
The Non-Invasive Assessment of Circulating D-Loop and mt-ccf Levels Opens an Intriguing Spyhole into Novel Approaches for the Tricky Diagnosis of NASH



Abstract
1. Introduction
2. NAFLD Diagnosis and Prognosis: Current Drawbacks and Newly Fashioned Strategies
Genetics and Metabolic Factors Co-Aid NAFLD Diagnosis
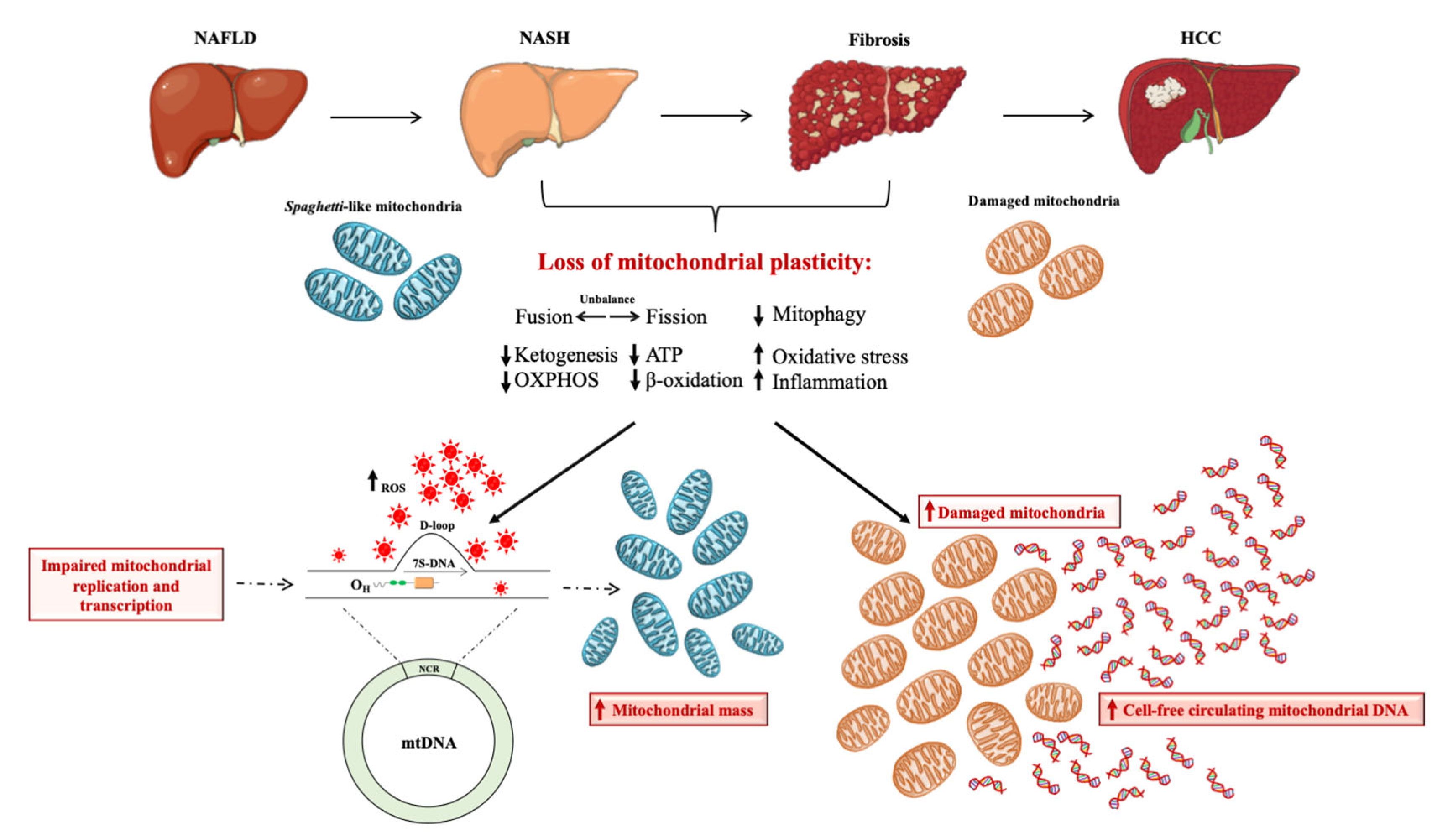
3. Mitochondria as the Hepatocytes’ “Powerhouse”: How do They Contribute to NAFLD?
4. Brief Overview on Mitochondrial DNA Content as Biomarker of Mitochondrial Mass
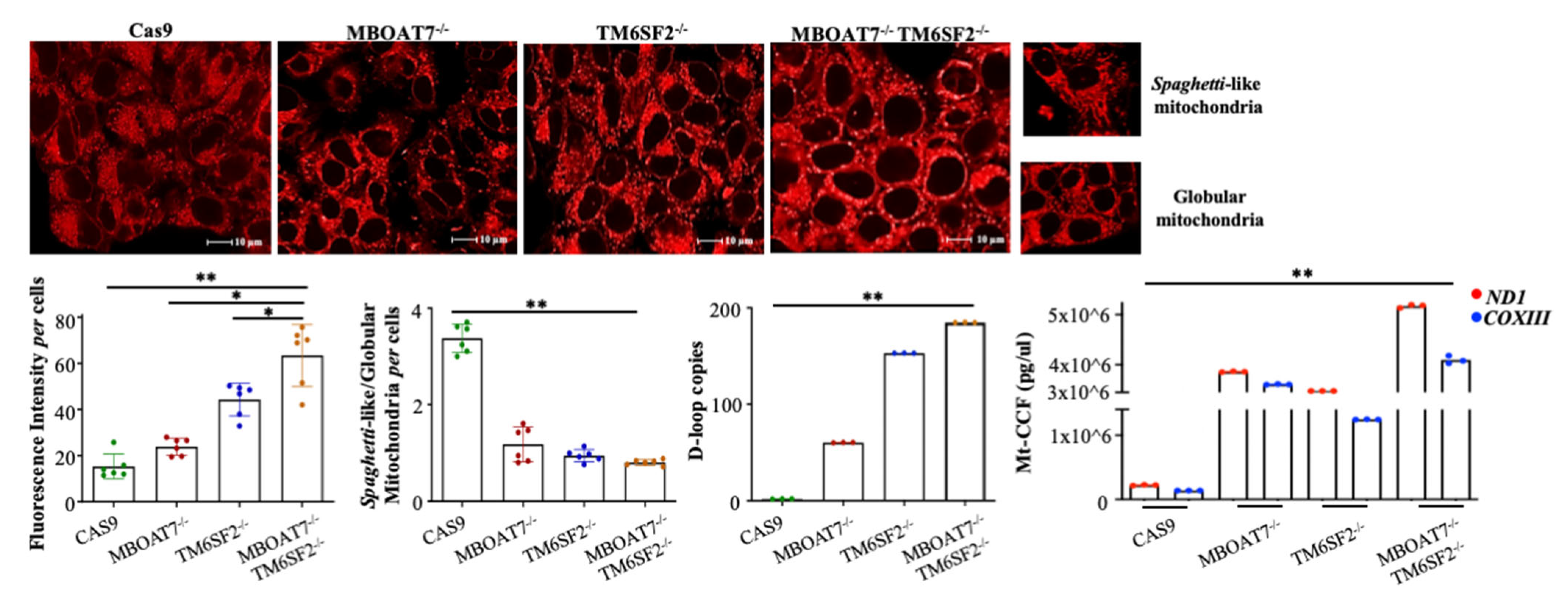
5. The Prognostic Power of Mitochondrial D-Loop Region during NAFLD
6. Clinical Application of Mt-ccf: A Promising Candidate Biomarker in NAFLD Diagnosis
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Marchesini, G.; Pinto-Cortez, H.; Petta, S. Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: Implications for Liver Transplantation. Transplantation 2019, 103, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Paolini, E.; Corsini, A.; Sirtori, C.R.; Ruscica, M. Nonalcoholic fatty liver disease or metabolic dysfunction-associated fatty liver disease diagnoses and cardiovascular diseases: From epidemiology to drug approaches. Eur. J. Clin. Investig. 2021, 51, e13519. [Google Scholar] [CrossRef] [PubMed]
- Meroni, M.; Longo, M. Genetics Is of the Essence to Face NAFLD. Biomedicines 2021, 9, 1359. [Google Scholar] [CrossRef] [PubMed]
- Meroni, M.; Dongiovanni, P.; Longo, M.; Carli, F.; Baselli, G.; Rametta, R.; Pelusi, S.; Badiali, S.; Maggioni, M.; Gaggini, M.; et al. Mboat7 down-regulation by hyper-insulinemia induces fat accumulation in hepatocytes. EBioMedicine 2020, 52, 102658. [Google Scholar] [CrossRef]
- Longo, M.; Meroni, M.; Paolini, E.; Erconi, V.; Carli, F.; Fortunato, F.; Ronchi, D.; Piciotti, R.; Sabatini, S.; Macchi, C.; et al. TM6SF2/PNPLA3/MBOAT7 Loss-of-Function Genetic Variants Impact on NAFLD Development and Progression Both in Patients and in In Vitro Models. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 759–788. [Google Scholar] [CrossRef]
- Longo, M.; Meroni, M.; Paolini, E.; Macchi, C.; Dongiovanni, P. Mitochondrial dynamics and nonalcoholic fatty liver disease (NAFLD): New perspectives for a fairy-tale ending? Metab. Clin. Exp. 2021, 117, 154708. [Google Scholar] [CrossRef]
- Longo, M.; Paolini, E.; Meroni, M.; Dongiovanni, P. Remodeling of Mitochondrial Plasticity: The Key Switch from NAFLD/NASH to HCC. Int. J. Mol. Sci. 2021, 22, 4173. [Google Scholar] [CrossRef]
- Léveillé, M.; Estall, J.L. Mitochondrial Dysfunction in the Transition from NASH to HCC. Metabolites 2019, 9, 233. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef]
- Clayton, D.A. Transcription and replication of mitochondrial DNA. Hum. Reprod. 2000, 15, 11–17. [Google Scholar] [CrossRef]
- Hasturk, B.; Yilmaz, Y.; Eren, F. Potential clinical variants detected in mitochondrial DNA D-loop hypervariable region I of patients with non-alcoholic steatohepatitis. Hormones 2019, 18, 463–475. [Google Scholar] [CrossRef]
- Abd Radzak, S.M.; Mohd Khair, S.Z.N.; Ahmad, F.; Patar, A.; Idris, Z.; Mohamed Yusoff, A.A. Insights regarding mitochondrial DNA copy number alterations in human cancer (Review). Int. J. Mol. Med. 2022, 50, 104. [Google Scholar] [CrossRef]
- Garcia-Martinez, I.; Santoro, N.; Chen, Y.; Hoque, R.; Ouyang, X.; Caprio, S.; Shlomchik, M.J.; Coffman, R.L.; Candia, A.; Mehal, W.Z. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Investig. 2016, 126, 859–864. [Google Scholar] [CrossRef]
- An, P.; Wei, L.L.; Zhao, S.; Sverdlov, D.Y.; Vaid, K.A.; Miyamoto, M.; Kuramitsu, K.; Lai, M.; Popov, Y.V. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun. 2020, 11, 2362. [Google Scholar] [CrossRef]
- Cormio, A.; Milella, F.; Marra, M.; Pala, M.; Lezza, A.M.; Bonfigli, A.R.; Franceschi, C.; Cantatore, P.; Gadaleta, M.N. Variations at the H-strand replication origins of mitochondrial DNA and mitochondrial DNA content in the blood of type 2 diabetes patients. Biochim. Biophys. Acta 2009, 1787, 547–552. [Google Scholar] [CrossRef]
- Kamfar, S.; Alavian, S.M.; Houshmand, M.; Yadegarazari, R.; Seifi Zarei, B.; Khalaj, A.; Shabab, N.; Saidijam, M. Liver Mitochondrial DNA Copy Number and Deletion Levels May Contribute to Nonalcoholic Fatty Liver Disease Susceptibility. Hepat. Mon. 2016, 16, e40774. [Google Scholar] [CrossRef]
- Pirola, C.J.; Gianotti, T.F.; Burgueño, A.L.; Rey-Funes, M.; Loidl, C.F.; Mallardi, P.; Martino, J.S.; Castaño, G.O.; Sookoian, S. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 2013, 62, 1356. [Google Scholar] [CrossRef]
- Lee, H.C.; Yin, P.H.; Lu, C.Y.; Chi, C.W.; Wei, Y.H. Increase of mitochondria and mitochondrial DNA in response to oxidative stress in human cells. Biochem. J. 2000, 348, 425–432. [Google Scholar] [CrossRef]
- Wong, L.J.; Tan, D.J.; Bai, R.K.; Yeh, K.T.; Chang, J. Molecular alterations in mitochondrial DNA of hepatocellular carcinomas: Is there a correlation with clinicopathological profile? J. Med. Genet. 2004, 41, e65. [Google Scholar] [CrossRef]
- Qiao, L.; Ru, G.; Mao, Z.; Wang, C.; Nie, Z.; Li, Q.; Huang-yang, Y.; Zhu, L.; Liang, X.; Yu, J.; et al. Mitochondrial DNA depletion, mitochondrial mutations and high TFAM expression in hepatocellular carcinoma. Oncotarget 2017, 8, 84373–84383. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Flichman, D.; Scian, R.; Rohr, C.; Dopazo, H.; Gianotti, T.F.; Martino, J.S.; Castaño, G.O.; Pirola, C.J. Mitochondrial genome architecture in non-alcoholic fatty liver disease. J. Pathol. 2016, 240, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Qi, Y.; Cui, X.; Sun, Y.; Huo, Q.; Yang, Y.; Wen, X.; Tan, M.; Du, S.; Zhang, H.; et al. Heteroplasmy and Copy Number Variations of Mitochondria in 88 Hepatocellular Carcinoma Individuals. J. Cancer 2017, 8, 4011–4017. [Google Scholar] [CrossRef] [PubMed]
- Tsai, E.; Lee, T.P. Diagnosis and Evaluation of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis, Including Noninvasive Biomarkers and Transient Elastography. Clin. Liver Dis. 2018, 22, 73–92. [Google Scholar] [CrossRef]
- Shirazi, F.; Wang, J.; Wong, R.J. Nonalcoholic Steatohepatitis Becomes the Leading Indication for Liver Transplant Registrants Among US Adults Born Between 1945 and 1965. J. Clin. Exp. Hepatol. 2020, 10, 30–36. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378.e365; quiz e314–e365. [Google Scholar] [CrossRef]
- Nalbantoglu, I.L.; Brunt, E.M. Role of liver biopsy in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 9026–9037. [Google Scholar] [CrossRef]
- Ferraioli, G.; Soares Monteiro, L.B. Ultrasound-based techniques for the diagnosis of liver steatosis. World J. Gastroenterol. 2019, 25, 6053–6062. [Google Scholar] [CrossRef]
- European Association for Study of Liver. EASL-ALEH Clinical Practice Guidelines: Non-invasive tests for evaluation of liver disease severity and prognosis. J. Hepatol. 2015, 63, 237–264. [Google Scholar] [CrossRef]
- Coco, B.; Oliveri, F.; Maina, A.M.; Ciccorossi, P.; Sacco, R.; Colombatto, P.; Bonino, F.; Brunetto, M.R. Transient elastography: A new surrogate marker of liver fibrosis influenced by major changes of transaminases. J. Viral Hepat. 2007, 14, 360–369. [Google Scholar] [CrossRef]
- Foucher, J.; Castéra, L.; Bernard, P.H.; Adhoute, X.; Laharie, D.; Bertet, J.; Couzigou, P.; de Lédinghen, V. Prevalence and factors associated with failure of liver stiffness measurement using FibroScan in a prospective study of 2114 examinations. Eur. J. Gastroenterol. Hepatol. 2006, 18, 411–412. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 14205–14218. [Google Scholar] [CrossRef]
- Malik, A.N.; Czajka, A. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion 2013, 13, 481–492. [Google Scholar] [CrossRef]
- Ma, C.; Liu, Y.; He, S.; Zeng, J.; Li, P.; Ma, C.; Ping, F.; Zhang, H.; Xu, L.; Li, W.; et al. Association Between Leukocyte Mitochondrial DNA Copy Number and Non-alcoholic Fatty Liver Disease in a Chinese Population Is Mediated by 8-Oxo-2′-Deoxyguanosine. Front. Med. 2020, 7, 536. [Google Scholar] [CrossRef]
- Lewis, C.M.; Vassos, E. Polygenic risk scores: From research tools to clinical instruments. Genome Med. 2020, 12, 44. [Google Scholar] [CrossRef]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Paolini, E.; Lombardi, R.; Piciotti, R.; Francione, P.; Badiali, S.; Maggioni, M.; Fracanzani, A.L. MAFLD definition underestimates the risk to develop HCC in genetically predisposed patients. J. Intern. Med. 2022, 291, 374–376. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Stender, S.; Pietrelli, A.; Mancina, R.M.; Cespiati, A.; Petta, S.; Pelusi, S.; Pingitore, P.; Badiali, S.; Maggioni, M.; et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J. Intern. Med. 2018, 283, 356–370. [Google Scholar] [CrossRef]
- Di Costanzo, A.; Pacifico, L.; Chiesa, C.; Perla, F.M.; Ceci, F.; Angeloni, A.; D’Erasmo, L.; Di Martino, M.; Arca, M. Genetic and metabolic predictors of hepatic fat content in a cohort of Italian children with obesity. Pediatr. Res. 2019, 85, 671–677. [Google Scholar] [CrossRef]
- Suomela, E.; Oikonen, M.; Pitkänen, N.; Ahola-Olli, A.; Virtanen, J.; Parkkola, R.; Jokinen, E.; Laitinen, T.; Hutri-Kähönen, N.; Kähönen, M.; et al. Childhood predictors of adult fatty liver. The Cardiovascular Risk in Young Finns Study. J. Hepatol. 2016, 65, 784–790. [Google Scholar] [CrossRef]
- Bianco, C.; Jamialahmadi, O.; Pelusi, S.; Baselli, G.; Dongiovanni, P.; Zanoni, I.; Santoro, L.; Maier, S.; Liguori, A.; Meroni, M.; et al. Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. J. Hepatol. 2021, 74, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Dessein, A. Clinical utility of polygenic risk scores for predicting NAFLD disorders. J. Hepatol. 2021, 74, 769–770. [Google Scholar] [CrossRef] [PubMed]
- Shum, M.; Ngo, J.; Shirihai, O.S.; Liesa, M. Mitochondrial oxidative function in NAFLD: Friend or foe? Mol. Metab. 2021, 50, 101134. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, Y.; Zhang, H.X.; Guo, J.R.; Lam, C.W.K.; Wang, C.Y.; Zhang, W. Mitochondria-Mediated Pathogenesis and Therapeutics for Non-Alcoholic Fatty Liver Disease. Mol. Nutr. Food Res. 2019, 63, e1900043. [Google Scholar] [CrossRef] [PubMed]
- Bonekamp, N.A.; Larsson, N.G. SnapShot: Mitochondrial Nucleoid. Cell 2018, 172, 388. [Google Scholar] [CrossRef]
- Nicholls, T.J.; Minczuk, M. In D-loop: 40 years of mitochondrial 7S DNA. Exp. Gerontol. 2014, 56, 175–181. [Google Scholar] [CrossRef]
- Filograna, R.; Mennuni, M. Mitochondrial DNA copy number in human disease: The more the better? FEBS Lett. 2021, 595, 976–1002. [Google Scholar] [CrossRef]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Shami, G.J.; Cheng, D.; Verhaegh, P.; Koek, G.; Wisse, E.; Braet, F. Three-dimensional ultrastructure of giant mitochondria in human non-alcoholic fatty liver disease. Sci. Rep. 2021, 11, 3319. [Google Scholar] [CrossRef]
- Straub, B.K.; Herpel, E.; Singer, S.; Zimbelmann, R.; Breuhahn, K.; Macher-Goeppinger, S.; Warth, A.; Lehmann-Koch, J.; Longerich, T.; Heid, H.; et al. Lipid droplet-associated PAT-proteins show frequent and differential expression in neoplastic steatogenesis. Mod. Pathol. 2010, 23, 480–492. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Paolini, E.; Tria, G.; Ripolone, M.; Napoli, L.; Moggio, M.; Fracanzani, A.L.; Dongiovanni, P. Expanding the phenotypic spectrum of non-alcoholic fatty liver disease and hypertriglyceridemia. Front. Nutr. 2022, 9, 967899. [Google Scholar] [CrossRef]
- Pessayre, D. Role of mitochondria in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2007, 22, S20–S27. [Google Scholar] [CrossRef]
- Chinnery, P.F.; Thorburn, D.R.; Samuels, D.C.; White, S.L.; Dahl, H.M.; Turnbull, D.M.; Lightowlers, R.N.; Howell, N. The inheritance of mitochondrial DNA heteroplasmy: Random drift, selection or both? Trends Genet. TIG 2000, 16, 500–505. [Google Scholar] [CrossRef]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef]
- Dornas, W.; Schuppan, D. Mitochondrial oxidative injury: A key player in nonalcoholic fatty liver disease. Am. J. Physiol. -Gastrointest. Liver Physiol. 2020, 319, G400–G411. [Google Scholar] [CrossRef]
- Pérez-Amado, C.J.; Bazan-Cordoba, A.; Hidalgo-Miranda, A. Mitochondrial Heteroplasmy Shifting as a Potential Biomarker of Cancer Progression. Int. J. Mol. Sci. 2021, 22, 7369. [Google Scholar] [CrossRef]
- Handy, D.E.; Loscalzo, J. Redox regulation of mitochondrial function. Antioxid. Redox Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef]
- Malik, A.N.; Simões, I.C.M.; Rosa, H.S.; Khan, S.; Karkucinska-Wieckowska, A.; Wieckowski, M.R. A Diet Induced Maladaptive Increase in Hepatic Mitochondrial DNA Precedes OXPHOS Defects and May Contribute to Non-Alcoholic Fatty Liver Disease. Cells 2019, 8, 1222. [Google Scholar] [CrossRef]
- Lee, H.C.; Wei, Y.H. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int. J. Biochem. Cell Biol. 2005, 37, 822–834. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Platek, M.; Mahasneh, A.; Ambrosone, C.B.; Zhao, H. Mitochondrial copy number and risk of breast cancer: A pilot study. Mitochondrion 2010, 10, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.W.; Masayesva, B.; Zahurak, M.; Carvalho, A.L.; Rosenbaum, E.; Mambo, E.; Zhou, S.; Minhas, K.; Benoit, N.; Westra, W.H.; et al. Increased mitochondrial DNA content in saliva associated with head and neck cancer. Clin. Cancer Res. 2005, 11, 2486–2491. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Zhou, Y.; Shi, Y.; Ning, L.; Yang, Y.; Wei, X.; Zhang, N.; Hao, X.; Niu, R. Reduced mitochondrial DNA copy number is correlated with tumor progression and prognosis in Chinese breast cancer patients. IUBMB Life 2007, 59, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Fan, A.X.; Radpour, R.; Haghighi, M.M.; Kohler, C.; Xia, P.; Hahn, S.; Holzgreve, W.; Zhong, X.Y. Mitochondrial DNA content in paired normal and cancerous breast tissue samples from patients with breast cancer. J. Cancer Res. Clin. Oncol. 2009, 135, 983–989. [Google Scholar] [CrossRef]
- Nomoto, S.; Yamashita, K.; Koshikawa, K.; Nakao, A.; Sidransky, D. Mitochondrial D-loop mutations as clonal markers in multicentric hepatocellular carcinoma and plasma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 481–487. [Google Scholar]
- Okochi, O.; Hibi, K.; Uemura, T.; Inoue, S.; Takeda, S.; Kaneko, T.; Nakao, A. Detection of mitochondrial DNA alterations in the serum of hepatocellular carcinoma patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 2875–2878. [Google Scholar]
- Lee, H.K.; Song, J.H.; Shin, C.S.; Park, D.J.; Park, K.S.; Lee, K.U.; Koh, C.S. Decreased mitochondrial DNA content in peripheral blood precedes the development of non-insulin-dependent diabetes mellitus. Diabetes Res. Clin. Pract. 1998, 42, 161–167. [Google Scholar] [CrossRef]
- Wong, J.; McLennan, S.V.; Molyneaux, L.; Min, D.; Twigg, S.M.; Yue, D.K. Mitochondrial DNA content in peripheral blood monocytes: Relationship with age of diabetes onsetand diabetic complications. Diabetologia 2009, 52, 1953–1961. [Google Scholar] [CrossRef]
- Cho, S.B.; Koh, I.; Nam, H.-Y.; Jeon, J.-P.; Lee, H.K.; Han, B.-G. Mitochondrial DNA copy number augments performance of A1C and oral glucose tolerance testing in the prediction of type 2 diabetes. Sci. Rep. 2017, 7, 43203. [Google Scholar] [CrossRef]
- Weng, S.W.; Lin, T.K.; Liou, C.W.; Chen, S.D.; Wei, Y.H.; Lee, H.C.; Chen, I.Y.; Hsieh, C.J.; Wang, P.W. Peripheral blood mitochondrial DNA content and dysregulation of glucose metabolism. Diabetes Res. Clin. Pract. 2009, 83, 94–99. [Google Scholar] [CrossRef]
- Malik, A.N.; Shahni, R.; Iqbal, M.M. Increased peripheral blood mitochondrial DNA in type 2 diabetic patients with nephropathy. Diabetes Res. Clin. Pract. 2009, 86, e22–e24. [Google Scholar] [CrossRef]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.G. Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Miralles Fusté, J.; Shi, Y.; Wanrooij, S.; Zhu, X.; Jemt, E.; Persson, Ö.; Sabouri, N.; Gustafsson, C.M.; Falkenberg, M. In vivo occupancy of mitochondrial single-stranded DNA binding protein supports the strand displacement mode of DNA replication. PLoS Genet. 2014, 10, e1004832. [Google Scholar] [CrossRef]
- Arnberg, A.; van Bruggen, E.F.; Borst, P. The presence of DNA molecules with a displacement loop in standard mitochondrial DNA preparations. Biochim. Et Biophys. Acta 1971, 246, 353–357. [Google Scholar] [CrossRef]
- He, J.; Mao, C.C.; Reyes, A.; Sembongi, H.; Di Re, M.; Granycome, C.; Clippingdale, A.B.; Fearnley, I.M.; Harbour, M.; Robinson, A.J.; et al. The AAA+ protein ATAD3 has displacement loop binding properties and is involved in mitochondrial nucleoid organization. J. Cell Biol. 2007, 176, 141–146. [Google Scholar] [CrossRef]
- Polyak, K.; Li, Y.; Zhu, H.; Lengauer, C.; Willson, J.K.; Markowitz, S.D.; Trush, M.A.; Kinzler, K.W.; Vogelstein, B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat. Genet. 1998, 20, 291–293. [Google Scholar] [CrossRef]
- Fliss, M.S.; Usadel, H.; Caballero, O.L.; Wu, L.; Buta, M.R.; Eleff, S.M.; Jen, J.; Sidransky, D. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science 2000, 287, 2017–2019. [Google Scholar] [CrossRef]
- Lee, H.C.; Li, S.H.; Lin, J.C.; Wu, C.C.; Yeh, D.C.; Wei, Y.H. Somatic mutations in the D-loop and decrease in the copy number of mitochondrial DNA in human hepatocellular carcinoma. Mutat. Res. 2004, 547, 71–78. [Google Scholar] [CrossRef]
- Tamori, A.; Nishiguchi, S.; Nishikawa, M.; Kubo, S.; Koh, N.; Hirohashi, K.; Shiomi, S.; Inoue, M. Correlation between clinical characteristics and mitochondrial D-loop DNA mutations in hepatocellular carcinoma. J. Gastroenterol. 2004, 39, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Kamfar, S.; Alavian, S.M.; Hasrak, K.; Houshmand, M.; Seifi Zarei, B.; Khalaj, A.; Homaunpur, F.; Saidijam, M. Analysis of Mitochondrial 4977-bp Deletion and D-Loop Variation in Iranian Non-Alcoholic Fatty Liver Disease Patients. Hepat. Mon. 2019, 19, e84553. [Google Scholar] [CrossRef]
- Thurairajah, K.; Briggs, G.D.; Balogh, Z.J. The source of cell-free mitochondrial DNA in trauma and potential therapeutic strategies. Eur. J. Trauma Emerg. Surg. 2018, 44, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Nakahira, K.; Guo, X.; Choi, A.M.; Gu, Z. Very Short Mitochondrial DNA Fragments and Heteroplasmy in Human Plasma. Sci. Rep. 2016, 6, 36097. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, K.; Guo, S.; Wang, Y.; Ji, X.; Yuan, Q.; Su, L.; Guo, X.; Gu, X.; Xing, J. NGS-based accurate and efficient detection of circulating cell-free mitochondrial DNA in cancer patients. Mol. Ther. Nucleic Acids 2021, 23, 657–666. [Google Scholar] [CrossRef]
- Kohler, C.; Radpour, R.; Barekati, Z.; Asadollahi, R.; Bitzer, J.; Wight, E.; Bürki, N.; Diesch, C.; Holzgreve, W.; Zhong, X.Y. Levels of plasma circulating cell free nuclear and mitochondrial DNA as potential biomarkers for breast tumors. Mol. Cancer 2009, 8, 105. [Google Scholar] [CrossRef]
- Rainer, T.H.; Wong, L.K.; Lam, W.; Yuen, E.; Lam, N.Y.; Metreweli, C.; Lo, Y.M. Prognostic use of circulating plasma nucleic acid concentrations in patients with acute stroke. Clin. Chem. 2003, 49, 562–569. [Google Scholar] [CrossRef]
- Ellinger, J.; Müller, S.C.; Wernert, N.; von Ruecker, A.; Bastian, P.J. Mitochondrial DNA in serum of patients with prostate cancer: A predictor of biochemical recurrence after prostatectomy. BJU Int. 2008, 102, 628–632. [Google Scholar] [CrossRef]
- Zhou, G.; Li, Y.; Li, S.; Liu, H.; Xu, F.; Lai, X.; Zhang, Q.; Xu, J.; Wan, S. Circulating Cell-Free mtDNA Content as a Non-invasive Prognostic Biomarker in HCC Patients Receiving TACE and Traditional Chinese Medicine. Front. Genet. 2021, 12, 719451. [Google Scholar] [CrossRef]
- Heitzer, E.; Haque, I.S. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef]
- Karlas, T.; Weise, L.; Kuhn, S.; Krenzien, F.; Mehdorn, M.; Petroff, D.; Linder, N.; Schaudinn, A.; Busse, H.; Keim, V.; et al. Correlation of cell-free DNA plasma concentration with severity of non-alcoholic fatty liver disease. J. Transl. Med. 2017, 15, 106. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| mt-DNA Region | Mutations | NAFLD Stages | Number of Cases | Other Diseases | References |
|---|---|---|---|---|---|
| -loop | 94 variations: 2 deletions, 4 insertions and 88 single nucleotide polymorphisms | Steatosis, NASH, fibrosis (NAFLD) | 43 NAFLD patients | - | Kamfar S. et al., 2019 [17] |
| D-loop | m.16318 A>C, CC | NASH | 150 NASH patients | MetS, T2DM, and hypothyroidism | Hasturk B et al., 2019 [12] |
| D-loop | cytosine methylation (5mC) | NASH | 45 NAFLD patients | - | Pirola CJ et al., 2013 [18] |
| D-loop | m.16129 G>A, AA | Fibrosis Cirrhosis | 150 NASH patients | - | Hasturk B et al., 2019 [12] |
| D-loop | m.16249 T>C, CC | Steatosis Lobular inflammation | 150 NASH patients | - | Hasturk B et al., 2019 [12] |
| D-loop | polycytidine stretch between np 303–309 | HCC | 61 HCC patients: 40.7% | - | Lee HC et al., 2004 [19] |
| D-loop | (CA)n dinucleotide repeat at np 514 | HCC | 61 HCC patients: 37.0% (10/27) | - | Lee HC et al., 2004 [19] |
| D-loop | 50bp deletion between np 298/306 | HCC | 61 HCC patients: 78% | - | Lee HC et al., 2004 [19] |
| D-loop | 23 somatic mutations | HCC | 20 HCC patients: 50% | Other tumors (breast, colon, thyroid cancers) | Wong LJ et al., 2004 [20] |
| D-loop | 21 substituitions CCCC insertion at 573 position | HCC | 86 HCC patients: 30.43% (14/46) | - | Qiao L et al., 2017 [21] |
| NADH dehydrogenase | MT-ND6 (cytosine methylation (5mC)) | NASH | 45 NAFLD patients | - | Pirola CJ et al., 2013 [18] |
| NADH dehydrogenase | MT-ND4 (m.11040 T > C) MT-ND2 (m.4769 A > G) | NASH | 64 NAFLD patients | - | Sookoian S et al., 2016 [22] |
| MT-ATP6, MT-CYB, MT-CO1 | MT-ATP6 (m.9101 T > C) MT-CYB (m.14766 C > T) MT-CO1 (m.7028 C > T) | NASH | 64 NAFLD patients | - | Sookoian S et al., 2016 [22] |
| NADH dehydrogenase | MT-ND5 (m.13676 A>G) | HCC | 86 HCC patients: 19.57% (9/46) | - | Qiao L.+ et al., 2017 [21] |
| NADH dehydrogenase | Heteroplasmic mutations (MT-ND1, MT-ND3, MT-ND4, MT-ND5, MT-ND6) | HCC | 88 HCC patients | - | Li W et al.,2017 [23] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paolini, E.; Longo, M.; Corsini, A.; Dongiovanni, P. The Non-Invasive Assessment of Circulating D-Loop and mt-ccf Levels Opens an Intriguing Spyhole into Novel Approaches for the Tricky Diagnosis of NASH. Int. J. Mol. Sci. 2023, 24, 2331. https://doi.org/10.3390/ijms24032331
Paolini E, Longo M, Corsini A, Dongiovanni P. The Non-Invasive Assessment of Circulating D-Loop and mt-ccf Levels Opens an Intriguing Spyhole into Novel Approaches for the Tricky Diagnosis of NASH. International Journal of Molecular Sciences. 2023; 24(3):2331. https://doi.org/10.3390/ijms24032331
Chicago/Turabian StylePaolini, Erika, Miriam Longo, Alberto Corsini, and Paola Dongiovanni. 2023. "The Non-Invasive Assessment of Circulating D-Loop and mt-ccf Levels Opens an Intriguing Spyhole into Novel Approaches for the Tricky Diagnosis of NASH" International Journal of Molecular Sciences 24, no. 3: 2331. https://doi.org/10.3390/ijms24032331
APA StylePaolini, E., Longo, M., Corsini, A., & Dongiovanni, P. (2023). The Non-Invasive Assessment of Circulating D-Loop and mt-ccf Levels Opens an Intriguing Spyhole into Novel Approaches for the Tricky Diagnosis of NASH. International Journal of Molecular Sciences, 24(3), 2331. https://doi.org/10.3390/ijms24032331