
Role of Glycolytic and Glutamine Metabolism Reprogramming on the Proliferation, Invasion, and Apoptosis Resistance through Modulation of Signaling Pathways in Glioblastoma

 , ,
, ,
Abstract
:Simple Summary
Abstract

1. Introduction
2. Glycolysis in Cancer Cells
2.1. The Warburg Effect and Glucose Avidity in Glioblastoma
2.2. Glycolytic Enzymes in Glioma
2.2.1. Hexokinase 2
2.2.2. Glucose-6-Phosphate Isomerase (G6PI)
2.2.3. Phosphofructokinase
2.2.4. Aldolase
2.2.5. Glyceraldehyde 3 Phosphate Dehydrogenase
2.2.6. Phosphoglycerate Kinase (PGK)
2.2.7. Phosphoglycerate Mutase (PGM)
2.2.8. Enolase
2.2.9. Pyruvate Kinase
2.2.10. Lactate Dehydrogenase
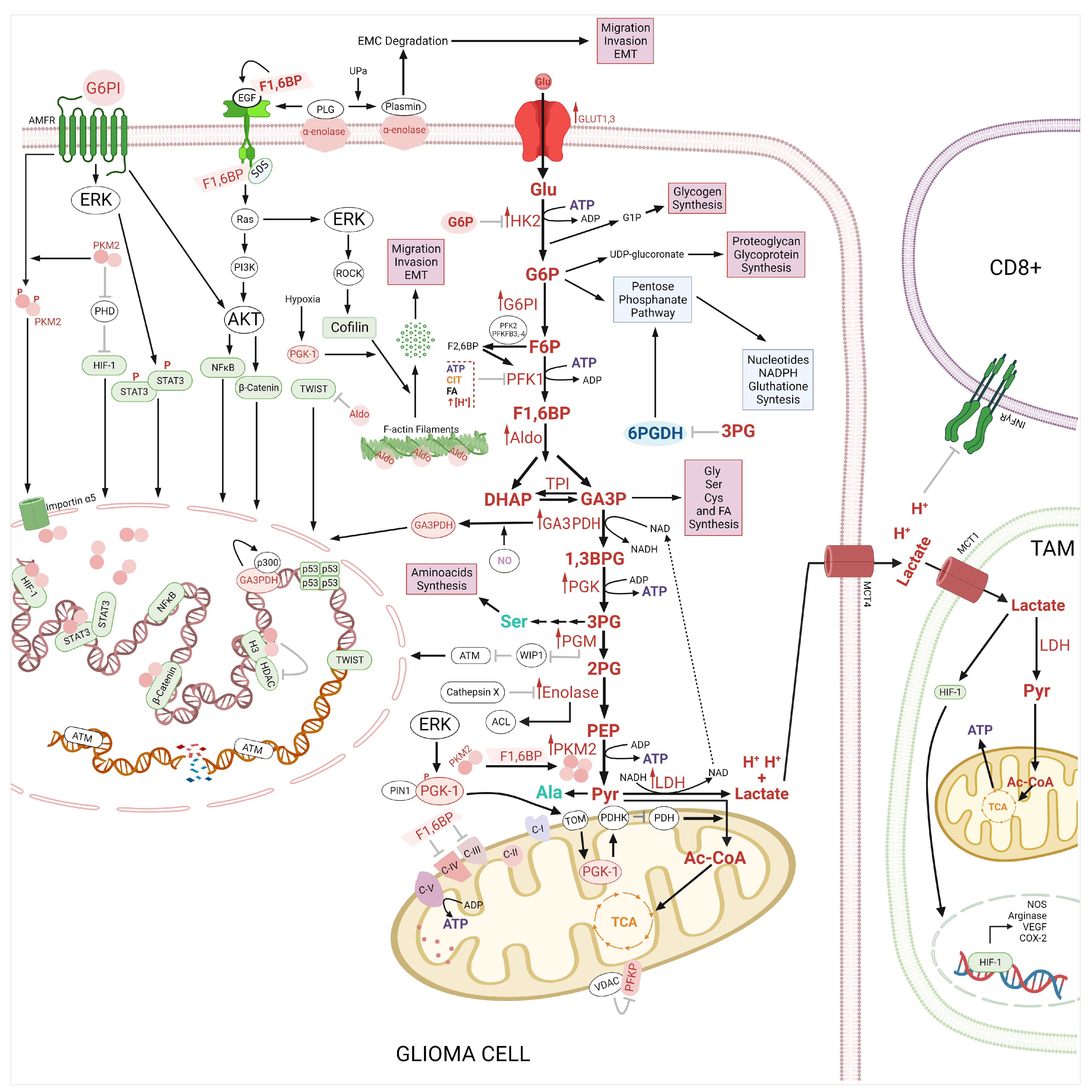
2.3. Therapeutic Implications of Glycolysis and Lipid Metabolism in the Apoptosis Regulation in Glioma
2.4. Regulation of the Autophagy by Glycolysis and Lipid Metabolism
2.5. Glycolysis as a Target of Therapeutic Drugs in Gliomas
3. Glutaminase Pathway
3.1. Glutaminase Pathway in Glioblastoma
3.1.1. Glutamine Synthetase
3.1.2. Glutaminase
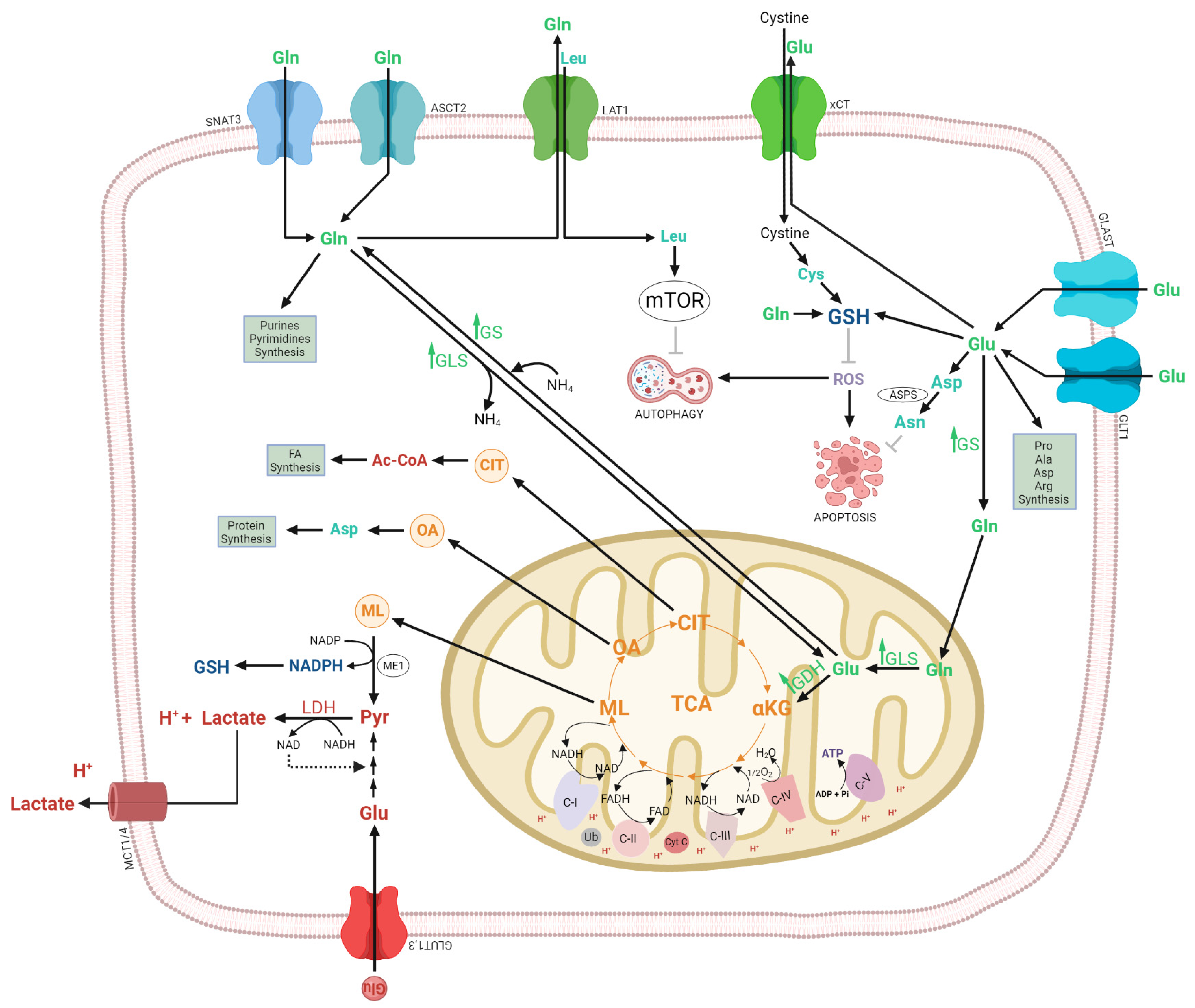
3.1.3. Glutamate Dehydrogenase
3.2. Regulation of the Autophagy by Glutamine
3.3. Glutaminolisis as a Target of Therapeutic Drugs in Gliomas
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crocetti, E.; Trama, A.; Stiller, C.; Caldarella, A.; Soffietti, R.; Jaal, J.; Weber, D.C.; Ricardi, U.; Slowinski, J.; Brandes, A.; et al. Epidemiology of glial and non-glial brain tumours in Europe. Eur. J. Cancer 2012, 48, 1532–1542. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Grobben, B.; De Deyn, P.P.; Slegers, H. Rat C6 glioma as experimental model system for the study of glioblastoma growth and invasion. Cell Tissue Res. 2002, 310, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.J.; Lyssiotis, C.A.; Wahl, D.R. Clinical Targeting of Altered Metabolism in High-Grade Glioma. Cancer J. 2021, 27, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Mischel, P.S.; Cloughesy, T.F. Targeted molecular therapy of GBM. Brain Pathol. 2003, 13, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Oprita, A.; Baloi, S.C.; Staicu, G.A.; Alexandru, O.; Tache, D.E.; Danoiu, S.; Micu, E.S.; Sevastre, A.S. Updated Insights on EGFR Signaling Pathways in Glioma. Int. J. Mol. Sci. 2021, 22, 587. [Google Scholar] [CrossRef]
- Huang, P.H.; Xu, A.M.; White, F.M. Oncogenic EGFR signaling networks in glioma. Sci. Signal 2009, 2, re6. [Google Scholar] [CrossRef]
- Bjorkblom, B.; Wibom, C.; Eriksson, M.; Bergenheim, A.T.; Sjoberg, R.L.; Jonsson, P.; Brannstrom, T.; Antti, H.; Sandstrom, M.; Melin, B. Distinct metabolic hallmarks of WHO classified adult glioma subtypes. Neuro Oncol. 2022, 24, 1454–1468. [Google Scholar] [CrossRef]
- Sampetrean, O.; Saya, H. What can metabolites tell us about gliomas? Neuro Oncol. 2022, 24, 1469–1470. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Shao, F.; Bian, X.; Meng, Y.; Liang, T.; Lu, Z. The Evolving Landscape of Noncanonical Functions of Metabolic Enzymes in Cancer and Other Pathologies. Cell Metab. 2021, 33, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Hunter, T. Metabolic Kinases Moonlighting as Protein Kinases. Trends Biochem. Sci. 2018, 43, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; Enerback, S. Lactate: The ugly duckling of energy metabolism. Nat. Metab. 2020, 2, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Marin-Hernandez, A.; Gallardo-Perez, J.C.; Rodriguez-Enriquez, S.; Encalada, R.; Moreno-Sanchez, R.; Saavedra, E. Modeling cancer glycolysis. Biochim. Biophys. Acta 2011, 1807, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Orozco, J.M.; Krawczyk, P.A.; Scaria, S.M.; Cangelosi, A.L.; Chan, S.H.; Kunchok, T.; Lewis, C.A.; Sabatini, D.M. Dihydroxyacetone phosphate signals glucose availability to mTORC1. Nat. Metab. 2020, 2, 893–901. [Google Scholar] [CrossRef]
- Suzuki, M.; Sasabe, J.; Miyoshi, Y.; Kuwasako, K.; Muto, Y.; Hamase, K.; Matsuoka, M.; Imanishi, N.; Aiso, S. Glycolytic flux controls D-serine synthesis through glyceraldehyde-3-phosphate dehydrogenase in astrocytes. Proc. Natl. Acad. Sci. USA 2015, 112, E2217–E2224. [Google Scholar] [CrossRef]
- Wiese, E.K.; Hitosugi, T. Tyrosine Kinase Signaling in Cancer Metabolism: PKM2 Paradox in the Warburg Effect. Front. Cell Dev. Biol. 2018, 6, 79. [Google Scholar] [CrossRef]
- Icard, P.; Simula, L.; Zahn, G.; Alifano, M.; Mycielska, M.E. The dual role of citrate in cancer. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188987. [Google Scholar] [CrossRef]
- Icard, P.; Poulain, L.; Lincet, H. Understanding the central role of citrate in the metabolism of cancer cells. Biochim. Biophys. Acta 2012, 1825, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Icard, P.; Lincet, H. The reduced concentration of citrate in cancer cells: An indicator of cancer aggressiveness and a possible therapeutic target. Drug Resist. Updat. 2016, 29, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Ruiz, R.; Averet, N.; Araiza, D.; Pinson, B.; Uribe-Carvajal, S.; Devin, A.; Rigoulet, M. Mitochondrial oxidative phosphorylation is regulated by fructose 1,6-bisphosphate. A possible role in Crabtree effect induction? J. Biol. Chem. 2008, 283, 26948–26955. [Google Scholar] [CrossRef] [PubMed]
- Peeters, K.; Van Leemputte, F.; Fischer, B.; Bonini, B.M.; Quezada, H.; Tsytlonok, M.; Haesen, D.; Vanthienen, W.; Bernardes, N.; Gonzalez-Blas, C.B.; et al. Fructose-1,6-bisphosphate couples glycolytic flux to activation of Ras. Nat. Commun. 2017, 8, 922. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.O.; Li, C.W.; Xia, W.; Lee, H.H.; Chang, S.S.; Shen, J.; Hsu, J.L.; Raftery, D.; Djukovic, D.; Gu, H.; et al. EGFR Signaling Enhances Aerobic Glycolysis in Triple-Negative Breast Cancer Cells to Promote Tumor Growth and Immune Escape. Cancer Res. 2016, 76, 1284–1296. [Google Scholar] [CrossRef] [PubMed]
- Das, J.; Ho, M.; Zikherman, J.; Govern, C.; Yang, M.; Weiss, A.; Chakraborty, A.K.; Roose, J.P. Digital signaling and hysteresis characterize ras activation in lymphoid cells. Cell 2009, 136, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Minchenko, O.; Opentanova, I.; Caro, J. Hypoxic regulation of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene family (PFKFB-1-4) expression in vivo. FEBS Lett. 2003, 554, 264–270. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Glycolysis in cancer: A potential target for therapy. Int. J. Biochem. Cell Biol. 2007, 39, 1358–1366. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.; Camisaschi, C.; Berzi, A.; Ferro, S.; Lugini, L.; Triulzi, T.; Tuccitto, A.; Tagliabue, E.; Castelli, C.; Rivoltini, L. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 2017, 43, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Forbes, R.A.; Verma, A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J. Biol. Chem. 2002, 277, 23111–23115. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat. Rev. Cancer 2008, 8, 865–873. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef]
- Ryan, D.G.; Murphy, M.P.; Frezza, C.; Prag, H.A.; Chouchani, E.T.; O’Neill, L.A.; Mills, E.L. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat. Metab. 2019, 1, 16–33. [Google Scholar] [CrossRef]
- Marquez, J.; Flores, J.; Kim, A.H.; Nyamaa, B.; Nguyen, A.T.T.; Park, N.; Han, J. Rescue of TCA Cycle Dysfunction for Cancer Therapy. J. Clin. Med. 2019, 8, 2161. [Google Scholar] [CrossRef]
- Xu, R.H.; Pelicano, H.; Zhou, Y.; Carew, J.S.; Feng, L.; Bhalla, K.N.; Keating, M.J.; Huang, P. Inhibition of glycolysis in cancer cells: A novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005, 65, 613–621. [Google Scholar] [CrossRef]
- Strickland, M.; Stoll, E.A. Metabolic Reprogramming in Glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef]
- Sheraj, I.; Guray, N.T.; Banerjee, S. A pan-cancer transcriptomic study showing tumor specific alterations in central metabolism. Sci. Rep. 2021, 11, 13637. [Google Scholar] [CrossRef] [PubMed]
- Oudard, S.; Arvelo, F.; Miccoli, L.; Apiou, F.; Dutrillaux, A.M.; Poisson, M.; Dutrillaux, B.; Poupon, M.F. High glycolysis in gliomas despite low hexokinase transcription and activity correlated to chromosome 10 loss. Br. J. Cancer 1996, 74, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Santra, A.; Kumar, R.; Sharma, P.; Bal, C.; Kumar, A.; Julka, P.K.; Malhotra, A. F-18 FDG PET-CT in patients with recurrent glioma: Comparison with contrast enhanced MRI. Eur. J. Radiol. 2012, 81, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.A.; Marin-Valencia, I.; Bachoo, R.M.; Mashimo, T.; Raisanen, J.; Hatanpaa, K.J.; Jindal, A.; Jeffrey, F.M.; Choi, C.; Madden, C.; et al. Metabolism of [U-13 C]glucose in human brain tumors in vivo. NMR Biomed. 2012, 25, 1234–1244. [Google Scholar] [CrossRef]
- Corbin, Z.A. New metabolic imaging tools in neuro-oncology. Curr. Opin. Neurol. 2019, 32, 872–877. [Google Scholar] [CrossRef]
- Valdes-Rives, S.A.; Casique-Aguirre, D.; German-Castelan, L.; Velasco-Velazquez, M.A.; Gonzalez-Arenas, A. Apoptotic Signaling Pathways in Glioblastoma and Therapeutic Implications. Biomed. Res. Int. 2017, 2017, 7403747. [Google Scholar] [CrossRef]
- Jelluma, N.; Yang, X.; Stokoe, D.; Evan, G.I.; Dansen, T.B.; Haas-Kogan, D.A. Glucose withdrawal induces oxidative stress followed by apoptosis in glioblastoma cells but not in normal human astrocytes. Mol. Cancer Res. 2006, 4, 319–330. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, Y.; Shingu, T.; Feng, L.; Chen, Z.; Ogasawara, M.; Keating, M.J.; Kondo, S.; Huang, P. Metabolic alterations in highly tumorigenic glioblastoma cells: Preference for hypoxia and high dependency on glycolysis. J. Biol. Chem. 2011, 286, 32843–32853. [Google Scholar] [CrossRef]
- Miyai, M.; Kanayama, T.; Hyodo, F.; Kinoshita, T.; Ishihara, T.; Okada, H.; Suzuki, H.; Takashima, S.; Wu, Z.; Hatano, Y.; et al. Glucose transporter Glut1 controls diffuse invasion phenotype with perineuronal satellitosis in diffuse glioma microenvironment. Neurooncol. Adv. 2021, 3, vdaa150. [Google Scholar] [CrossRef]
- Guda, M.R.; Labak, C.M.; Omar, S.I.; Asuthkar, S.; Airala, S.; Tuszynski, J.; Tsung, A.J.; Velpula, K.K. GLUT1 and TUBB4 in Glioblastoma Could be Efficacious Targets. Cancers 2019, 11, 1308. [Google Scholar] [CrossRef]
- Kesarwani, P.; Kant, S.; Prabhu, A.; Chinnaiyan, P. The interplay between metabolic remodeling and immune regulation in glioblastoma. Neuro Oncol. 2017, 19, 1308–1315. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Stanke, K.M.; Wilson, C.; Kidambi, S. High Expression of Glycolytic Genes in Clinical Glioblastoma Patients Correlates With Lower Survival. Front. Mol. Biosci. 2021, 8, 752404. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, M.W.; Park, Y.J.; Ahn, J.W.; Sim, J.M.; Kim, S.; Heo, J.; Jeong, J.H.; Lee, M.; Lim, J.; et al. miR-542-3p Contributes to the HK2-Mediated High Glycolytic Phenotype in Human Glioma Cells. Genes 2021, 12, 633. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.; Golbourn, B.; Huang, X.; Remke, M.; Younger, S.; Cairns, R.A.; Chalil, A.; Smith, C.A.; Krumholtz, S.L.; Mackenzie, D.; et al. PINK1 Is a Negative Regulator of Growth and the Warburg Effect in Glioblastoma. Cancer Res. 2016, 76, 4708–4719. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, P.L. Voltage dependent anion channels (VDACs): A brief introduction with a focus on the outer mitochondrial compartment’s roles together with hexokinase-2 in the "Warburg effect" in cancer. J. Bioenerg. Biomembr. 2008, 40, 123–126. [Google Scholar] [CrossRef]
- Li, R.; Yang, W. Gomisin J inhibits the glioma progression by inducing apoptosis and reducing HKII-regulated glycolysis. Biochem. Biophys. Res. Commun. 2020, 529, 15–22. [Google Scholar] [CrossRef]
- Arif, T.; Stern, O.; Pittala, S.; Chalifa-Caspi, V.; Shoshan-Barmatz, V. Rewiring of Cancer Cell Metabolism by Mitochondrial VDAC1 Depletion Results in Time-Dependent Tumor Reprogramming: Glioblastoma as a Proof of Concept. Cells 2019, 8, 1330. [Google Scholar] [CrossRef]
- Arif, T.; Krelin, Y.; Nakdimon, I.; Benharroch, D.; Paul, A.; Dadon-Klein, D.; Shoshan-Barmatz, V. VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro Oncol. 2017, 19, 951–964. [Google Scholar] [CrossRef]
- Shteinfer-Kuzmine, A.; Arif, T.; Krelin, Y.; Tripathi, S.S.; Paul, A.; Shoshan-Barmatz, V. Mitochondrial VDAC1-based peptides: Attacking oncogenic properties in glioblastoma. Oncotarget 2017, 8, 31329–31346. [Google Scholar] [CrossRef]
- Pastorino, J.G.; Shulga, N.; Hoek, J.B. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J. Biol. Chem. 2002, 277, 7610–7618. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Dang, C.V. Multifaceted roles of glycolytic enzymes. Trends Biochem. Sci. 2005, 30, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Majewski, N.; Nogueira, V.; Bhaskar, P.; Coy, P.E.; Skeen, J.E.; Gottlob, K.; Chandel, N.S.; Thompson, C.B.; Robey, R.B.; Hay, N. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol. Cell 2004, 16, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Tan, V.P.; Miyamoto, S. HK2/hexokinase-II integrates glycolysis and autophagy to confer cellular protection. Autophagy 2015, 11, 963–964. [Google Scholar] [CrossRef] [PubMed]
- Escamilla-Ramirez, A.; Castillo-Rodriguez, R.A.; Zavala-Vega, S.; Jimenez-Farfan, D.; Anaya-Rubio, I.; Briseno, E.; Palencia, G.; Guevara, P.; Cruz-Salgado, A.; Sotelo, J.; et al. Autophagy as a Potential Therapy for Malignant Glioma. Pharmaceuticals 2020, 13, 156. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, S.; He, C.; Wang, C.; Wang, L.; Piao, M.; Chi, G.; Luo, Y.; Ge, P. RSL3 induced autophagic death in glioma cells via causing glycolysis dysfunction. Biochem. Biophys. Res. Commun. 2019, 518, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, C.J.; Bahnson, B.J.; Chien, W.; Ringe, D.; Petsko, G.A. Crystal structure of rabbit phosphoglucose isomerase, a glycolytic enzyme that moonlights as neuroleukin, autocrine motility factor, and differentiation mediator. Biochemistry 2000, 39, 955–964. [Google Scholar] [CrossRef]
- Kathagen-Buhmann, A.; Maire, C.L.; Weller, J.; Schulte, A.; Matschke, J.; Holz, M.; Ligon, K.L.; Glatzel, M.; Westphal, M.; Lamszus, K. The secreted glycolytic enzyme GPI/AMF stimulates glioblastoma cell migration and invasion in an autocrine fashion but can have anti-proliferative effects. Neuro Oncol. 2018, 20, 1594–1605. [Google Scholar] [CrossRef]
- Watanabe, H.; Carmi, P.; Hogan, V.; Raz, T.; Silletti, S.; Nabi, I.R.; Raz, A. Purification of human tumor cell autocrine motility factor and molecular cloning of its receptor. J. Biol. Chem. 1991, 266, 13442–13448. [Google Scholar] [CrossRef]
- Jeoung, N.H.; Jo, A.L.; Park, H.S. Synergistic effects of autocrine motility factor and methyl jasmonate on human breast cancer cells. Biochem. Biophys. Res. Commun. 2021, 558, 22–28. [Google Scholar] [CrossRef]
- Funasaka, T.; Hogan, V.; Raz, A. Phosphoglucose isomerase/autocrine motility factor mediates epithelial and mesenchymal phenotype conversions in breast cancer. Cancer Res. 2009, 69, 5349–5356. [Google Scholar] [CrossRef] [PubMed]
- Lincet, H.; Icard, P. How do glycolytic enzymes favour cancer cell proliferation by nonmetabolic functions? Oncogene 2015, 34, 3751–3759. [Google Scholar] [CrossRef] [PubMed]
- Tanizaki, Y.; Sato, Y.; Oka, H.; Utsuki, S.; Kondo, K.; Miyajima, Y.; Nagashio, R.; Fujii, K. Expression of autocrine motility factor mRNA is a poor prognostic factor in high-grade astrocytoma. Pathol. Int. 2006, 56, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wei, Z.; Dong, B.; Lian, Z.; Xu, Y. Silencing of phosphoglucose isomerase/autocrine motility factor decreases U87 human glioblastoma cell migration. Int. J. Mol. Med. 2016, 37, 998–1004. [Google Scholar] [CrossRef]
- Al Hasawi, N.; Alkandari, M.F.; Luqmani, Y.A. Phosphofructokinase: A mediator of glycolytic flux in cancer progression. Crit. Rev. Oncol. Hematol. 2014, 92, 312–321. [Google Scholar] [CrossRef]
- Mor, I.; Cheung, E.C.; Vousden, K.H. Control of glycolysis through regulation of PFK1: Old friends and recent additions. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 211–216. [Google Scholar] [CrossRef]
- Van Schaftingen, E.; Jett, M.F.; Hue, L.; Hers, H.G. Control of liver 6-phosphofructokinase by fructose 2,6-bisphosphate and other effectors. Proc. Natl. Acad. Sci. USA 1981, 78, 3483–3486. [Google Scholar] [CrossRef]
- Uyeda, K.; Furuya, E.; Luby, L.J. The effect of natural and synthetic D-fructose 2,6-bisphosphate on the regulatory kinetic properties of liver and muscle phosphofructokinases. J. Biol. Chem. 1981, 256, 8394–8399. [Google Scholar] [CrossRef]
- Okar, D.A.; Manzano, A.; Navarro-Sabate, A.; Riera, L.; Bartrons, R.; Lange, A.J. PFK-2/FBPase-2: Maker and breaker of the essential biofactor fructose-2,6-bisphosphate. Trends Biochem. Sci. 2001, 26, 30–35. [Google Scholar] [CrossRef]
- Yalcin, A.; Telang, S.; Clem, B.; Chesney, J. Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp. Mol. Pathol. 2009, 86, 174–179. [Google Scholar] [CrossRef]
- Kotowski, K.; Rosik, J.; Machaj, F.; Supplitt, S.; Wiczew, D.; Jablonska, K.; Wiechec, E.; Ghavami, S.; Dziegiel, P. Role of PFKFB3 and PFKFB4 in Cancer: Genetic Basis, Impact on Disease Development/Progression, and Potential as Therapeutic Targets. Cancers 2021, 13, 909. [Google Scholar] [CrossRef] [PubMed]
- Staal, G.E.; Kalff, A.; Heesbeen, E.C.; van Veelen, C.W.; Rijksen, G. Subunit composition, regulatory properties, and phosphorylation of phosphofructokinase from human gliomas. Cancer Res. 1987, 47, 5047–5051. [Google Scholar] [PubMed]
- Kessler, R.; Fleischer, M.; Springsguth, C.; Bigl, M.; Warnke, J.P.; Eschrich, K. Prognostic Value of PFKFB3 to PFKFB4 mRNA Ratio in Patients With Primary Glioblastoma (IDH-Wildtype). J. Neuropathol. Exp. Neurol. 2019, 78, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Goidts, V.; Bageritz, J.; Puccio, L.; Nakata, S.; Zapatka, M.; Barbus, S.; Toedt, G.; Campos, B.; Korshunov, A.; Momma, S.; et al. RNAi screening in glioma stem-like cells identifies PFKFB4 as a key molecule important for cancer cell survival. Oncogene 2012, 31, 3235–3243. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.B.; Zhang, C.H.; Wang, S.Q.; Ai, P.H.; Chen, K.; Zhu, L.; Sun, Z.L.; Feng, D.F. Transforming growth factor beta induced (TGFBI) is a potential signature gene for mesenchymal subtype high-grade glioma. J. Neurooncol. 2018, 137, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Garcia, A.; Samso, P.; Fontova, P.; Simon-Molas, H.; Manzano, A.; Castano, E.; Rosa, J.L.; Martinez-Outshoorn, U.; Ventura, F.; Navarro-Sabate, A.; et al. TGF-beta1 targets Smad, p38 MAPK, and PI3K/Akt signaling pathways to induce PFKFB3 gene expression and glycolysis in glioblastoma cells. FEBS J. 2017, 284, 3437–3454. [Google Scholar] [CrossRef] [PubMed]
- Constam, D.B.; Philipp, J.; Malipiero, U.V.; ten Dijke, P.; Schachner, M.; Fontana, A. Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. J. Immunol. 1992, 148, 1404–1410. [Google Scholar] [CrossRef]
- Gong, L.; Ji, L.; Xu, D.; Wang, J.; Zou, J. TGF-beta links glycolysis and immunosuppression in glioblastoma. Histol. Histopathol. 2021, 36, 1111–1124. [Google Scholar] [CrossRef]
- Nusblat, L.M.; Carroll, M.J.; Roth, C.M. Crosstalk between M2 macrophages and glioma stem cells. Cell Oncol. 2017, 40, 471–482. [Google Scholar] [CrossRef]
- Zhou, K.; Yao, Y.L.; He, Z.C.; Chen, C.; Zhang, X.N.; Yang, K.D.; Liu, Y.Q.; Liu, Q.; Fu, W.J.; Chen, Y.P.; et al. VDAC2 interacts with PFKP to regulate glucose metabolism and phenotypic reprogramming of glioma stem cells. Cell Death Dis. 2018, 9, 988. [Google Scholar] [CrossRef]
- Chang, Y.C.; Yang, Y.C.; Tien, C.P.; Yang, C.J.; Hsiao, M. Roles of Aldolase Family Genes in Human Cancers and Diseases. Trends Endocrinol. Metab. 2018, 29, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; He, D.; Fu, Y.; Zhang, R.; Guo, H.; Wang, Z.; Wang, Y.; Gao, T.; Wei, Y.; Guo, Y.; et al. A novel lncRNA ARST represses glioma progression by inhibiting ALDOA-mediated actin cytoskeleton integrity. J. Exp. Clin. Cancer Res. 2021, 40, 187. [Google Scholar] [CrossRef] [PubMed]
- Kathagen, A.; Schulte, A.; Balcke, G.; Phillips, H.S.; Martens, T.; Matschke, J.; Gunther, H.S.; Soriano, R.; Modrusan, Z.; Sandmann, T.; et al. Hypoxia and oxygenation induce a metabolic switch between pentose phosphate pathway and glycolysis in glioma stem-like cells. Acta Neuropathol. 2013, 126, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Tsai, H.F.; Huang, S.P.; Chen, C.L.; Hsiao, M.; Tsai, W.C. Enrichment of Aldolase C Correlates with Low Non-Mutated IDH1 Expression and Predicts a Favorable Prognosis in Glioblastomas. Cancers 2019, 11, 1238. [Google Scholar] [CrossRef] [PubMed]
- Akram, M. Mini-review on glycolysis and cancer. J. Cancer Educ. 2013, 28, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Appelskog, I.B.; Ammerpohl, O.; Svechnikova, I.G.; Lui, W.O.; Almqvist, P.M.; Ekstrom, T.J. Histone deacetylase inhibitor 4-phenylbutyrate suppresses GAPDH mRNA expression in glioma cells. Int. J. Oncol. 2004, 24, 1419–1425. [Google Scholar] [PubMed]
- Lu, J.; Xu, Z.; Duan, H.; Ji, H.; Zhen, Z.; Li, B.; Wang, H.; Tang, H.; Zhou, J.; Guo, T.; et al. Tumor-associated macrophage interleukin-beta promotes glycerol-3-phosphate dehydrogenase activation, glycolysis and tumorigenesis in glioma cells. Cancer Sci. 2020, 111, 1979–1990. [Google Scholar] [CrossRef]
- Sen, N.; Hara, M.R.; Kornberg, M.D.; Cascio, M.B.; Bae, B.I.; Shahani, N.; Thomas, B.; Dawson, T.M.; Dawson, V.L.; Snyder, S.H.; et al. Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat. Cell Biol. 2008, 10, 866–873. [Google Scholar] [CrossRef]
- Mikeladze, M.A.; Dutysheva, E.A.; Kartsev, V.G.; Margulis, B.A.; Guzhova, I.V.; Lazarev, V.F. Disruption of the Complex between GAPDH and Hsp70 Sensitizes C6 Glioblastoma Cells to Hypoxic Stress. Int. J. Mol. Sci. 2021, 22, 1520. [Google Scholar] [CrossRef]
- Lazarev, V.F.; Nikotina, A.D.; Mikhaylova, E.R.; Nudler, E.; Polonik, S.G.; Guzhova, I.V.; Margulis, B.A. Hsp70 chaperone rescues C6 rat glioblastoma cells from oxidative stress by sequestration of aggregating GAPDH. Biochem. Biophys. Res. Commun. 2016, 470, 766–771. [Google Scholar] [CrossRef]
- Wang, F.; Dai, A.Y.; Tao, K.; Xiao, Q.; Huang, Z.L.; Gao, M.; Li, H.; Wang, X.; Cao, W.X.; Feng, W.L. Heat shock protein-70 neutralizes apoptosis inducing factor in Bcr/Abl expressing cells. Cell Signal 2015, 27, 1949–1955. [Google Scholar] [CrossRef] [PubMed]
- Sirover, M.A. Subcellular dynamics of multifunctional protein regulation: Mechanisms of GAPDH intracellular translocation. J. Cell Biochem. 2012, 113, 2193–2200. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, E.J.; Kelly, C.; Artalejo, C.R. Glyceraldehyde-3-phosphate dehydrogenase interacts with Rab2 and plays an essential role in endoplasmic reticulum to Golgi transport exclusive of its glycolytic activity. J. Biol. Chem. 2004, 279, 54046–54052. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Huang, M.; Xu, P.; Wang, M.; Ye, S.; Wang, Q.; Zeng, S.; Chen, X.; Gao, W.; Chen, J.; et al. Microcystins-LR induced apoptosis via S-nitrosylation of GAPDH in colorectal cancer cells. Ecotoxicol. Environ. Saf. 2020, 190, 110096. [Google Scholar] [CrossRef] [PubMed]
- Sirover, M.A. Structural analysis of glyceraldehyde-3-phosphate dehydrogenase functional diversity. Int. J. Biochem. Cell Biol. 2014, 57, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Polyakova, O.V.; Roitel, O.; Asryants, R.A.; Poliakov, A.A.; Branlant, G.; Muronetz, V.I. Misfolded forms of glyceraldehyde-3-phosphate dehydrogenase interact with GroEL and inhibit chaperonin-assisted folding of the wild-type enzyme. Protein Sci. 2005, 14, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Canarelli, S.E.; Swalm, B.M.; Larson, E.T.; Morrison, M.J.; Weerapana, E. Monitoring GAPDH activity and inhibition with cysteine-reactive chemical probes. RSC Chem. Biol. 2022, 3, 972–982. [Google Scholar] [CrossRef]
- Martinez-Val, A.; Bekker-Jensen, D.B.; Steigerwald, S.; Koenig, C.; Ostergaard, O.; Mehta, A.; Tran, T.; Sikorski, K.; Torres-Vega, E.; Kwasniewicz, E.; et al. Spatial-proteomics reveals phospho-signaling dynamics at subcellular resolution. Nat. Commun. 2021, 12, 7113. [Google Scholar] [CrossRef]
- Sawant Dessai, A.; Kalhotra, P.; Novickis, A.T.; Dasgupta, S. Regulation of tumor metabolism by post translational modifications on metabolic enzymes. Cancer Gene Ther. 2022, 30, 548–558. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Hol, W.G. Crystal structures of substrates and products bound to the phosphoglycerate kinase active site reveal the catalytic mechanism. Biochemistry 1998, 37, 4429–4436. [Google Scholar] [CrossRef]
- Yan, H.; Yang, K.; Xiao, H.; Zou, Y.J.; Zhang, W.B.; Liu, H.Y. Over-expression of cofilin-1 and phosphoglycerate kinase 1 in astrocytomas involved in pathogenesis of radioresistance. CNS Neurosci. Ther. 2012, 18, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, Y.; Meisenhelder, J.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Aldape, K.; He, J.; Hunter, T.; et al. Mitochondria-Translocated PGK1 Functions as a Protein Kinase to Coordinate Glycolysis and the TCA Cycle in Tumorigenesis. Mol. Cell 2016, 61, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jia, W.; Wu, C.; Chen, L.; Sun, K.; Wang, J.; Ding, B.; Liu, N.; Xu, R. The Neurogenic Compound P7C3 Regulates the Aerobic Glycolysis by Targeting Phosphoglycerate Kinase 1 in Glioma. Front. Oncol. 2021, 11, 644492. [Google Scholar] [CrossRef] [PubMed]
- Irshad, K.; Mohapatra, S.K.; Srivastava, C.; Garg, H.; Mishra, S.; Dikshit, B.; Sarkar, C.; Gupta, D.; Chandra, P.S.; Chattopadhyay, P.; et al. A combined gene signature of hypoxia and notch pathway in human glioblastoma and its prognostic relevance. PLoS ONE 2015, 10, e0118201. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Li, X.; Cai, Q.; Zhang, C.; Yu, Q.; Jiang, Y.; Lee, J.H.; Hawke, D.; Wang, Y.; Xia, Y.; et al. Phosphoglycerate Kinase 1 Phosphorylates Beclin1 to Induce Autophagy. Mol. Cell 2017, 65, 917–931.e916. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Li, X.; Lu, Z. Protein kinase activity of the glycolytic enzyme PGK1 regulates autophagy to promote tumorigenesis. Autophagy 2017, 13, 1246–1247. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Wang, S.; Jin, L.; Weng, M.; Zhou, D.; Wang, J.; Tang, Z.; Quan, Z. Long non-coding RNA GBCDRlnc1 induces chemoresistance of gallbladder cancer cells by activating autophagy. Mol. Cancer 2019, 18, 82. [Google Scholar] [CrossRef]
- Ding, H.; Cheng, Y.J.; Yan, H.; Zhang, R.; Zhao, J.B.; Qian, C.F.; Zhang, W.B.; Xiao, H.; Liu, H.Y. Phosphoglycerate kinase 1 promotes radioresistance in U251 human glioma cells. Oncol. Rep. 2014, 31, 894–900. [Google Scholar] [CrossRef]
- Odreman, F.; Vindigni, M.; Gonzales, M.L.; Niccolini, B.; Candiano, G.; Zanotti, B.; Skrap, M.; Pizzolitto, S.; Stanta, G.; Vindigni, A. Proteomic studies on low- and high-grade human brain astrocytomas. J. Proteome Res. 2005, 4, 698–708. [Google Scholar] [CrossRef]
- Sun, W.; Yan, H.; Qian, C.; Wang, C.; Zhao, M.; Liu, Y.; Zhong, Y.; Liu, H.; Xiao, H. Cofilin-1 and phosphoglycerate kinase 1 as promising indicators for glioma radiosensibility and prognosis. Oncotarget 2017, 8, 55073–55083. [Google Scholar] [CrossRef]
- Sharif, F.; Rasul, A.; Ashraf, A.; Hussain, G.; Younis, T.; Sarfraz, I.; Chaudhry, M.A.; Bukhari, S.A.; Ji, X.Y.; Selamoglu, Z.; et al. Phosphoglycerate mutase 1 in cancer: A promising target for diagnosis and therapy. IUBMB Life 2019, 71, 1418–1427. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.G.; Ding, J.; Du, C.; Xu, N.; Wang, E.L.; Li, J.Y.; Wang, Y.Y.; Yu, J.M. Phosphoglycerate mutase 1 is highly expressed in C6 glioma cells and human astrocytoma. Oncol. Lett. 2018, 15, 8935–8940. [Google Scholar] [CrossRef] [PubMed]
- Ohba, S.; Johannessen, T.A.; Chatla, K.; Yang, X.; Pieper, R.O.; Mukherjee, J. Phosphoglycerate Mutase 1 Activates DNA Damage Repair via Regulation of WIP1 Activity. Cell Rep. 2020, 31, 107518. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Gong, J.; Wang, C.; Wang, Y.; Song, Y.; Xu, W.; Liu, Z.; Liu, Y. The diagnostic value and functional roles of phosphoglycerate mutase 1 in glioma. Oncol. Rep. 2016, 36, 2236–2244. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Roig, I.; Knobel, P.A.; Marjanovic, M. The ATM signaling network in development and disease. Front. Genet. 2013, 4, 37. [Google Scholar] [CrossRef]
- Johannessen, T.A.; Mukherjee, J. Phosphoglycerate mutase 1 (PGAM1) overexpression promotes radio- and chemoresistance in gliomas by activating the DNA damage response. Mol. Cell Oncol. 2021, 8, 1875804. [Google Scholar] [CrossRef]
- Sanzey, M.; Abdul Rahim, S.A.; Oudin, A.; Dirkse, A.; Kaoma, T.; Vallar, L.; Herold-Mende, C.; Bjerkvig, R.; Golebiewska, A.; Niclou, S.P. Comprehensive analysis of glycolytic enzymes as therapeutic targets in the treatment of glioblastoma. PLoS ONE 2015, 10, e0123544. [Google Scholar] [CrossRef]
- Vos, M.J.; Postma, T.J.; Martens, F.; Uitdehaag, B.M.; Blankenstein, M.A.; Vandertop, W.P.; Slotman, B.J.; Heimans, J.J. Serum levels of S-100B protein and neuron-specific enolase in glioma patients: A pilot study. Anticancer Res. 2004, 24, 2511–2514. [Google Scholar]
- Vinores, S.A.; Marangos, P.J.; Bonnin, J.M.; Rubinstein, L.J. Immunoradiometric and immunohistochemical demonstration of neuron-specific enolase in experimental rat gliomas. Cancer Res. 1984, 44, 2595–2599. [Google Scholar]
- Majc, B.; Habic, A.; Novak, M.; Rotter, A.; Porcnik, A.; Mlakar, J.; Zupunski, V.; Pecar Fonovic, U.; Knez, D.; Zidar, N.; et al. Upregulation of Cathepsin X in Glioblastoma: Interplay with gamma-Enolase and the Effects of Selective Cathepsin X Inhibitors. Int. J. Mol. Sci. 2022, 23, 1784. [Google Scholar] [CrossRef]
- Song, Y.; Luo, Q.; Long, H.; Hu, Z.; Que, T.; Zhang, X.; Li, Z.; Wang, G.; Yi, L.; Liu, Z.; et al. Alpha-enolase as a potential cancer prognostic marker promotes cell growth, migration, and invasion in glioma. Mol. Cancer 2014, 13, 65. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, Y.; Wang, H.; Zeng, Y.Y.; Li, Z.; Li, M.L.; Li, F.F.; You, J.; Zhang, Z.M.; Tzeng, C.M. WW domain-binding protein 2 acts as an oncogene by modulating the activity of the glycolytic enzyme ENO1 in glioma. Cell Death Dis. 2018, 9, 347. [Google Scholar] [CrossRef] [PubMed]
- Schofield, L.; Lincz, L.F.; Skelding, K.A. Unlikely role of glycolytic enzyme α-enolase in cancer metastasis and its potential as a prognostic biomarker. J. Cancer Metastasis Treat. 2020, 6, 10. [Google Scholar] [CrossRef]
- Zheng, R.; Yao, Q.; Li, X.; Xu, B. Long Noncoding Ribonucleic Acid SNHG18 Promotes Glioma Cell Motility via Disruption of alpha-Enolase Nucleocytoplasmic Transport. Front. Genet. 2019, 10, 1140. [Google Scholar] [CrossRef] [PubMed]
- Beckner, M.E.; Fellows-Mayle, W.; Zhang, Z.; Agostino, N.R.; Kant, J.A.; Day, B.W.; Pollack, I.F. Identification of ATP citrate lyase as a positive regulator of glycolytic function in glioblastomas. Int. J. Cancer 2010, 126, 2282–2295. [Google Scholar] [CrossRef] [PubMed]
- Khadka, S.; Arthur, K.; Barekatain, Y.; Behr, E.; Washington, M.; Ackroyd, J.; Crowley, K.; Suriyamongkol, P.; Lin, Y.H.; Pham, C.D.; et al. Impaired anaplerosis is a major contributor to glycolysis inhibitor toxicity in glioma. Cancer Metab. 2021, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Yu-Hsi, L.; Nikunj, S.; Naima, H.; Jeffrey, J.A.; Sunada, K.; Victoria, C.Y.; Dimitra, K.G.; Yuting, S.; Rafal, Z.; Theresa, T.; et al. Eradication of ENO1-deleted Glioblastoma through Collateral Lethality. BioRxiv 2018, 331538. [Google Scholar] [CrossRef]
- Satani, N.; Lin, Y.H.; Hammoudi, N.; Raghavan, S.; Georgiou, D.K.; Muller, F.L. ENOblock Does Not Inhibit the Activity of the Glycolytic Enzyme Enolase. PLoS ONE 2016, 11, e0168739. [Google Scholar] [CrossRef]
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980. [Google Scholar] [CrossRef]
- Liu, V.M.; Vander Heiden, M.G. The Role of Pyruvate Kinase M2 in Cancer Metabolism. Brain Pathol. 2015, 25, 781–783. [Google Scholar] [CrossRef]
- Liang, J.; Cao, R.; Wang, X.; Zhang, Y.; Wang, P.; Gao, H.; Li, C.; Yang, F.; Zeng, R.; Wei, P.; et al. Mitochondrial PKM2 regulates oxidative stress-induced apoptosis by stabilizing Bcl2. Cell Res. 2017, 27, 329–351. [Google Scholar] [CrossRef] [PubMed]
- Verma, H.; Cholia, R.P.; Kaur, S.; Dhiman, M.; Mantha, A.K. A short review on cross-link between pyruvate kinase (PKM2) and Glioblastoma Multiforme. Metab. Brain Dis. 2021, 36, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Alfred Yung, W.K.; Lu, Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 2011, 480, 118–122. [Google Scholar] [CrossRef]
- Gao, M.; Yang, J.; Gong, H.; Lin, Y.; Liu, J. Trametinib Inhibits the Growth and Aerobic Glycolysis of Glioma Cells by Targeting the PKM2/c-Myc Axis. Front. Pharmacol. 2021, 12, 760055. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Cao, R.; Zhang, Y.; Xia, Y.; Zheng, Y.; Li, X.; Wang, L.; Yang, W.; Lu, Z. PKM2 dephosphorylation by Cdc25A promotes the Warburg effect and tumorigenesis. Nat. Commun. 2016, 7, 12431. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Li, Z.; Fu, R.; Wu, H.; Li, Z. Pyruvate kinase M2 facilitates colon cancer cell migration via the modulation of STAT3 signalling. Cell Signal. 2014, 26, 1853–1862. [Google Scholar] [CrossRef]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The Regulation and Function of Lactate Dehydrogenase A: Therapeutic Potential in Brain Tumor. Brain Pathol. 2016, 26, 3–17. [Google Scholar] [CrossRef]
- Li, J.; Zhu, S.; Tong, J.; Hao, H.; Yang, J.; Liu, Z.; Wang, Y. Suppression of lactate dehydrogenase A compromises tumor progression by downregulation of the Warburg effect in glioblastoma. Neuroreport 2016, 27, 110–115. [Google Scholar] [CrossRef]
- Inoue, A.; Nishikawa, M.; Ohnishi, T.; Yano, H.; Kanemura, Y.; Ohtsuka, Y.; Ozaki, S.; Nakamura, Y.; Matsumoto, S.; Suehiro, S.; et al. Prediction of Glioma Stemlike Cell Infiltration in the Non-Contrast-Enhancing Area by Quantitative Measurement of Lactate on Magnetic Resonance Spectroscopy in Glioblastoma. World Neurosurg. 2021, 153, e76–e95. [Google Scholar] [CrossRef]
- Baumann, F.; Leukel, P.; Doerfelt, A.; Beier, C.P.; Dettmer, K.; Oefner, P.J.; Kastenberger, M.; Kreutz, M.; Nickl-Jockschat, T.; Bogdahn, U.; et al. Lactate promotes glioma migration by TGF-beta2-dependent regulation of matrix metalloproteinase-2. Neuro Oncol. 2009, 11, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Smith, C.P.; Wilbur, R.R.; Cain, C.P.; Kallu, S.R.; Valasapalli, S.; Sahoo, A.; Guda, M.R.; Tsung, A.J.; Velpula, K.K. An overview of MCT1 and MCT4 in GBM: Small molecule transporters with large implications. Am. J. Cancer Res. 2018, 8, 1967–1976. [Google Scholar] [PubMed]
- Miranda-Goncalves, V.; Goncalves, C.S.; Granja, S.; Vieira de Castro, J.; Reis, R.M.; Costa, B.M.; Baltazar, F. MCT1 Is a New Prognostic Biomarker and Its Therapeutic Inhibition Boosts Response to Temozolomide in Human Glioblastoma. Cancers 2021, 13, 3468. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.S.; Lim, K.J.; Price, A.C.; Orr, B.A.; Eberhart, C.G.; Bar, E.E. Inhibition of monocarboxylate transporter-4 depletes stem-like glioblastoma cells and inhibits HIF transcriptional response in a lactate-independent manner. Oncogene 2014, 33, 4433–4441. [Google Scholar] [CrossRef] [PubMed]
- Duan, K.; Liu, Z.J.; Hu, S.Q.; Huo, H.Y.; Xu, Z.R.; Ruan, J.F.; Sun, Y.; Dai, L.P.; Yan, C.B.; Xiong, W.; et al. Lactic acid induces lactate transport and glycolysis/OXPHOS interconversion in glioblastoma. Biochem. Biophys. Res. Commun. 2018, 503, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Kubelt, C.; Peters, S.; Ahmeti, H.; Huhndorf, M.; Huber, L.; Cohrs, G.; Hovener, J.B.; Jansen, O.; Synowitz, M.; Held-Feindt, J. Intratumoral Distribution of Lactate and the Monocarboxylate Transporters 1 and 4 in Human Glioblastoma Multiforme and Their Relationships to Tumor Progression-Associated Markers. Int. J. Mol. Sci. 2020, 21, 6254. [Google Scholar] [CrossRef] [PubMed]
- Takada, T.; Takata, K.; Ashihara, E. Inhibition of monocarboxylate transporter 1 suppresses the proliferation of glioblastoma stem cells. J. Physiol. Sci. 2016, 66, 387–396. [Google Scholar] [CrossRef]
- Fan, J.; Hitosugi, T.; Chung, T.W.; Xie, J.; Ge, Q.; Gu, T.L.; Polakiewicz, R.D.; Chen, G.Z.; Boggon, T.J.; Lonial, S.; et al. Tyrosine phosphorylation of lactate dehydrogenase A is important for NADH/NAD(+) redox homeostasis in cancer cells. Mol. Cell Biol. 2011, 31, 4938–4950. [Google Scholar] [CrossRef]
- Li, S.; Zhao, C.; Gao, J.; Zhuang, X.; Liu, S.; Xing, X.; Liu, Q.; Chen, C.; Wang, S.; Luo, Y. Cyclin G2 reverses immunosuppressive tumor microenvironment and potentiates PD-1 blockade in glioma. J. Exp. Clin. Cancer Res. 2021, 40, 273. [Google Scholar] [CrossRef]
- Trejo-Solis, C.; Serrano-Garcia, N.; Escamilla-Ramirez, A.; Castillo-Rodriguez, R.A.; Jimenez-Farfan, D.; Palencia, G.; Calvillo, M.; Alvarez-Lemus, M.A.; Flores-Najera, A.; Cruz-Salgado, A.; et al. Autophagic and Apoptotic Pathways as Targets for Chemotherapy in Glioblastoma. Int. J. Mol. Sci. 2018, 19, 3773. [Google Scholar] [CrossRef]
- Alizadeh, J.; Kavoosi, M.; Singh, N.; Lorzadeh, S.; Ravandi, A.; Kidane, B.; Ahmed, N.; Mraiche, F.; Mowat, M.R.; Ghavami, S. Regulation of Autophagy via Carbohydrate and Lipid Metabolism in Cancer. Cancers 2023, 15, 2195. [Google Scholar] [CrossRef]
- Chu, Y.; Chang, Y.; Lu, W.; Sheng, X.; Wang, S.; Xu, H.; Ma, J. Regulation of Autophagy by Glycolysis in Cancer. Cancer Manag. Res. 2020, 12, 13259–13271. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xiong, H.; Wu, F.; Zhang, Y.; Wang, J.; Zhao, L.; Guo, X.; Chang, L.J.; Zhang, Y.; You, M.J.; et al. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014, 8, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.; Marayati, R.; Moffitt, R.; Yeh, J.J. Hexokinase 2 promotes tumor growth and metastasis by regulating lactate production in pancreatic cancer. Oncotarget 2017, 8, 56081–56094. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.Q.; Tan, X.Y.; Zhang, B.W.; Wu, T.; Liu, P.; Sun, S.J.; Cao, Y.G. 3-Bromopyruvate inhibits cell proliferation and induces apoptosis in CD133+ population in human glioma. Tumour Biol. 2016, 37, 3543–3548. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y. Strategies of temozolomide in future glioblastoma treatment. Onco Targets Ther. 2017, 10, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Shoshan, M.C. 3-Bromopyruvate: Targets and outcomes. J. Bioenerg. Biomembr. 2012, 44, 7–15. [Google Scholar] [CrossRef]
- Ihrlund, L.S.; Hernlund, E.; Khan, O.; Shoshan, M.C. 3-Bromopyruvate as inhibitor of tumour cell energy metabolism and chemopotentiator of platinum drugs. Mol. Oncol. 2008, 2, 94–101. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Q.; Huang, Z.; Li, B.; Nice, E.C.; Huang, C.; Wei, L.; Zou, B. Targeting Glucose Metabolism Enzymes in Cancer Treatment: Current and Emerging Strategies. Cancers 2022, 14, 4568. [Google Scholar] [CrossRef]
- Ko, Y.H.; Verhoeven, H.A.; Lee, M.J.; Corbin, D.J.; Vogl, T.J.; Pedersen, P.L. A translational study "case report" on the small molecule "energy blocker" 3-bromopyruvate (3BP) as a potent anticancer agent: From bench side to bedside. J. Bioenerg. Biomembr. 2012, 44, 163–170. [Google Scholar] [CrossRef]
- Di Cosimo, S.; Ferretti, G.; Papaldo, P.; Carlini, P.; Fabi, A.; Cognetti, F. Lonidamine: Efficacy and safety in clinical trials for the treatment of solid tumors. Drugs Today 2003, 39, 157–174. [Google Scholar] [CrossRef] [PubMed]
- De Lena, M.; Lorusso, V.; Bottalico, C.; Brandi, M.; De Mitrio, A.; Catino, A.; Guida, M.; Latorre, A.; Leone, B.; Vallejo, C.; et al. Revertant and potentiating activity of lonidamine in patients with ovarian cancer previously treated with platinum. J. Clin. Oncol. 1997, 15, 3208–3213. [Google Scholar] [CrossRef] [PubMed]
- De Marinis, F.; Rinaldi, M.; Ardizzoni, A.; Bruzzi, P.; Pennucci, M.C.; Portalone, L.; D’Aprile, M.; Ripanti, P.; Romano, F.; Belli, M.; et al. The role of vindesine and lonidamine in the treatment of elderly patients with advanced non-small cell lung cancer: A phase III randomized FONICAP trial. Italian Lung Cancer Task Force. Tumori 1999, 85, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Berruti, A.; Bitossi, R.; Gorzegno, G.; Bottini, A.; Alquati, P.; De Matteis, A.; Nuzzo, F.; Giardina, G.; Danese, S.; De Lena, M.; et al. Time to progression in metastatic breast cancer patients treated with epirubicin is not improved by the addition of either cisplatin or lonidamine: Final results of a phase III study with a factorial design. J. Clin. Oncol. 2002, 20, 4150–4159. [Google Scholar] [CrossRef] [PubMed]
- Price, G.S.; Page, R.L.; Riviere, J.E.; Cline, J.M.; Thrall, D.E. Pharmacokinetics and toxicity of oral and intravenous lonidamine in dogs. Cancer Chemother. Pharmacol. 1996, 38, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.E.; Daum, G. Lipids of mitochondria. Prog. Lipid Res. 2013, 52, 590–614. [Google Scholar] [CrossRef]
- Harwood, J.L. [44] Phosphoglycerides of mitochondrial membranes. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1987; Volume 148, pp. 475–485. [Google Scholar]
- Ganesan, V.; Perera, M.N.; Colombini, D.; Datskovskiy, D.; Chadha, K.; Colombini, M. Ceramide and activated Bax act synergistically to permeabilize the mitochondrial outer membrane. Apoptosis 2010, 15, 553–562. [Google Scholar] [CrossRef]
- Siskind, L.J.; Feinstein, L.; Yu, T.; Davis, J.S.; Jones, D.; Choi, J.; Zuckerman, J.E.; Tan, W.; Hill, R.B.; Hardwick, J.M.; et al. Anti-apoptotic Bcl-2 Family Proteins Disassemble Ceramide Channels. J. Biol. Chem. 2008, 283, 6622–6630. [Google Scholar] [CrossRef]
- Sorice, M.; Circella, A.; Cristea, I.M.; Garofalo, T.; Di Renzo, L.; Alessandri, C.; Valesini, G.; Esposti, M.D. Cardiolipin and its metabolites move from mitochondria to other cellular membranes during death receptor-mediated apoptosis. Cell Death Differ. 2004, 11, 1133–1145. [Google Scholar] [CrossRef]
- Siskind, L.J. Mitochondrial ceramide and the induction of apoptosis. J. Bioenerg. Biomembr. 2005, 37, 143–153. [Google Scholar] [CrossRef]
- Chang, C.Y.; Li, J.R.; Wu, C.C.; Wang, J.D.; Yang, C.P.; Chen, W.Y.; Wang, W.Y.; Chen, C.J. Indomethacin induced glioma apoptosis involving ceramide signals. Exp. Cell Res. 2018, 365, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.C.; Ali, T.; Ramanadham, S.; Hjelmeland, A.B. Sphingolipid Metabolism in Glioblastoma and Metastatic Brain Tumors: A Review of Sphingomyelinases and Sphingosine-1-Phosphate. Biomolecules 2020, 10, 1357. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; McStay, G.P.; Bharti, A.; Kuwana, T.; Clarke, C.J.; Siskind, L.J.; Obeid, L.M.; Green, D.R. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 2012, 148, 988–1000. [Google Scholar] [CrossRef] [PubMed]
- Amaegberi, N.V.; Semenkova, G.N.; Kvacheva, Z.B.; Lisovskaya, A.G.; Pinchuk, S.V.; Shadyro, O.I. 2-Hexadecenal inhibits growth of C6 glioma cells. Cell Biochem. Funct. 2019, 37, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Lucken-Ardjomande, S.; Montessuit, S.; Martinou, J.C. Bax activation and stress-induced apoptosis delayed by the accumulation of cholesterol in mitochondrial membranes. Cell Death Differ. 2008, 15, 484–493. [Google Scholar] [CrossRef]
- Montero, J.; Morales, A.; Llacuna, L.; Lluis, J.M.; Terrones, O.; Basanez, G.; Antonsson, B.; Prieto, J.; Garcia-Ruiz, C.; Colell, A.; et al. Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma. Cancer Res. 2008, 68, 5246–5256. [Google Scholar] [CrossRef]
- Zhang, C.; Yu, H.; Shen, Y.; Ni, X.; Shen, S.; Das, U.N. Polyunsaturated fatty acids trigger apoptosis of colon cancer cells through a mitochondrial pathway. Arch. Med. Sci. 2015, 11, 1081–1094. [Google Scholar]
- Kwon, J.I.; Kim, G.Y.; Park, K.Y.; Ryu, C.H.; Choi, Y.H. Induction of apoptosis by linoleic acid is associated with the modulation of Bcl-2 family and Fas/FasL system and activation of caspases in AGS human gastric adenocarcinoma cells. J. Med. Food 2008, 11, 1–8. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, W.; He, Q.; Wu, Y.; Lu, Z.; Sun, J.; Liu, Z.; Shao, Y.; Wang, A. Oleic acid induces apoptosis and autophagy in the treatment of Tongue Squamous cell carcinomas. Sci. Rep. 2017, 7, 11277. [Google Scholar] [CrossRef]
- Jatyan, R.; Sahel, D.K.; Singh, P.; Sakhuja, R.; Mittal, A.; Chitkara, D. Temozolomide-fatty acid conjugates for glioblastoma multiforme: In vitro and in vivo evaluation. J. Control Release 2023, 359, 161–174. [Google Scholar] [CrossRef]
- Lin, Z.; Long, F.; Kang, R.; Klionsky, D.J.; Yang, M.; Tang, D. The lipid basis of cell death and autophagy. Autophagy 2023, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Kridel, S.; Thorburn, A.; Kooshki, M.; Little, J.; Hebbar, S.; Robbins, M. Fatty acid synthase: A novel target for antiglioma therapy. Br. J. Cancer 2006, 95, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Knizhnik, A.V.; Roos, W.P.; Nikolova, T.; Quiros, S.; Tomaszowski, K.H.; Christmann, M.; Kaina, B. Survival and death strategies in glioma cells: Autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS ONE 2013, 8, e55665. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef]
- Mazurek, M.; Litak, J.; Kamieniak, P.; Kulesza, B.; Jonak, K.; Baj, J.; Grochowski, C. Metformin as Potential Therapy for High-Grade Glioma. Cancers 2020, 12, 210. [Google Scholar] [CrossRef]
- Sesen, J.; Dahan, P.; Scotland, S.J.; Saland, E.; Dang, V.T.; Lemarie, A.; Tyler, B.M.; Brem, H.; Toulas, C.; Cohen-Jonathan Moyal, E.; et al. Metformin inhibits growth of human glioblastoma cells and enhances therapeutic response. PLoS ONE 2015, 10, e0123721. [Google Scholar] [CrossRef]
- Marini, C.; Salani, B.; Massollo, M.; Amaro, A.; Esposito, A.I.; Orengo, A.M.; Capitanio, S.; Emionite, L.; Riondato, M.; Bottoni, G.; et al. Direct inhibition of hexokinase activity by metformin at least partially impairs glucose metabolism and tumor growth in experimental breast cancer. Cell Cycle 2013, 12, 3490–3499. [Google Scholar] [CrossRef]
- Sato, A.; Sunayama, J.; Okada, M.; Watanabe, E.; Seino, S.; Shibuya, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem Cells Transl. Med. 2012, 1, 811–824. [Google Scholar] [CrossRef]
- Klarer, A.C.; O’Neal, J.; Imbert-Fernandez, Y.; Clem, A.; Ellis, S.R.; Clark, J.; Clem, B.; Chesney, J.; Telang, S. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) induces autophagy as a survival mechanism. Cancer Metab. 2014, 2, 2. [Google Scholar] [CrossRef]
- Blum, R.; Jacob-Hirsch, J.; Amariglio, N.; Rechavi, G.; Kloog, Y. Ras inhibition in glioblastoma down-regulates hypoxia-inducible factor-1alpha, causing glycolysis shutdown and cell death. Cancer Res. 2005, 65, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Su, H.; Zhang, D.; Wang, Y.; Shen, Q.; Liu, B.; Huang, R.; Zhou, T.; Peng, C.; Wong, C.C.; et al. AMPK-Dependent Phosphorylation of GAPDH Triggers Sirt1 Activation and Is Necessary for Autophagy upon Glucose Starvation. Mol. Cell 2015, 60, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.N.; Ha, S.H.; Kim, J.; Koh, A.; Lee, C.S.; Kim, J.H.; Jeon, H.; Kim, D.H.; Suh, P.G.; Ryu, S.H. Glycolytic flux signals to mTOR through glyceraldehyde-3-phosphate dehydrogenase-mediated regulation of Rheb. Mol. Cell Biol. 2009, 29, 3991–4001. [Google Scholar] [CrossRef] [PubMed]
- Dando, I.; Pacchiana, R.; Pozza, E.D.; Cataldo, I.; Bruno, S.; Conti, P.; Cordani, M.; Grimaldi, A.; Butera, G.; Caraglia, M.; et al. UCP2 inhibition induces ROS/Akt/mTOR axis: Role of GAPDH nuclear translocation in genipin/everolimus anticancer synergism. Free Radic. Biol. Med. 2017, 113, 176–189. [Google Scholar] [CrossRef]
- He, C.L.; Bian, Y.Y.; Xue, Y.; Liu, Z.X.; Zhou, K.Q.; Yao, C.F.; Lin, Y.; Zou, H.F.; Luo, F.X.; Qu, Y.Y.; et al. Pyruvate Kinase M2 Activates mTORC1 by Phosphorylating AKT1S1. Sci. Rep. 2016, 6, 21524. [Google Scholar] [CrossRef]
- Meng, M.B.; Wang, H.H.; Guo, W.H.; Wu, Z.Q.; Zeng, X.L.; Zaorsky, N.G.; Shi, H.S.; Qian, D.; Niu, Z.M.; Jiang, B.; et al. Targeting pyruvate kinase M2 contributes to radiosensitivity of non-small cell lung cancer cells in vitro and in vivo. Cancer Lett. 2015, 356, 985–993. [Google Scholar] [CrossRef]
- Wang, L.; Yang, L.; Yang, Z.; Tang, Y.; Tao, Y.; Zhan, Q.; Lei, L.; Jing, Y.; Jiang, X.; Jin, H.; et al. Glycolytic Enzyme PKM2 Mediates Autophagic Activation to Promote Cell Survival in NPM1-Mutated Leukemia. Int. J. Biol. Sci. 2019, 15, 882–894. [Google Scholar] [CrossRef]
- Huang, X.; Qi, Q.; Hua, X.; Li, X.; Zhang, W.; Sun, H.; Li, S.; Wang, X.; Li, B. Beclin 1, an autophagy-related gene, augments apoptosis in U87 glioblastoma cells. Oncol. Rep. 2014, 31, 1761–1767. [Google Scholar] [CrossRef]
- Shi, L.; Yan, H.; An, S.; Shen, M.; Jia, W.; Zhang, R.; Zhao, L.; Huang, G.; Liu, J. SIRT5-mediated deacetylation of LDHB promotes autophagy and tumorigenesis in colorectal cancer. Mol. Oncol. 2019, 13, 358–375. [Google Scholar] [CrossRef]
- Dall’Armi, C.; Devereaux, K.A.; Di Paolo, G. The role of lipids in the control of autophagy. Curr. Biol. 2013, 23, R33–R45. [Google Scholar] [CrossRef] [PubMed]
- Vicinanza, M.; Korolchuk, V.I.; Ashkenazi, A.; Puri, C.; Menzies, F.M.; Clarke, J.H.; Rubinsztein, D.C. PI(5)P regulates autophagosome biogenesis. Mol. Cell 2015, 57, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, M.; Obara, K.; Kihara, A. Sphingolipid synthesis is involved in autophagy in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 2011, 410, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Li, D.; Wang, L.; Xia, L.; Ma, J.; Guan, Z.; Feng, G.; Zhu, X. c-Jun NH2-terminal kinase activation is essential for up-regulation of LC3 during ceramide-induced autophagy in human nasopharyngeal carcinoma cells. J. Transl. Med. 2011, 9, 161. [Google Scholar] [CrossRef] [PubMed]
- Li, D.D.; Wang, L.L.; Deng, R.; Tang, J.; Shen, Y.; Guo, J.F.; Wang, Y.; Xia, L.P.; Feng, G.K.; Liu, Q.Q.; et al. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene 2009, 28, 886–898. [Google Scholar] [CrossRef]
- Kim, S.; Jing, K.; Shin, S.; Jeong, S.; Han, S.H.; Oh, H.; Yoo, Y.S.; Han, J.; Jeon, Y.J.; Heo, J.Y.; et al. omega3-polyunsaturated fatty acids induce cell death through apoptosis and autophagy in glioblastoma cells: In vitro and in vivo. Oncol. Rep. 2018, 39, 239–246. [Google Scholar] [CrossRef]
- Humbert, M.; Seiler, K.; Mosimann, S.; Rentsch, V.; Sharma, K.; Pandey, A.V.; McKenna, S.L.; Tschan, M.P. Reducing FASN expression sensitizes acute myeloid leukemia cells to differentiation therapy. Cell Death Differ. 2021, 28, 2465–2481. [Google Scholar] [CrossRef]
- Grube, S.; Dunisch, P.; Freitag, D.; Klausnitzer, M.; Sakr, Y.; Walter, J.; Kalff, R.; Ewald, C. Overexpression of fatty acid synthase in human gliomas correlates with the WHO tumor grade and inhibition with Orlistat reduces cell viability and triggers apoptosis. J. Neurooncol. 2014, 118, 277–287. [Google Scholar] [CrossRef]
- Zhou, Y.; Jin, G.; Mi, R.; Zhang, J.; Zhang, J.; Xu, H.; Cheng, S.; Zhang, Y.; Song, W.; Liu, F. Inhibition of fatty acid synthase suppresses neovascularization via regulating the expression of VEGF-A in glioma. J. Cancer Res. Clin. Oncol. 2016, 142, 2447–2459. [Google Scholar] [CrossRef]
- Misirkic, M.; Janjetovic, K.; Vucicevic, L.; Tovilovic, G.; Ristic, B.; Vilimanovich, U.; Harhaji-Trajkovic, L.; Sumarac-Dumanovic, M.; Micic, D.; Bumbasirevic, V.; et al. Inhibition of AMPK-dependent autophagy enhances in vitro antiglioma effect of simvastatin. Pharmacol. Res. 2012, 65, 111–119. [Google Scholar] [CrossRef]
- Almejun, M.B.; Borge, M.; Colado, A.; Elias, E.E.; Podaza, E.; Risnik, D.; De Brasi, C.D.; Stanganelli, C.; Slavutsky, I.; Cabrejo, M.; et al. Sphingosine kinase 1 participates in the activation, proliferation and survival of chronic lymphocytic leukemia cells. Haematologica 2017, 102, e257–e260. [Google Scholar] [CrossRef] [PubMed]
- Poteet, E.; Choudhury, G.R.; Winters, A.; Li, W.; Ryou, M.G.; Liu, R.; Tang, L.; Ghorpade, A.; Wen, Y.; Yuan, F.; et al. Reversing the Warburg effect as a treatment for glioblastoma. J. Biol. Chem. 2013, 288, 9153–9164. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.M.; Shen, H.; McKelvey, K.J.; Gee, H.E.; Hau, E. Targeting Glucose Metabolism of Cancer Cells with Dichloroacetate to Radiosensitize High-Grade Gliomas. Int. J. Mol. Sci. 2021, 22, 7265. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010, 2, 31ra34. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xue, W.; Xu, K.; Yi, L.; Guo, Y.; Xie, T.; Tong, H.; Zhou, B.; Wang, S.; Li, Q.; et al. Dual inhibition of PFKFB3 and VEGF normalizes tumor vasculature, reduces lactate production, and improves chemotherapy in glioblastoma: Insights from protein expression profiling and MRI. Theranostics 2020, 10, 7245–7259. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Xue, D.; Chen, Z.; Ou-Yang, Y.; Zhang, J.; Mai, J.; Gu, J.; Lu, W.; Liu, X.; Liu, W.; et al. R406 elicits anti-Warburg effect via Syk-dependent and -independent mechanisms to trigger apoptosis in glioma stem cells. Cell Death Dis. 2019, 10, 358. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Goncalves, V.; Honavar, M.; Pinheiro, C.; Martinho, O.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; Costa, P.; Palmeirim, I.; et al. Monocarboxylate transporters (MCTs) in gliomas: Expression and exploitation as therapeutic targets. Neuro Oncol. 2013, 15, 172–188. [Google Scholar] [CrossRef]
- Voss, D.M.; Spina, R.; Carter, D.L.; Lim, K.S.; Jeffery, C.J.; Bar, E.E. Disruption of the monocarboxylate transporter-4-basigin interaction inhibits the hypoxic response, proliferation, and tumor progression. Sci. Rep. 2017, 7, 4292. [Google Scholar] [CrossRef]
- Radoul, M.; Najac, C.; Viswanath, P.; Mukherjee, J.; Kelly, M.; Gillespie, A.M.; Chaumeil, M.M.; Eriksson, P.; Delos Santos, R.; Pieper, R.O.; et al. HDAC inhibition in glioblastoma monitored by hyperpolarized (13) C MRSI. NMR Biomed. 2019, 32, e4044. [Google Scholar] [CrossRef]
- Dranoff, G.; Elion, G.B.; Friedman, H.S.; Campbell, G.L.; Bigner, D.D. Influence of glutamine on the growth of human glioma and medulloblastoma in culture. Cancer Res. 1985, 45, 4077–4081. [Google Scholar]
- Ohba, S.; Hirose, Y. L-asparaginase and 6-diazo-5-oxo-L-norleucine synergistically inhibit the growth of glioblastoma cells. J. Neurooncol. 2020, 146, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, U.D.; Cheng, M.; Koch, K.; Marchionni, L.; Fan, X.; Raabe, E.H.; Maciaczyk, J.; Glunde, K.; Eberhart, C.G. Alterations in cellular metabolome after pharmacological inhibition of Notch in glioblastoma cells. Int. J. Cancer 2016, 138, 1246–1255. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, M.J.; Bennett, B.D.; Joshi, A.D.; Gao, P.; Thomas, A.G.; Ferraris, D.V.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Rabinowitz, J.D.; et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010, 70, 8981–8987. [Google Scholar] [CrossRef]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116.e125. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.H.; Qiu, Y.; Stamatatos, O.; Janowitz, T.; Lukey, M.J. Enhancing the Efficacy of Glutamine Metabolism Inhibitors in Cancer Therapy. Trends Cancer 2021, 7, 790–804. [Google Scholar] [CrossRef]
- Szeliga, M.; Bogacinska-Karas, M.; Kuzmicz, K.; Rola, R.; Albrecht, J. Downregulation of GLS2 in glioblastoma cells is related to DNA hypermethylation but not to the p53 status. Mol. Carcinog. 2016, 55, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.J.; Wu, M.; Chen, H.N.; Wen, T.T.; Lyu, J.X.; Shen, Y. Carnosine suppresses human glioma cells under normoxic and hypoxic conditions partly via inhibiting glutamine metabolism. Acta Pharmacol. Sin. 2021, 42, 767–779. [Google Scholar] [CrossRef]
- Yamashita, A.S.; da Costa Rosa, M.; Stumpo, V.; Rais, R.; Slusher, B.S.; Riggins, G.J. The glutamine antagonist prodrug JHU-083 slows malignant glioma growth and disrupts mTOR signaling. Neurooncol. Adv. 2021, 3, vdaa149. [Google Scholar] [CrossRef]
- De Groot, J.F.; Liu, T.J.; Fuller, G.; Yung, W.K. The excitatory amino acid transporter-2 induces apoptosis and decreases glioma growth in vitro and in vivo. Cancer Res. 2005, 65, 1934–1940. [Google Scholar] [CrossRef]
- Robe, P.A.; Martin, D.H.; Nguyen-Khac, M.T.; Artesi, M.; Deprez, M.; Albert, A.; Vanbelle, S.; Califice, S.; Bredel, M.; Bours, V. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 2009, 9, 372. [Google Scholar] [CrossRef]
- Corbetta, C.; Di Ianni, N.; Bruzzone, M.G.; Patane, M.; Pollo, B.; Cantini, G.; Cominelli, M.; Zucca, I.; Pisati, F.; Poliani, P.L.; et al. Altered function of the glutamate-aspartate transporter GLAST, a potential therapeutic target in glioblastoma. Int. J. Cancer 2019, 144, 2539–2554. [Google Scholar] [CrossRef] [PubMed]
- Marchiq, I.; Le Floch, R.; Roux, D.; Simon, M.P.; Pouyssegur, J. Genetic disruption of lactate/H+ symporters (MCTs) and their subunit CD147/BASIGIN sensitizes glycolytic tumor cells to phenformin. Cancer Res. 2015, 75, 171–180. [Google Scholar] [CrossRef]
- Chaumeil, M.M.; Radoul, M.; Najac, C.; Eriksson, P.; Viswanath, P.; Blough, M.D.; Chesnelong, C.; Luchman, H.A.; Cairncross, J.G.; Ronen, S.M. Hyperpolarized 13C MR imaging detects no lactate production in mutant IDH1 gliomas: Implications for diagnosis and response monitoring. NeuroImage Clin. 2016, 12, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Viswanath, P.; Najac, C.; Izquierdo-Garcia, J.L.; Pankov, A.; Hong, C.; Eriksson, P.; Costello, J.F.; Pieper, R.O.; Ronen, S.M. Mutant IDH1 expression is associated with down-regulation of monocarboxylate transporters. Oncotarget 2016, 7, 34942–34955. [Google Scholar] [CrossRef] [PubMed]
- Spehalski, E.I.; Lee, J.A.; Peters, C.; Tofilon, P.; Camphausen, K. The Quiescent Metabolic Phenotype of Glioma Stem Cells. J. Proteom. Bioinform. 2019, 12, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Joshi, K.; Li, J.; Kim, S.H.; Li, P.; Santana-Santos, L.; Luthra, S.; Chandran, U.R.; Benos, P.V.; Smith, L.; et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA 2013, 110, 8644–8649. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Duraj, T.; Garcia-Romero, N.; Carrion-Navarro, J.; Madurga, R.; Mendivil, A.O.; Prat-Acin, R.; Garcia-Canamaque, L.; Ayuso-Sacido, A. Beyond the Warburg Effect: Oxidative and Glycolytic Phenotypes Coexist within the Metabolic Heterogeneity of Glioblastoma. Cells 2021, 10, 202. [Google Scholar] [CrossRef]
- Bergstrom, J.; Furst, P.; Noree, L.O.; Vinnars, E. Intracellular free amino acid concentration in human muscle tissue. J. Appl. Physiol. 1974, 36, 693–697. [Google Scholar] [CrossRef]
- Curi, R.; Lagranha, C.J.; Doi, S.Q.; Sellitti, D.F.; Procopio, J.; Pithon-Curi, T.C.; Corless, M.; Newsholme, P. Molecular mechanisms of glutamine action. J. Cell Physiol. 2005, 204, 392–401. [Google Scholar] [CrossRef]
- Lee, J.S.; Adler, L.; Karathia, H.; Carmel, N.; Rabinovich, S.; Auslander, N.; Keshet, R.; Stettner, N.; Silberman, A.; Agemy, L.; et al. Urea Cycle Dysregulation Generates Clinically Relevant Genomic and Biochemical Signatures. Cell 2018, 174, 1559–1570.e1522. [Google Scholar] [CrossRef] [PubMed]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Feng, Z.; Hoos, A.; Klimberg, V.S. Glutamine enhances gut glutathione production. JPEN J. Parenter. Enter. Nutr. 1998, 22, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Neu, J.; Shenoy, V.; Chakrabarti, R. Glutamine nutrition and metabolism: Where do we go from here ? FASEB J. 1996, 10, 829–837. [Google Scholar] [CrossRef]
- Chen, L.; Cui, H. Targeting Glutamine Induces Apoptosis: A Cancer Therapy Approach. Int. J. Mol. Sci. 2015, 16, 22830–22855. [Google Scholar] [CrossRef]
- Martins, F.; Goncalves, L.G.; Pojo, M.; Serpa, J. Take Advantage of Glutamine Anaplerosis, the Kernel of the Metabolic Rewiring in Malignant Gliomas. Biomolecules 2020, 10, 1370. [Google Scholar] [CrossRef]
- Martinez-Hernandez, A.; Bell, K.P.; Norenberg, M.D. Glutamine synthetase: Glial localization in brain. Science 1977, 195, 1356–1358. [Google Scholar] [CrossRef]
- Albrecht, J.; Sidoryk-Wegrzynowicz, M.; Zielinska, M.; Aschner, M. Roles of glutamine in neurotransmission. Neuron Glia Biol. 2010, 6, 263–276. [Google Scholar] [CrossRef]
- Albrecht, J.; Zielinska, M. Exchange-mode glutamine transport across CNS cell membranes. Neuropharmacology 2019, 161, 107560. [Google Scholar] [CrossRef]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Tapiero, H.; Mathe, G.; Couvreur, P.; Tew, K.D., II. Glutamine and glutamate. Biomed. Pharmacother. 2002, 56, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Masisi, B.K.; El Ansari, R.; Alfarsi, L.; Rakha, E.A.; Green, A.R.; Craze, M.L. The role of glutaminase in cancer. Histopathology 2020, 76, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Obara-Michlewska, M.; Szeliga, M. Targeting Glutamine Addiction in Gliomas. Cancers 2020, 12, 310. [Google Scholar] [CrossRef]
- Dringen, R.; Hirrlinger, J. Glutathione pathways in the brain. Biol. Chem. 2003, 384, 505–516. [Google Scholar] [CrossRef]
- Lange, F.; Hornschemeyer, J.; Kirschstein, T. Glutamatergic Mechanisms in Glioblastoma and Tumor-Associated Epilepsy. Cells 2021, 10, 1226. [Google Scholar] [CrossRef]
- Behrens, P.F.; Langemann, H.; Strohschein, R.; Draeger, J.; Hennig, J. Extracellular glutamate and other metabolites in and around RG2 rat glioma: An intracerebral microdialysis study. J. Neurooncol. 2000, 47, 11–22. [Google Scholar] [CrossRef]
- Atlante, A.; Calissano, P.; Bobba, A.; Giannattasio, S.; Marra, E.; Passarella, S. Glutamate neurotoxicity, oxidative stress and mitochondria. FEBS Lett. 2001, 497, 1–5. [Google Scholar] [CrossRef]
- Habib, E.; Linher-Melville, K.; Lin, H.X.; Singh, G. Expression of xCT and activity of system xc(-) are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 2015, 5, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.J.; Lyons, S.A.; Nelson, G.M.; Hamza, H.; Gladson, C.L.; Gillespie, G.Y.; Sontheimer, H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J. Neurosci. 2005, 25, 7101–7110. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Z.; Xu, H.; Liao, Y.; Sun, C.; Chen, Y.; Sheng, M.; Lan, Q.; Wang, Z. PSMD12 promotes glioma progression by upregulating the expression of Nrf2. Ann. Transl. Med. 2021, 9, 700. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.; Sandhu, J.K.; Harper, M.E.; Cuperlovic-Culf, M. Role of Glutathione in Cancer: From Mechanisms to Therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.K.; Venneti, S. Glutamine Metabolism in Brain Tumors. Cancers 2019, 11, 1628. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Marquez, J.; Alonso, F.J.; Mates, J.M.; Segura, J.A.; Martin-Rufian, M.; Campos-Sandoval, J.A. Glutamine Addiction In Gliomas. Neurochem. Res. 2017, 42, 1735–1746. [Google Scholar] [CrossRef]
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.L.; Rajagopalan, K.N.; Maddie, M.; Vemireddy, V.; Zhao, Z.; et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012, 15, 827–837. [Google Scholar] [CrossRef]
- Tanaka, K.; Sasayama, T.; Irino, Y.; Takata, K.; Nagashima, H.; Satoh, N.; Kyotani, K.; Mizowaki, T.; Imahori, T.; Ejima, Y.; et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J. Clin. Investig. 2015, 125, 1591–1602. [Google Scholar] [CrossRef]
- Dolinska, M.; Dybel, A.; Hilgier, W.; Zielinska, M.; Zablocka, B.; Buzanska, L.; Albrecht, J. Glutamine transport in C6 glioma cells: Substrate specificity and modulation in a glutamine deprived culture medium. J. Neurosci. Res. 2001, 66, 959–966. [Google Scholar] [CrossRef]
- Sidoryk, M.; Matyja, E.; Dybel, A.; Zielinska, M.; Bogucki, J.; Jaskolski, D.J.; Liberski, P.P.; Kowalczyk, P.; Albrecht, J. Increased expression of a glutamine transporter SNAT3 is a marker of malignant gliomas. Neuroreport 2004, 15, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Dolinska, M.; Dybel, A.; Zablocka, B.; Albrecht, J. Glutamine transport in C6 glioma cells shows ASCT2 system characteristics. Neurochem. Int. 2003, 43, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.C.; Rothstein, J.D.; Sontheimer, H. Compromised glutamate transport in human glioma cells: Reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J. Neurosci. 1999, 19, 10767–10777. [Google Scholar] [CrossRef] [PubMed]
- Broer, A.; Brookes, N.; Ganapathy, V.; Dimmer, K.S.; Wagner, C.A.; Lang, F.; Broer, S. The astroglial ASCT2 amino acid transporter as a mediator of glutamine efflux. J. Neurochem. 1999, 73, 2184–2194. [Google Scholar] [PubMed]
- Lorin, S.; Tol, M.J.; Bauvy, C.; Strijland, A.; Pous, C.; Verhoeven, A.J.; Codogno, P.; Meijer, A.J. Glutamate dehydrogenase contributes to leucine sensing in the regulation of autophagy. Autophagy 2013, 9, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Haining, Z.; Kawai, N.; Miyake, K.; Okada, M.; Okubo, S.; Zhang, X.; Fei, Z.; Tamiya, T. Relation of LAT1/4F2hc expression with pathological grade, proliferation and angiogenesis in human gliomas. BMC Clin. Pathol. 2012, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Nawashiro, H.; Otani, N.; Uozumi, Y.; Ooigawa, H.; Toyooka, T.; Suzuki, T.; Katoh, H.; Tsuzuki, N.; Ohnuki, A.; Shima, K.; et al. High expression of L-type amino acid transporter 1 in infiltrating glioma cells. Brain Tumor Pathol. 2005, 22, 89–91. [Google Scholar] [CrossRef]
- Kobayashi, K.; Ohnishi, A.; Promsuk, J.; Shimizu, S.; Kanai, Y.; Shiokawa, Y.; Nagane, M. Enhanced tumor growth elicited by L-type amino acid transporter 1 in human malignant glioma cells. Neurosurgery 2008, 62, 493–503, discussion 503–494. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef]
- Wang, X.; Yang, K.; Wu, Q.; Kim, L.J.Y.; Morton, A.R.; Gimple, R.C.; Prager, B.C.; Shi, Y.; Zhou, W.; Bhargava, S.; et al. Targeting pyrimidine synthesis accentuates molecular therapy response in glioblastoma stem cells. Sci. Transl. Med. 2019, 11, eaau4972. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.H.; Coloff, J.L. The Diverse Functions of Non-Essential Amino Acids in Cancer. Cancers 2019, 11, 675. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Luengo, A.; Danai, L.V.; Bush, L.N.; Diehl, F.F.; Hosios, A.M.; Lau, A.N.; Elmiligy, S.; Malstrom, S.; Lewis, C.A.; et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nat. Cell Biol. 2018, 20, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Li, B.S.; Gu, L.J.; Luo, C.Y.; Li, W.S.; Jiang, L.M.; Shen, S.H.; Jiang, H.; Shen, S.H.; Zhang, B.; Chen, J.; et al. The downregulation of asparagine synthetase expression can increase the sensitivity of cells resistant to l-asparaginase. Leukemia 2006, 20, 2199–2201. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Stampouloglou, E.; Kingston, N.M.; Zhang, L.; Monti, S.; Varelas, X. Glutamine-utilizing transaminases are a metabolic vulnerability of TAZ/YAP-activated cancer cells. EMBO Rep. 2018, 19, e43577. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fan, J.; Venneti, S.; Cross, J.R.; Takagi, T.; Bhinder, B.; Djaballah, H.; Kanai, M.; Cheng, E.H.; Judkins, A.R.; et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol. Cell 2014, 56, 205–218. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Lopes-Coelho, F.; Gouveia-Fernandes, S.; Goncalves, L.G.; Nunes, C.; Faustino, I.; Silva, F.; Felix, A.; Pereira, S.A.; Serpa, J. HNF1beta drives glutathione (GSH) synthesis underlying intrinsic carboplatin resistance of ovarian clear cell carcinoma (OCCC). Tumour Biol. 2016, 37, 4813–4829. [Google Scholar] [CrossRef] [PubMed]
- McCormick, D.; McQuaid, S.; McCusker, C.; Allen, I.V. A study of glutamine synthetase in normal human brain and intracranial tumours. Neuropathol. Appl. Neurobiol. 1990, 16, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.O.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Rosati, A.; Poliani, P.L.; Todeschini, A.; Cominelli, M.; Medicina, D.; Cenzato, M.; Simoncini, E.L.; Magrini, S.M.; Buglione, M.; Grisanti, S.; et al. Glutamine synthetase expression as a valuable marker of epilepsy and longer survival in newly diagnosed glioblastoma multiforme. Neuro Oncol. 2013, 15, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Rosati, A.; Marconi, S.; Pollo, B.; Tomassini, A.; Lovato, L.; Maderna, E.; Maier, K.; Schwartz, A.; Rizzuto, N.; Padovani, A.; et al. Epilepsy in glioblastoma multiforme: Correlation with glutamine synthetase levels. J. Neurooncol. 2009, 93, 319–324. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Shi, X.; Zhang, L.; Yi, C.; Zhang, X.; Zhang, X. De Novo Glutamine Synthesis: Importance for the Proliferation of Glioma Cells and Potentials for Its Detection With 13N-Ammonia. Mol. Imaging 2016, 15, 1536012116645440. [Google Scholar] [CrossRef] [PubMed]
- Bagga, P.; Behar, K.L.; Mason, G.F.; De Feyter, H.M.; Rothman, D.L.; Patel, A.B. Characterization of cerebral glutamine uptake from blood in the mouse brain: Implications for metabolic modeling of 13C NMR data. J. Cereb. Blood Flow. Metab. 2014, 34, 1666–1672. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Yin, Y.; Sun, W.; Xiang, J.; Deng, L.; Zhang, B.; Xie, P.; Qiao, W.; Zou, J.; Liu, C. Glutamine synthetase functions as a negative growth regulator in glioma. J. Neurooncol. 2013, 114, 59–69. [Google Scholar] [CrossRef]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes. Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef]
- Tong, X.; Zhao, F.; Thompson, C.B. The molecular determinants of de novo nucleotide biosynthesis in cancer cells. Curr. Opin. Genet. Dev. 2009, 19, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Mates, J.M.; Segura, J.A.; Martin-Rufian, M.; Campos-Sandoval, J.A.; Alonso, F.J.; Marquez, J. Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr. Mol. Med. 2013, 13, 514–534. [Google Scholar] [CrossRef]
- De la Rosa, V.; Campos-Sandoval, J.A.; Martin-Rufian, M.; Cardona, C.; Mates, J.M.; Segura, J.A.; Alonso, F.J.; Marquez, J. A novel glutaminase isoform in mammalian tissues. Neurochem. Int. 2009, 55, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Mates, J.M.; Campos-Sandoval, J.A.; Santos-Jimenez, J.L.; Marquez, J. Dysregulation of glutaminase and glutamine synthetase in cancer. Cancer Lett. 2019, 467, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Mates, J.M.; Campos-Sandoval, J.A.; de Los Santos-Jimenez, J.; Segura, J.A.; Alonso, F.J.; Marquez, J. Metabolic Reprogramming of Cancer by Chemicals that Target Glutaminase Isoenzymes. Curr. Med. Chem. 2020, 27, 5317–5339. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.; Hartmann, R.; Tsiampali, J.; Uhlmann, C.; Nickel, A.C.; He, X.; Kamp, M.A.; Sabel, M.; Barker, R.A.; Steiger, H.J.; et al. A comparative pharmaco-metabolomic study of glutaminase inhibitors in glioma stem-like cells confirms biological effectiveness but reveals differences in target-specificity. Cell Death Discov. 2020, 6, 20. [Google Scholar] [CrossRef]
- Martin-Rufian, M.; Nascimento-Gomes, R.; Higuero, A.; Crisma, A.R.; Campos-Sandoval, J.A.; Gomez-Garcia, M.C.; Cardona, C.; Cheng, T.; Lobo, C.; Segura, J.A.; et al. Both GLS silencing and GLS2 overexpression synergize with oxidative stress against proliferation of glioma cells. J. Mol. Med. 2014, 92, 277–290. [Google Scholar] [CrossRef]
- Nayak, P.; Chatterjee, A.K. Response of regional brain glutamate transaminases of rat to aluminum in protein malnutrition. BMC Neurosci. 2002, 3, 12. [Google Scholar] [CrossRef]
- Yang, C.; Sudderth, J.; Dang, T.; Bachoo, R.M.; McDonald, J.G.; DeBerardinis, R.J. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009, 69, 7986–7993. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Nishimura, M.C.; Kharbanda, S.; Peale, F.; Deng, Y.; Daemen, A.; Forrest, W.F.; Kwong, M.; Hedehus, M.; Hatzivassiliou, G.; et al. Hominoid-specific enzyme GLUD2 promotes growth of IDH1R132H glioma. Proc. Natl. Acad. Sci. USA 2014, 111, 14217–14222. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E. Friend or foe-IDH1 mutations in glioma 10 years on. Carcinogenesis 2019, 40, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Zhang, Y.; Jung, M.; Cruz, N.; Alas, B.; Commisso, C. EGFR-Pak Signaling Selectively Regulates Glutamine Deprivation-Induced Macropinocytosis. Dev. Cell 2019, 50, 381–392.e385. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Sasayama, T.; Nagashima, H.; Irino, Y.; Takahashi, M.; Izumi, Y.; Uno, T.; Satoh, N.; Kitta, A.; Kyotani, K.; et al. Glioma cells require one-carbon metabolism to survive glutamine starvation. Acta Neuropathol. Commun. 2021, 9, 16. [Google Scholar] [CrossRef]
- Fares, H.M.; Lyu, X.; Xu, X.; Dong, R.; Ding, M.; Mi, S.; Wang, Y.; Li, X.; Yuan, S.; Sun, L. Autophagy in cancer: The cornerstone during glutamine deprivation. Eur. J. Pharmacol. 2022, 916, 174723. [Google Scholar] [CrossRef]
- Van der Vos, K.E.; Eliasson, P.; Proikas-Cezanne, T.; Vervoort, S.J.; van Boxtel, R.; Putker, M.; van Zutphen, I.J.; Mauthe, M.; Zellmer, S.; Pals, C.; et al. Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nat. Cell Biol. 2012, 14, 829–837. [Google Scholar] [CrossRef]
- Van der Vos, K.E.; Coffer, P.J. Glutamine metabolism links growth factor signaling to the regulation of autophagy. Autophagy 2012, 8, 1862–1864. [Google Scholar] [CrossRef]
- Sa-Nongdej, W.; Chongthammakun, S.; Songthaveesin, C. Nutrient starvation induces apoptosis and autophagy in C6 glioma stem-like cells. Heliyon 2021, 7, e06352. [Google Scholar] [CrossRef]
- Ali Shah, S.W.; Zhang, S.; Ishfaq, M.; Tang, Y.; Teng, X. PTEN/AKT/mTOR pathway involvement in autophagy, mediated by miR-99a-3p and energy metabolism in ammonia-exposed chicken bursal lymphocytes. Poult. Sci. 2021, 100, 553–564. [Google Scholar] [CrossRef]
- Lu, K.; Zimmermann, M.; Gorg, B.; Bidmon, H.J.; Biermann, B.; Klocker, N.; Haussinger, D.; Reichert, A.S. Hepatic encephalopathy is linked to alterations of autophagic flux in astrocytes. EBioMedicine 2019, 48, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Villar, V.H.; Duran, R.V. Glutamoptosis: A new cell death mechanism inhibited by autophagy during nutritional imbalance. Autophagy 2017, 13, 1078–1079. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.; Giacomelli, C.; Zappelli, E.; Granchi, C.; Trincavelli, M.L.; Minutolo, F.; Martini, C. Lactate dehydrogenase-A inhibition induces human glioblastoma multiforme stem cell differentiation and death. Sci. Rep. 2015, 5, 15556. [Google Scholar] [CrossRef] [PubMed]
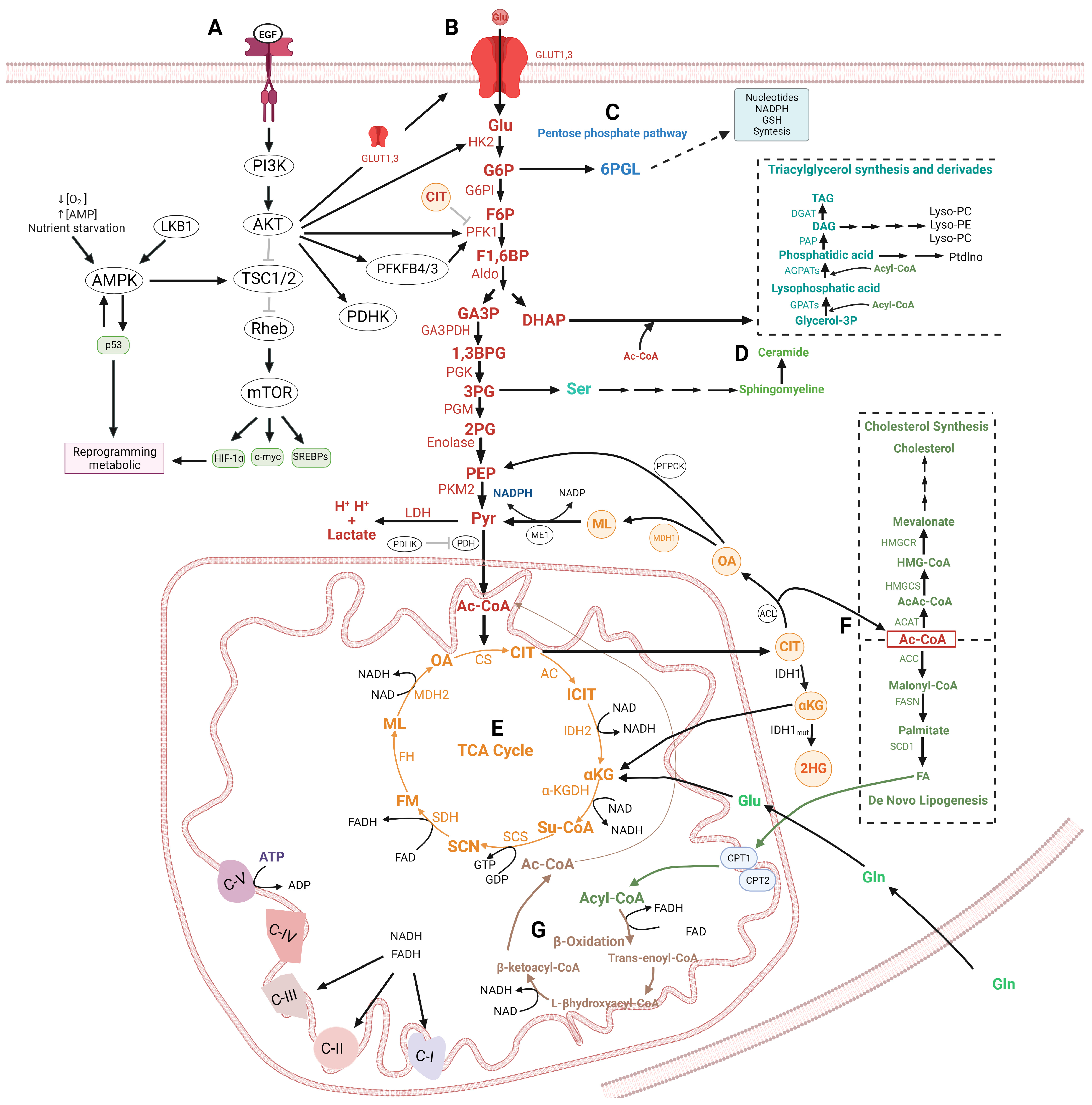
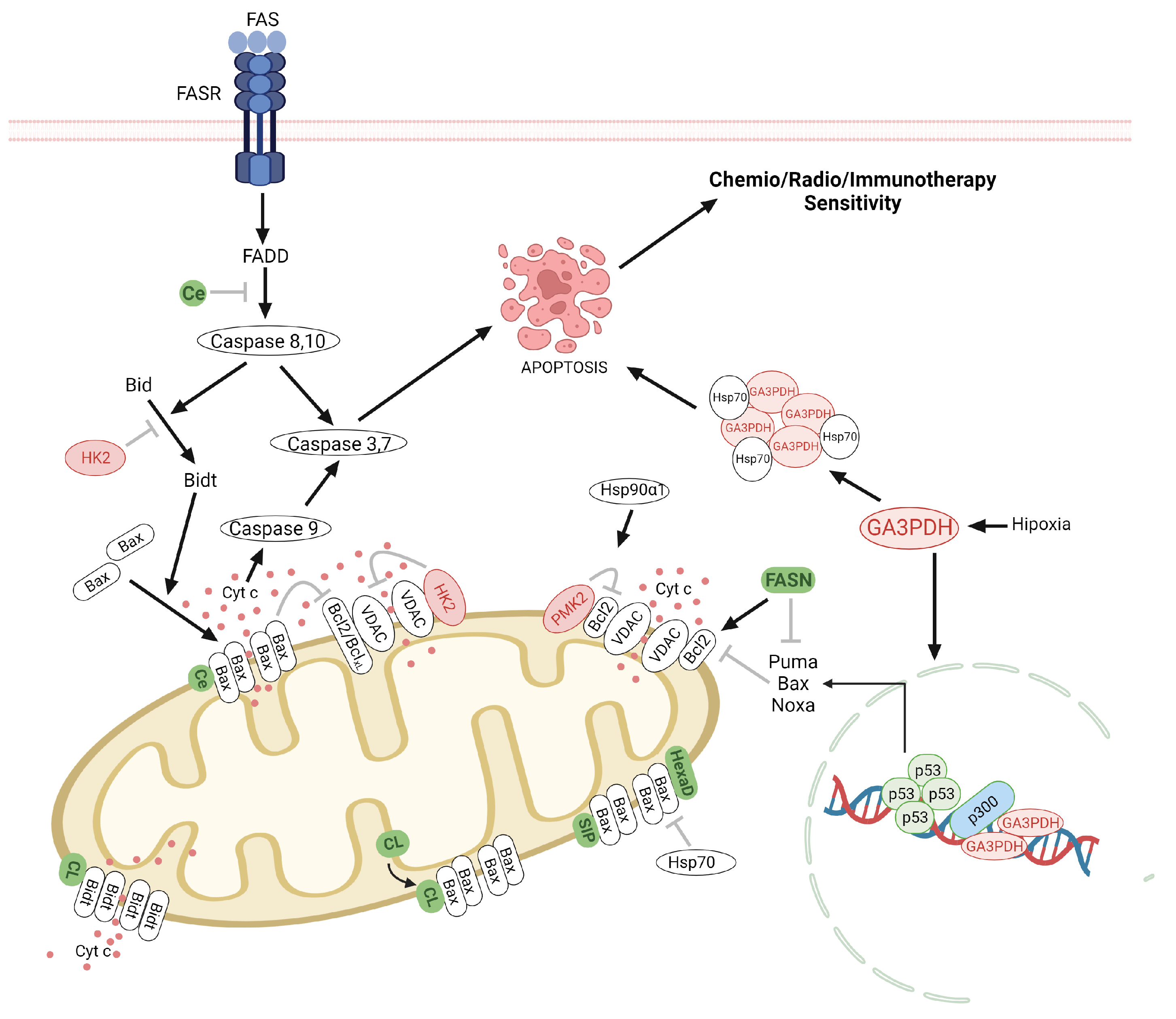
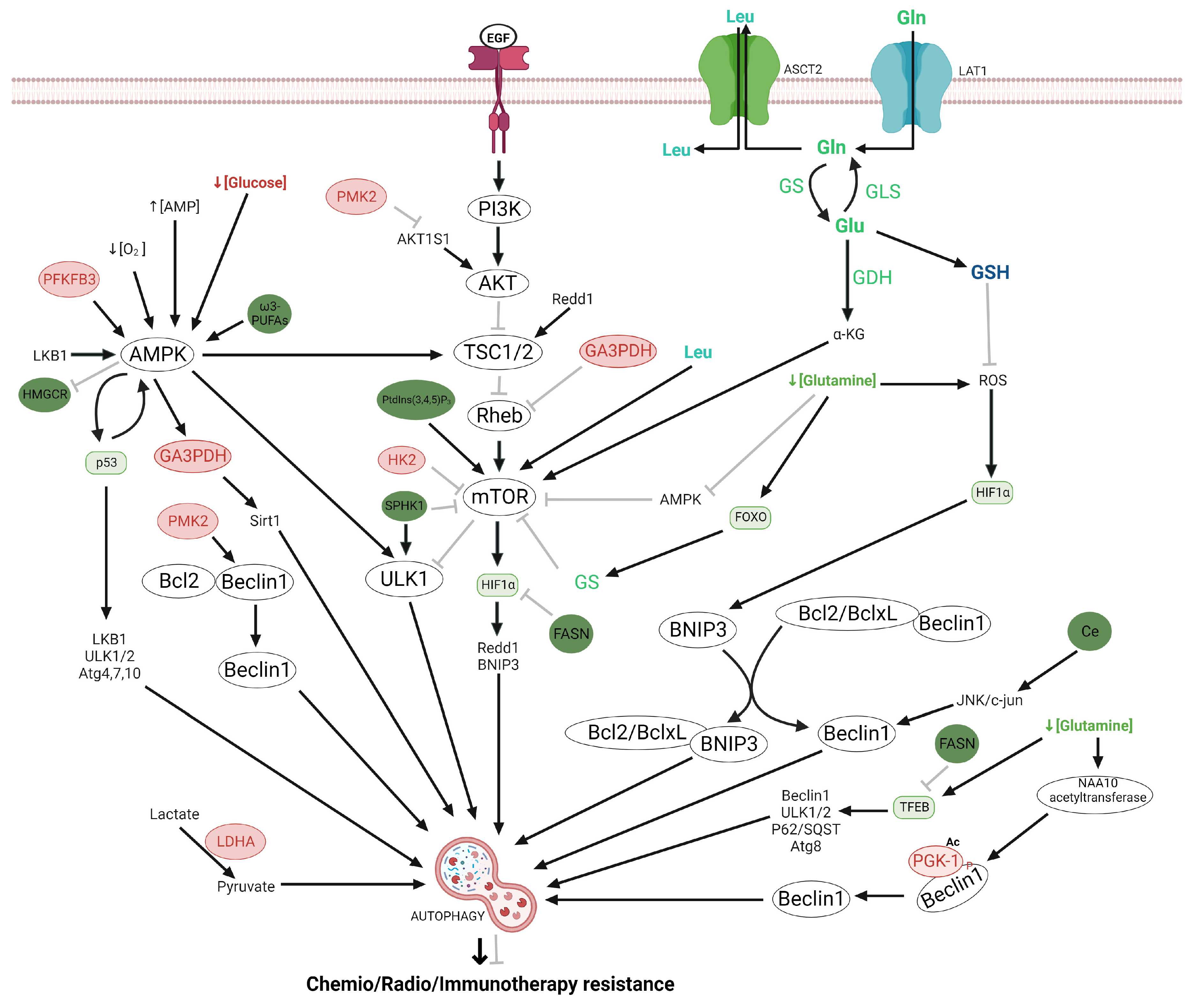

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Treatment | Clinical and Preclinical Trials |
|---|---|---|
| Glycolysis | ||
| Glycolysis pathway | Methylene blue | Increased OCR and decreased ECAR and lactate production in glioblastoma cell lines [223]. |
| Enolase | HEX and POMHEX | Decrease in TCA cycle metabolites using a glioma cell expressing ENO2 but no ENO1 [136]. |
| Pyruvate Dehydrogenase Kinases (PDHK) | Dichloroacetate (DCA) | Increased the oxidative phosphorylation in vitro. Also, induces cell death in glioma cells, through the generation of ROS with a decrease in p53, HIF-1, p21, proliferating cell nuclear antigen (PCNA), hexokinase 2, VEGF, and α-ketoglutarate [224,225]. |
| 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3). | 3-(3-Pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO) | In complement to bevacizumab, decreased cell proliferation and increased apoptosis in vitro, whereas delayed tumor growth and improved survival using in vivo [226] |
| Syk inhibitor | R406 | Shifts the glioma cells from a glycolytic profile toward OXPHOS metabolism, and increases apoptosis [227]. |
| Monocarboxylate transporter 4 (MCT4) | Acriflavine | Decrease cell proliferation and tumoral growth [228,229]. |
| Histone deacetylase (HDAC) | Vorinostat | Decrease intracellular lactate, cell viability, and tumor progression [230]. |
| Glutaminolysis | ||
| Glutaminase (GLS) | Compound 968 | Inhibit the glioma cell growth with the Notch pathway blocked [231,232,233] |
| bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl) ethyl sulfide (BPTES). | Slows GBM growth cell with IDH1mut [234]. | |
| CB-839 | Reduced proliferation, angiogenesis, and modestly increased apoptosis in glioma stem-like cells with IDH1mut [235,236,237]. | |
| Glutamine Synthetase (GS) | Actinomycin D, 5-azacytidine. | Decreased proliferation in glioma cells [231]. |
| Carnosine | Suppressed growth and migration in glioma cells [238]. | |
| Glutamine | Acivicin, 6-diazo-5-oxo-L-norleucine (DON). | Inhibit the growth of glioma cells [231]. |
| 6-diazo-5-oxo-L-norleucine (DON) plus L-asparaginase. | Induces apoptosis and autophagy in glioma cells by the depletion of asparagine via inhibition of asparagine synthetase [232]. | |
| JHU-083 | Inhibits cell growth and mTOR signaling [239]. | |
| Glutamate | GLT1 cDNA overexpression. | Inhibits growth proliferation in glioma cells [240]. |
| xCT | Sulfasalazine | Induce apoptosis in GBM cells [241]. |
| GLAST | shGLAST | Increases survival in glioma-bearing mice [242]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trejo-Solis, C.; Silva-Adaya, D.; Serrano-García, N.; Magaña-Maldonado, R.; Jimenez-Farfan, D.; Ferreira-Guerrero, E.; Cruz-Salgado, A.; Castillo-Rodriguez, R.A. Role of Glycolytic and Glutamine Metabolism Reprogramming on the Proliferation, Invasion, and Apoptosis Resistance through Modulation of Signaling Pathways in Glioblastoma. Int. J. Mol. Sci. 2023, 24, 17633. https://doi.org/10.3390/ijms242417633
Trejo-Solis C, Silva-Adaya D, Serrano-García N, Magaña-Maldonado R, Jimenez-Farfan D, Ferreira-Guerrero E, Cruz-Salgado A, Castillo-Rodriguez RA. Role of Glycolytic and Glutamine Metabolism Reprogramming on the Proliferation, Invasion, and Apoptosis Resistance through Modulation of Signaling Pathways in Glioblastoma. International Journal of Molecular Sciences. 2023; 24(24):17633. https://doi.org/10.3390/ijms242417633
Chicago/Turabian StyleTrejo-Solis, Cristina, Daniela Silva-Adaya, Norma Serrano-García, Roxana Magaña-Maldonado, Dolores Jimenez-Farfan, Elizabeth Ferreira-Guerrero, Arturo Cruz-Salgado, and Rosa Angelica Castillo-Rodriguez. 2023. "Role of Glycolytic and Glutamine Metabolism Reprogramming on the Proliferation, Invasion, and Apoptosis Resistance through Modulation of Signaling Pathways in Glioblastoma" International Journal of Molecular Sciences 24, no. 24: 17633. https://doi.org/10.3390/ijms242417633
APA StyleTrejo-Solis, C., Silva-Adaya, D., Serrano-García, N., Magaña-Maldonado, R., Jimenez-Farfan, D., Ferreira-Guerrero, E., Cruz-Salgado, A., & Castillo-Rodriguez, R. A. (2023). Role of Glycolytic and Glutamine Metabolism Reprogramming on the Proliferation, Invasion, and Apoptosis Resistance through Modulation of Signaling Pathways in Glioblastoma. International Journal of Molecular Sciences, 24(24), 17633. https://doi.org/10.3390/ijms242417633