286, 37813–37829. [Google Scholar] [CrossRef]
- Saleh, T.; Tyutyunyk-Massey, L.; Gewirtz, D.A. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Disease Recurrence after Dormancy. Cancer Res. 2019, 79, 1044–1046. [Google Scholar] [CrossRef]
- Pacifico, F.; Mellone, S.; D’Incalci, M.; Stornaiuolo, M.; Leonardi, A.; Crescenzi, E. Trabectedin suppresses escape from therapy-induced senescence in tumor cells by interfering with glutamine metabolism. Biochem. Pharmacol. 2022, 202, 115159. [Google Scholar] [CrossRef] [PubMed]
- Barrandon, Y.; Green, H. Three clonal types of keratinocyte with different capacities for multiplication. Proc. Natl. Acad. Sci. USA 1987, 84, 2302–2306. [Google Scholar] [CrossRef]
- Beaver, C.M.; Ahmed, A.; Masters, J.R. Clonogenicity: Holoclones and meroclones contain stem cells. PLoS ONE 2014, 9, e89834. [Google Scholar] [CrossRef]
- Aguilar, E.; Marin de Mas, I.; Zodda, E.; Marin, S.; Morrish, F.; Selivanov, V.; Meca-Cortés, Ó.; Delowar, H.; Pons, M.; Izquierdo, I.; et al. Metabolic Reprogramming and Dependencies Associated with Epithelial Cancer Stem Cells Independent of the Epithelial-Mesenchymal Transition Program. Stem Cells 2016, 34, 1163–1176. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Sun, Y.E. Epigenetic regulation of stem cell differentiation. Pediatr. Res. 2006, 59, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef]
- Yin, Y.; Morgunova, E.; Jolma, A.; Kaasinen, E.; Sahu, B.; Khund-Sayeed, S.; Das, P.K.; Kivioja, T.; Dave, K.; Zhong, F.; et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 2017, 356, eaaj2239. [Google Scholar] [CrossRef]
- Klose, R.J.; Zhang, Y. Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 307–318. [Google Scholar] [CrossRef]
- An, J.; Rao, A.; Ko, M. TET family dioxygenases and DNA demethylation in stem cells and cancers. Exp. Mol. Med. 2017, 49, e323. [Google Scholar] [CrossRef]
- Tran, T.Q.; Hanse, E.A.; Habowski, A.N.; Li, H.; Ishak Gabra, M.B.; Yang, Y.; Lowman, X.H.; Ooi, A.M.; Liao, S.Y.; Edwards, R.A.; et al. α-Ketoglutarate attenuates Wnt signaling and drives differentiation in colorectal cancer. Nat. Cancer 2020, 1, 345–358. [Google Scholar] [CrossRef]
- Shan, Y.; Liang, Z.; Xing, Q.; Zhang, T.; Wang, B.; Tian, S.; Huang, W.; Zhang, Y.; Yao, J.; Zhu, Y.; et al. PRC2 specifies ectoderm lineages and maintains pluripotency in primed but not naïve ESCs. Nat. Commun. 2017, 8, 672. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.; Kang, J.; Kim, T.I.; Joo, C.G. MRI assessment of glutamine uptake correlates with the distribution of glutamine transporters and cancer stem cell markers. Sci. Rep. 2022, 12, 5511. [Google Scholar] [CrossRef]
- Wang, V.M.; Ferreira, R.M.M.; Almagro, J.; Evan, T.; Legrave, N.; Zaw Thin, M.; Frith, D.; Carvalho, J.; Barry, D.J.; Snijders, A.P.; et al. CD9 identifies pancreatic cancer stem cells and modulates glutamine metabolism to fuel tumour growth. Nat. Cell Biol. 2019, 21, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- van Deventer, S.J.; Dunlock, V.E.; van Spriel, A.B. Molecular interactions shaping the tetraspanin web. Biochem. Soc. Trans. 2017, 45, 741–750. [Google Scholar] [CrossRef]
- Yoshida, S.; Fukumoto, S.; Kawaguchi, H.; Sato, S.; Ueda, R.; Furukawa, K. Ganglioside G(D2) in small cell lung cancer cell lines: Enhancement of cell proliferation and mediation of apoptosis. Cancer Res. 2001, 61, 4244–4252. [Google Scholar] [PubMed]
- Esaki, N.; Ohkawa, Y.; Hashimoto, N.; Tsuda, Y.; Ohmi, Y.; Bhuiyan, R.H.; Kotani, N.; Honke, K.; Enomoto, A.; Takahashi, M.; et al. ASC amino acid transporter 2, defined by enzyme-mediated activation of radical sources, enhances malignancy of GD2-positive small-cell lung cancer. Cancer Sci. 2018, 109, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Kamarajan, P.; Rajendiran, T.M.; Kinchen, J.; Bermúdez, M.; Danciu, T.; Kapila, Y.L. Head and Neck Squamous Cell Carcinoma Metabolism Draws on Glutaminolysis, and Stemness Is Specifically Regulated by Glutaminolysis via Aldehyde Dehydrogenase. J. Proteome Res. 2017, 16, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, Z.; Liu, W.; Ai, Z. CD133 promotes the self-renewal capacity of thyroid cancer stem cells through activation of glutamate aspartate transporter SLC1A3 expression. Biochem. Biophys. Res. Commun. 2019, 511, 87–91. [Google Scholar] [CrossRef]
- Tajan, M.; Hock, A.K.; Blagih, J.; Robertson, N.A.; Labuschagne, C.F.; Kruiswijk, F.; Humpton, T.J.; Adams, P.D.; Vousden, K.H. A Role for p53 in the Adaptation to Glutamine Starvation through the Expression of SLC1A3. Cell Metab. 2018, 28, 721–736.e6. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Zhang, Z.; Li, P.; Zhang, Z.; Zeng, M.; Liang, Z.; Li, D.; Wang, L.; Chen, Y.; Liang, Y.; et al. Reprogramming of glutamine metabolism and its impact on immune response in the tumor microenvironment. Cell Commun. Signal. 2022, 20, 114. [Google Scholar] [CrossRef]
- Yang, L.; Achreja, A.; Yeung, T.L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Q.; Du, A.; Li, Y.; Shi, Q.; Chen, Y.; Zhao, Y.; Wang, B.; Pan, F. Adipocytic Glutamine Synthetase Upregulation via Altered Histone Methylation Promotes 5FU Chemoresistance in Peritoneal Carcinomatosis of Colorectal Cancer. Front. Oncol. 2021, 11, 748730. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.H.; Lin, Z.; Flomenberg, N.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P.; Martinez-Outschoorn, U.E. Glutamine fuels a vicious cycle of autophagy in the tumor stroma and oxidative mitochondrial metabolism in epithelial cancer cells: Implications for preventing chemotherapy resistance. Cancer Biol. Ther. 2011, 12, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.H.; Yu, K.; Lucas, J.; White, E.; Abraham, R.T. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci. Signal. 2010, 3, ra31. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvi-ronment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Yan, W.; Wu, X.; Zhou, W.; Fong, M.Y.; Cao, M.; Liu, J.; Liu, X.; Chen, C.H.; Fadare, O.; Pizzo, D.P.; et al. Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells. Nat. Cell Biol. 2018, 20, 597–609. [Google Scholar] [CrossRef]
- Saretzki, G. Cellular senescence in the development and treatment of cancer. Curr. Pharm. Des. 2010, 16, 79–100. [Google Scholar] [CrossRef]
- Johmura, Y.; Yamanaka, T.; Omori, S.; Wang, T.W.; Sugiura, Y.; Matsumoto, M.; Suzuki, N.; Kumamoto, S.; Yamaguchi, K.; Hatakeyama, S.; et al. Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science 2021, 371, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Llop-Hernández, À.; Verdura, S.; Cuyàs, E.; Menendez, J.A. Nutritional Niches of Cancer Therapy-Induced Senescent Cells. Nutrients 2022, 14, 3636. [Google Scholar] [CrossRef]
- Jariyal, H.; Thakkar, H.; Kumar, A.S.; Bhattacharyya, M.; Shah, R.P.; Srivastava, A. Extrinsic hyaluronic acid induction differentially modulates intracellular glutamine metabolism in breast cancer stem cells. Int. J. Biol. Macromol. 2022, 218, 679–689. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cell Type | Gln-Dependent Metabolic Pathway | Impact on Stemness | Stemness Markers | References |
|---|---|---|---|---|
| Embryonal carcinoma (EC) | GSH biosynthesis | + | Oct4, Nanog, Sox2 Sphere formation | [35] |
| Triple-negative breast cancer (TNBC) | GSH biosynthesis | + | GD2 Sphere formation | [36] |
| Prostate cancer | GSH biosynthesis | + | ALDH activity Sphere formation Tumor initiation | [37] |
| Head and neck squamous cell carcinoma (HNSCC) | GSH biosynthesis | + | CD44v Sphere formation Tumor initiation | [38] |
| Pancreatic ductal adenocarcinoma (PDAC) | GOT1-dependent NADPH biosynthesis | + | CD44, CD133, ESA, ALDH1 Sphere formation Tumor initiation | [39] |
| Head and neck squamous cell carcinoma (HNSCC) | glutaminolysis and TCA cycle | + | CD44v | [42] |
| Non-small-cell lung cancer (NSCLC) | IDH2-dependent NADPH biosynthesis | + | Sphere formation | [44] |
| Glioblastoma (GBM) | IDH1-dependent NADPH biosynthesis | + | Sphere formation Tumor initiation | [48] |
| NSCLC, GBM | GSH-mediated β-catenin stability | + | Side population Sox2, ABCG2 Tumor initiation | [50] |
| Hepatocellular carcinoma (HCC) | GSH-mediated β-catenin nuclear translocation | + | Oct4, Nanog, Sox2, CD44, CD133, KLF4 Sphere formation | [51] |
| Epithelial ovarian cancer colorectal cancer (CRC) | ROS-mediated ERK1/2-dependent DRP1 activation | − | Oct4, Sox2, Nanog, ABCG2, CD44 ALDH activity Sphere formation | [52] |
| HCC | mitochondrial ATP production | + | P-glycoprotein CD49, CD99, CD34 | [53] |
| Ovarian clear cell adenocarcinoma (OCCA) Cervical squamous cell carcinoma (CSCC) | Amino acids biosynthesis | + | Sphere formation | [55] |
| GBM | TCA cycle and amino acids biosynthesis | + | Sphere formation | [56] |
| GBM | Amino acids biosynthesis | + | Sphere formation | [57] |
| GBM | Glutamate biosynthesis | + | Sphere formation | [61] |
| GBM | Nucleotides biosynthesis | + | CD133, Sox2, Olig2 | [63] |
| GBM | Nucleotides biosynthesis | + | Sox2, NES, Olig2 Sphere formation Tumor initiation | [65] |
| HCC | Nucleotides biosynthesis | − | Oct4, Sox2, KLF4, CD133 Sphere formation | [66] |
| PDAC | Nucleotides biosynthesis | − | Sphere formation Tumor initiation | [67] |
| Breast carcinoma NSCLC | Nucleotides biosynthesis | + | CD44, CD24 Clonogenicity assay | [68] |
| PDAC | NH4+ production | + | Sphere formation | [75] |
| GBM | IDH1-dependent αKG biosynthesis | + | Sphere formation Tumor initiation | [48] |
| GBM | αKG-dependent histone demethylation | + | Sphere formation Tumor initiation | [48] |
| Prostate cancer | αKG-dependent histone demethylation | + | ALDH activity Sphere formation Tumor initiation | [37] |
| CRC | αKG biosynthesis | − | Lgr5 Organoid formation Tumor initiation | [81] |
| Melanoma TNBC | αKG biosynthesis | − | CD271, ABCB5, CD133, Nanog, Sox2, NES, KLF4 | [1] |
| PDAC | TCA cycle | + | CD44 Organoid formation Tumor initiation | [84] |
| HNSCC | Glutamate biosynthesis and | + | CD44, ALDH | [88] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacifico, F.; Leonardi, A.; Crescenzi, E. Glutamine Metabolism in Cancer Stem Cells: A Complex Liaison in the Tumor Microenvironment. Int. J. Mol. Sci. 2023, 24, 2337. https://doi.org/10.3390/ijms24032337
Pacifico F, Leonardi A, Crescenzi E. Glutamine Metabolism in Cancer Stem Cells: A Complex Liaison in the Tumor Microenvironment. International Journal of Molecular Sciences. 2023; 24(3):2337. https://doi.org/10.3390/ijms24032337
Chicago/Turabian StylePacifico, Francesco, Antonio Leonardi, and Elvira Crescenzi. 2023. "Glutamine Metabolism in Cancer Stem Cells: A Complex Liaison in the Tumor Microenvironment" International Journal of Molecular Sciences 24, no. 3: 2337. https://doi.org/10.3390/ijms24032337
APA StylePacifico, F., Leonardi, A., & Crescenzi, E. (2023). Glutamine Metabolism in Cancer Stem Cells: A Complex Liaison in the Tumor Microenvironment. International Journal of Molecular Sciences, 24(3), 2337. https://doi.org/10.3390/ijms24032337