

ISGF3 and STAT2/IRF9 Control Basal and IFN-Induced Transcription through Genome-Wide Binding of Phosphorylated and Unphosphorylated Complexes to Common ISRE-Containing ISGs

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
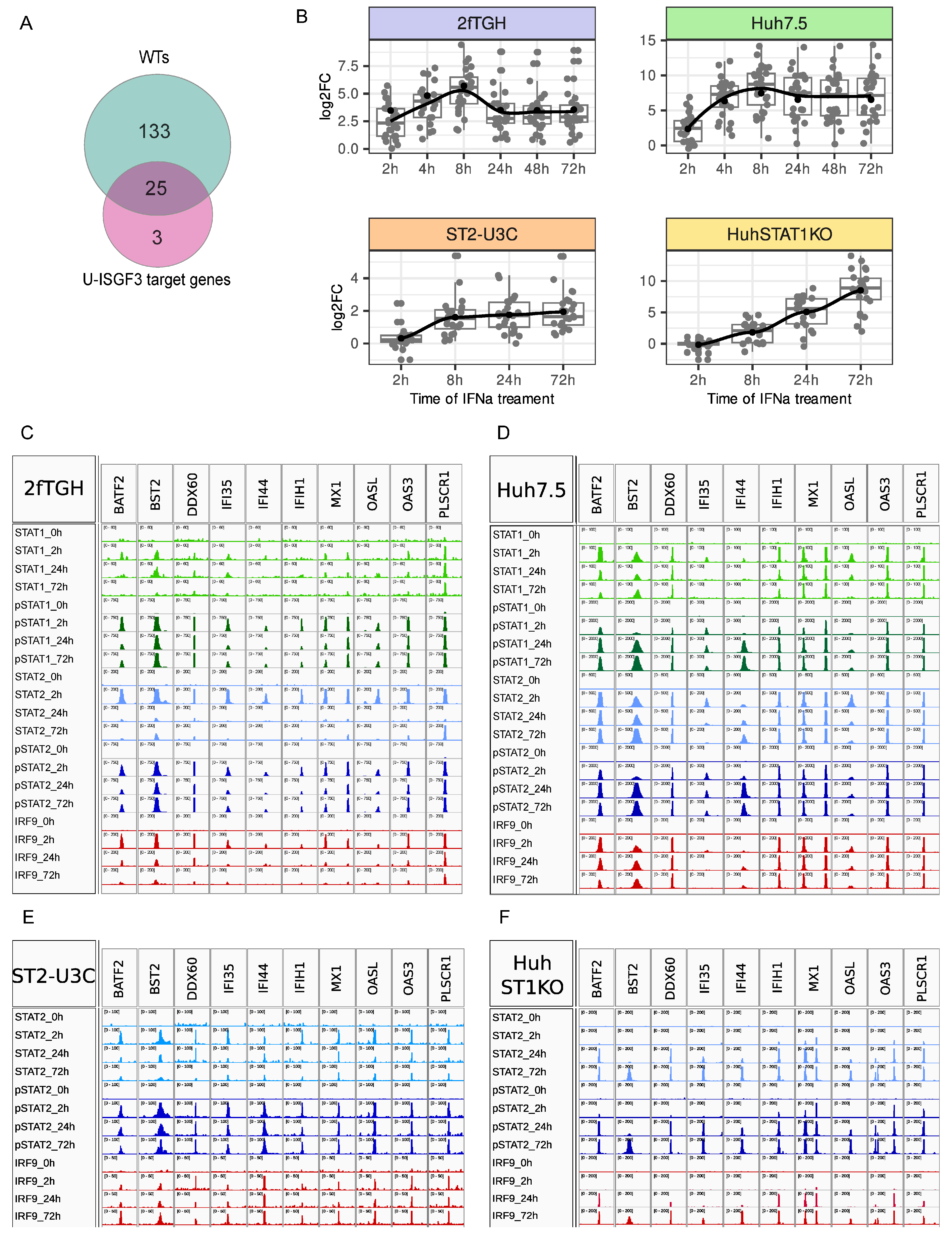
2.1. Characterization of Time-Dependent IFNα Responses in WT and STAT1-KO Cells
2.2. Genome-Wide Characterization of IFNα-Induced Transcription in WT vs. STAT1KO Cells
2.3. Genome-Wide Binding of Phosphorylated and Unphosphorylated ISGF3 Components to ISRE Sites of IFNα Upregulated Genes in WT vs. STAT1KO Cells
2.4. U-ISG Expression Correlates with pSTAT1 and pSTAT2 Expression and Long-Term Binding in Wild Type and STAT1KO Cells
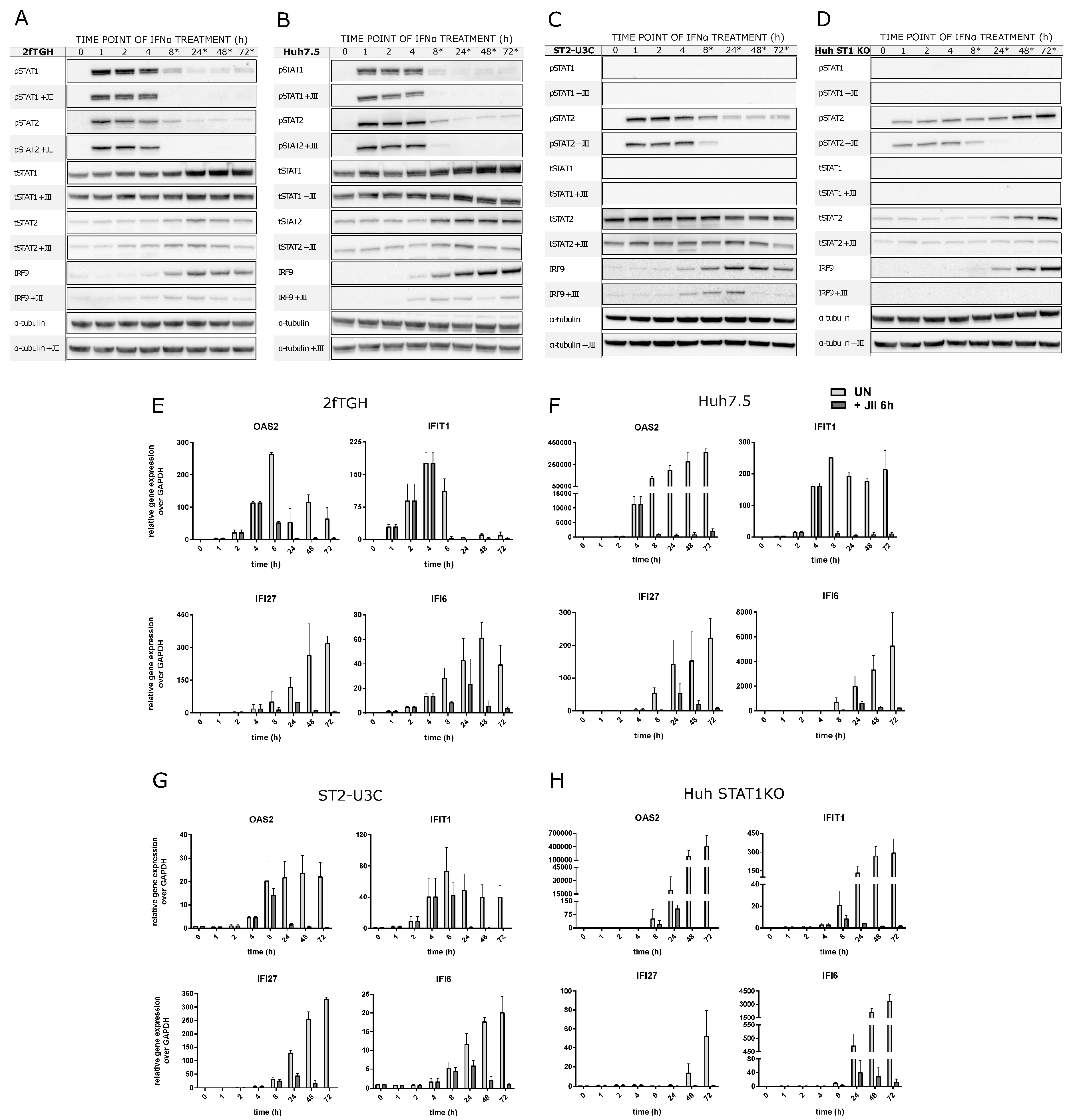
2.5. The Role of Phosphorylation of ISGF3 and STAT2/IRF9 in the Regulation of Prolonged ISG Expression in Wild Type and STAT1KO Cells
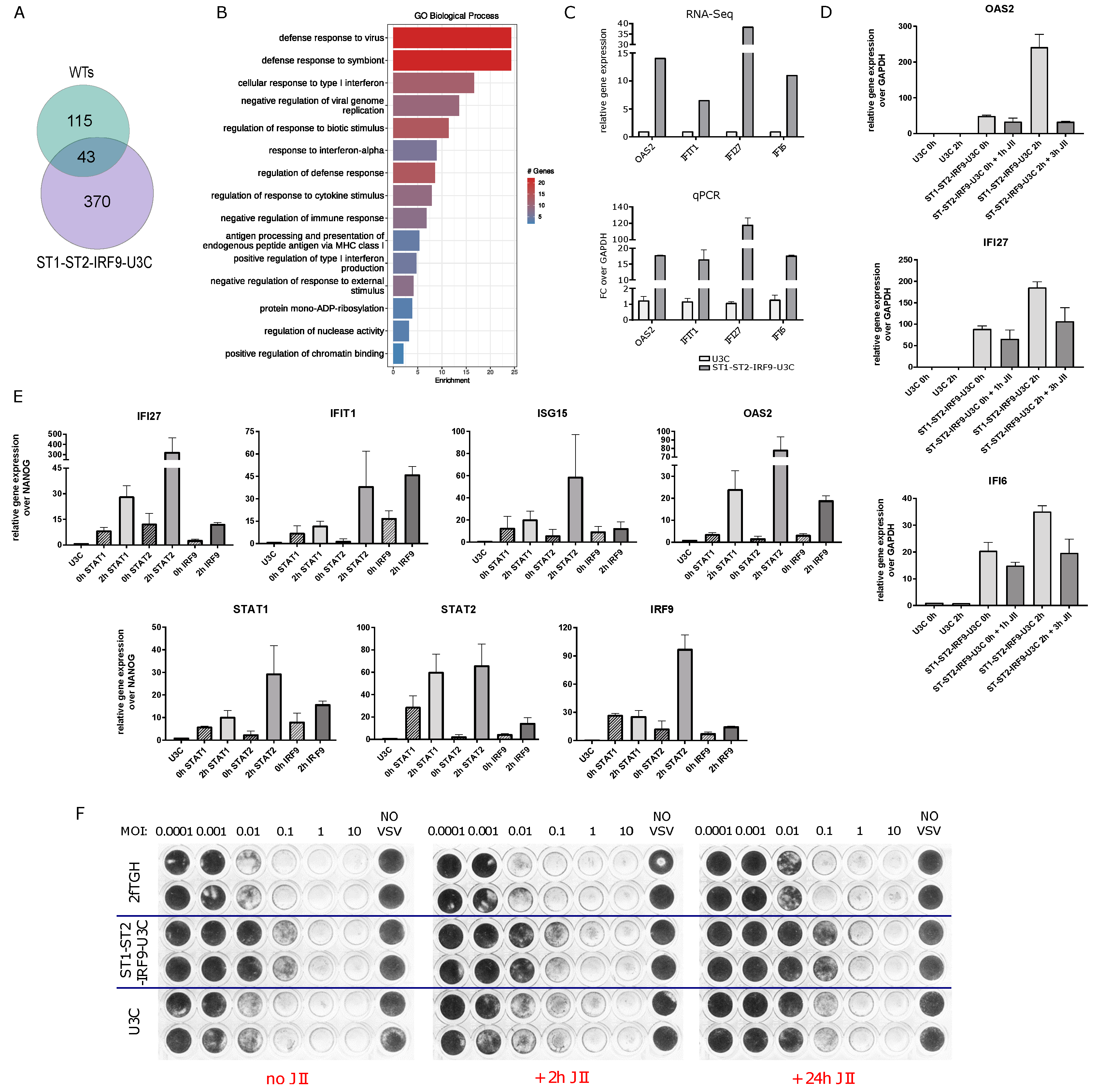
2.6. The Role of Unphosphorylated ISGF3 Components in the Regulation of Basal ISG Expression in Cells Overexpressing STAT1, STAT2 and IRF9
2.7. The Role of Unphosphorylated ISGF3 Components in Prolonged IFNα Signaling in Cells Overexpressing STAT1, STAT2 and IRF9
3. Discussion
4. Materials and Methods
4.1. Cell Lines
Cell Culture and Treatment
4.2. Western Blotting
4.3. RNA Isolation and qPCR
4.4. RNA-Seq Library Preparation and Sequencing
4.5. RNA-Seq Data Analysis
4.5.1. Differential Gene Expression Analysis (DEG)
4.5.2. Heatmap Generation
4.5.3. Gene Ontology Term Enrichment Analysis
4.5.4. Selection of Commonly Upregulated Genes
4.6. Chromatin Immunoprecipitation (ChIP) and Sequencing (ChIP-Seq)
4.7. ChIP-Seq Data Analysis
4.7.1. Visualization in the Integrative Genomics Viewer
4.7.2. Binding Profiles
4.7.3. Binding Site Motifs Identification
4.8. Deposited Sequencing Data
4.9. Antiviral Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blaszczyk, K.; Nowicka, H.; Kostyrko, K.; Antonczyk, A.; Wesoly, J.; Bluyssen, H.A.R. The Unique Role of STAT2 in Constitutive and IFN-Induced Transcription and Antiviral Responses. Cytokine Growth Factor Rev. 2016, 29, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Michalska, A.; Blaszczyk, K.; Wesoly, J.; Bluyssen, H.A.R. A Positive Feedback Amplifier Circuit That Regulates Interferon (IFN)-Stimulated Gene Expression and Controls Type I and Type II IFN Responses. Front. Immunol. 2018, 9, 1135. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Darnell, J.E. STATs: Transcriptional Control and Biological Impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Bluyssen, H.A.R.; Levy, D.E. Stat2 Is a Transcriptional Activator That Requires Sequence-Specific Contacts Provided by Stat1 and P48 for Stable Interaction with DNA. J. Biol. Chem. 1997, 272, 4600–4605. [Google Scholar] [CrossRef] [PubMed]
- Blaszczyk, K.; Olejnik, A.; Nowicka, H.; Ozgyin, L.; Chen, Y.L.; Chmielewski, S.; Kostyrko, K.; Wesoly, J.; Balint, B.L.; Lee, C.K.; et al. STAT2/IRF9 Directs a Prolonged ISGF3-like Transcriptional Response and Antiviral Activity in the Absence of STAT1. Biochem. J. 2015, 466, 511–524. [Google Scholar] [CrossRef]
- Lou, Y.J.; Pan, X.R.; Jia, P.M.; Li, D.; Xiao, S.; Zhang, Z.L.; Chen, S.J.; Chen, Z.; Tong, J.H. Ifr-9/Stat2 Functional Interaction Drives Retinoic Acid-Induced Gene g Expression Independently of Stat1. Cancer Res. 2009, 69, 3673–3680. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Sater, A.A.; Majoros, A.; Plumlee, C.R.; Perry, S.; Gu, A.-D.; Lee, C.; Shresta, S.; Decker, T.; Schindler, C. Different STAT Transcription Complexes Drive Early and Delayed Responses to Type I IFNs. J. Immunol. 2015, 195, 210–216. [Google Scholar] [CrossRef]
- Perry, S.T.; Buck, M.D.; Lada, S.M.; Schindler, C.; Shresta, S. STAT2 Mediates Innate Immunity to Dengue Virus in the Absence of STAT1 via the Type I Interferon Receptor. PLoS Pathog. 2011, 7, e1001297. [Google Scholar] [CrossRef]
- Yamauchi, S.; Takeuchi, K.; Chihara, K.; Honjoh, C.; Kato, Y.; Yoshiki, H.; Hotta, H.; Sada, K. STAT1 Is Essential for the Inhibition of Hepatitis C Virus Replication by Interferon-λ but Not by Interferon-α. Sci. Rep. 2016, 6, 38336. [Google Scholar] [CrossRef]
- Kraus, T.A.; Lau, J.F.; Parisien, J.P.; Horvath, C.M. A Hybrid IRF9-STAT2 Protein Recapitulates Interferon-Stimulated Gene Expression and Antiviral Response. J. Biol. Chem. 2003, 278, 13033–13038. [Google Scholar] [CrossRef]
- Poat, B.; Hazari, S.; Chandra, P.K.; Gunduz, F.; Alvarez, X.; Balart, L.A.; Garry, R.F.; Dash, S. Intracellular Expression of IRF9 Stat Fusion Protein Overcomes the Defective Jak-Stat Signaling and Inhibits HCV RNA Replication. Virol. J. 2010, 7, 265. [Google Scholar] [CrossRef] [PubMed]
- Cheon, H.; Holvey-Bates, E.G.; Schoggins, J.W.; Forster, S.; Hertzog, P.; Imanaka, N.; Rice, C.M.; Jackson, M.W.; Junk, D.J.; Stark, G.R. IFNβ-Dependent Increases in STAT1, STAT2, and IRF9 Mediate Resistance to Viruses and DNA Damage. EMBO J. 2013, 32, 2751–2763. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.S.; Cheon, H.J.; Cho, C.H.; Hong, S.H.; Park, D.Y.; Seo, H.I.; Park, S.H.; Yoon, S.K.; Stark, G.R.; Shin, E.C. Roles of Unphosphorylated ISGF3 in HCV Infection and Interferon Responsiveness. Proc. Natl. Acad. Sci. USA 2015, 112, 10443–10448. [Google Scholar] [CrossRef] [PubMed]
- Sekrecka, A.; Kluzek, K.; Sekrecki, M.; Boroujeni, M.E.; Hassani, S.; Yamauchi, S.; Sada, K.; Wesoly, J.; Bluyssen, H.A.R. Time-Dependent Recruitment of GAF, ISGF3 and IRF1 Complexes Shapes IFNα and IFNγ-Activated Transcriptional Responses and Explains Mechanistic and Functional Overlap. Cell. Mol. Life Sci. 2023, 80, 187. [Google Scholar] [CrossRef]
- Wang, W.; Yin, Y.; Xu, L.; Su, J.; Huang, F.; Wang, Y.; Boor, P.P.C.; Chen, K.; Wang, W.; Cao, W.; et al. Unphosphorylated ISGF3 Drives Constitutive Expression of Interferon-Stimulated Genes to Protect against Viral Infections. Sci. Signal. 2017, 10, eaah4248. [Google Scholar] [CrossRef]
- Platanitis, E.; Demiroz, D.; Schneller, A.; Fischer, K.; Capelle, C.; Hartl, M.; Gossenreiter, T.; Müller, M.; Novatchkova, M.; Decker, T. A Molecular Switch from STAT2-IRF9 to ISGF3 Underlies Interferon-Induced Gene Transcription. Nat. Commun. 2019, 10, 2921. [Google Scholar] [CrossRef]
- Cheon, H.J.; Stark, G.R. Unphosphorylated STAT1 Prolongs the Expression of Interferon-Induced Immune Regulatory Genes. Proc. Natl. Acad. Sci. USA 2009, 106, 9373–9378. [Google Scholar] [CrossRef]
- Majoros, A.; Platanitis, E.; Szappanos, D.; Cheon, H.; Vogl, C.; Shukla, P.; Stark, G.R.; Sexl, V.; Schreiber, R.; Schindler, C.; et al. Response to Interferons and Antibacterial Innate Immunity in the Absence of Tyrosine-Phosphorylated STAT1. EMBO Rep. 2016, 17, 367–382. [Google Scholar] [CrossRef]
- McKendry, R.; John, J.; Flavell, D.; Muller, M.; Kerr, I.M.; Stark, G.R. High-Frequency Mutagenesis of Human Cells and Characterization of a Mutant Unresponsive to Both α and γ Interferons. Proc. Natl. Acad. Sci. USA 1991, 88, 11455–11459. [Google Scholar] [CrossRef]
- Piaszyk-Borychowska, A.; Széles, L.; Csermely, A.; Chiang, H.C.; Wesoły, J.; Lee, C.K.; Nagy, L.; Bluyssen, H.A.R. Signal Integration of IFN-I and IFN-II with TLR4 Involves Sequential Recruitment of STAT1-Complexes and NFκB to Enhance pro-Inflammatory Transcription. Front. Immunol. 2019, 10, 1253. [Google Scholar] [CrossRef]
- Willems, E.; Leyns, L.; Vandesompele, J. Standardization of Real-Time PCR Gene Expression Data from Independent Biological Replicates. Anal. Biochem. 2008, 379, 127–129. [Google Scholar] [CrossRef]
- GraphPad Software, Inc. GraphPad Prism, Version 7.01; GraphPad Software, Inc.: La Jolla, CA, USA, 2016.
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; FastQC: San Diego, CA, USA, 2010. [Google Scholar]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Kolde, R. Pheatmap: Pretty Heatmaps, Version 1.0.12; R Package; CRAN: Vienna, Austria, 2019.
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. Jvenn: An Interactive Venn Diagram Viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [PubMed]
- Inkscape Project. Available online: https://inkscape.org/09.2020 (accessed on 13 October 2023).
- ENCODE Project Consortium Transcription Factor ChIP-Seq Data Standards and Processing Pipeline—ENCODE. Available online: https://www.encodeproject.org/chip-seq/transcription_factor/ (accessed on 13 October 2023).
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Jun, G.; Wing, M.K.; Abecasis, G.R.; Kang, H.M. An Efficient and Scalable Analysis Framework for Variant Extraction and Refinement from Population-Scale DNA Sequence Data. Genome Res. 2015, 25, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-Based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime Cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next Generation Web Server for Deep-Sequencing Data Analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genome Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Carroll, T.; Barrows, D. Profileplyr: Visualization and Annotation of Read Signal over Genomic Ranges with Profileplyr, Version 1.19.0; Bioconductor: Seattle, WA, USA, 2021.
- Tremblay, B.J.-M. Universalmotif: Import, Modify, and Export Motifs with R, Version 1.20.0; Bioconductor: Seattle, WA, USA, 2021.
- Wang, W.-B.; Levy, D.E.; Lee, C.-K. STAT3 Negatively Regulates Type I IFN-Mediated Antiviral Response. J. Immunol. 2011, 187, 2578–2585. [Google Scholar] [CrossRef]
- Costa-Pereira, A.P.; Williams, T.M.; Strobl, B.; Watling, D.; Briscoe, J.; Kerr, I.M. The Antiviral Response to Gamma Interferon. J. Virol. 2002, 76, 9060–9068. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nowicka, H.; Sekrecka, A.; Blaszczyk, K.; Kluzek, K.; Chang, C.-Y.; Wesoly, J.; Lee, C.-K.; Bluyssen, H.A.R. ISGF3 and STAT2/IRF9 Control Basal and IFN-Induced Transcription through Genome-Wide Binding of Phosphorylated and Unphosphorylated Complexes to Common ISRE-Containing ISGs. Int. J. Mol. Sci. 2023, 24, 17635. https://doi.org/10.3390/ijms242417635
Nowicka H, Sekrecka A, Blaszczyk K, Kluzek K, Chang C-Y, Wesoly J, Lee C-K, Bluyssen HAR. ISGF3 and STAT2/IRF9 Control Basal and IFN-Induced Transcription through Genome-Wide Binding of Phosphorylated and Unphosphorylated Complexes to Common ISRE-Containing ISGs. International Journal of Molecular Sciences. 2023; 24(24):17635. https://doi.org/10.3390/ijms242417635
Chicago/Turabian StyleNowicka, Hanna, Agata Sekrecka, Katarzyna Blaszczyk, Katarzyna Kluzek, Chan-Yu Chang, Joanna Wesoly, Chien-Kuo Lee, and Hans A. R. Bluyssen. 2023. "ISGF3 and STAT2/IRF9 Control Basal and IFN-Induced Transcription through Genome-Wide Binding of Phosphorylated and Unphosphorylated Complexes to Common ISRE-Containing ISGs" International Journal of Molecular Sciences 24, no. 24: 17635. https://doi.org/10.3390/ijms242417635
APA StyleNowicka, H., Sekrecka, A., Blaszczyk, K., Kluzek, K., Chang, C.-Y., Wesoly, J., Lee, C.-K., & Bluyssen, H. A. R. (2023). ISGF3 and STAT2/IRF9 Control Basal and IFN-Induced Transcription through Genome-Wide Binding of Phosphorylated and Unphosphorylated Complexes to Common ISRE-Containing ISGs. International Journal of Molecular Sciences, 24(24), 17635. https://doi.org/10.3390/ijms242417635