
Radiotracers for Imaging of Inflammatory Biomarkers TSPO and COX-2 in the Brain and in the Periphery

 ,
,  and
and 
Abstract
1. General Introduction
2. Translocator Protein (TSPO)
2.1. Introduction
2.2. First Generation of TSPO Radiotracer
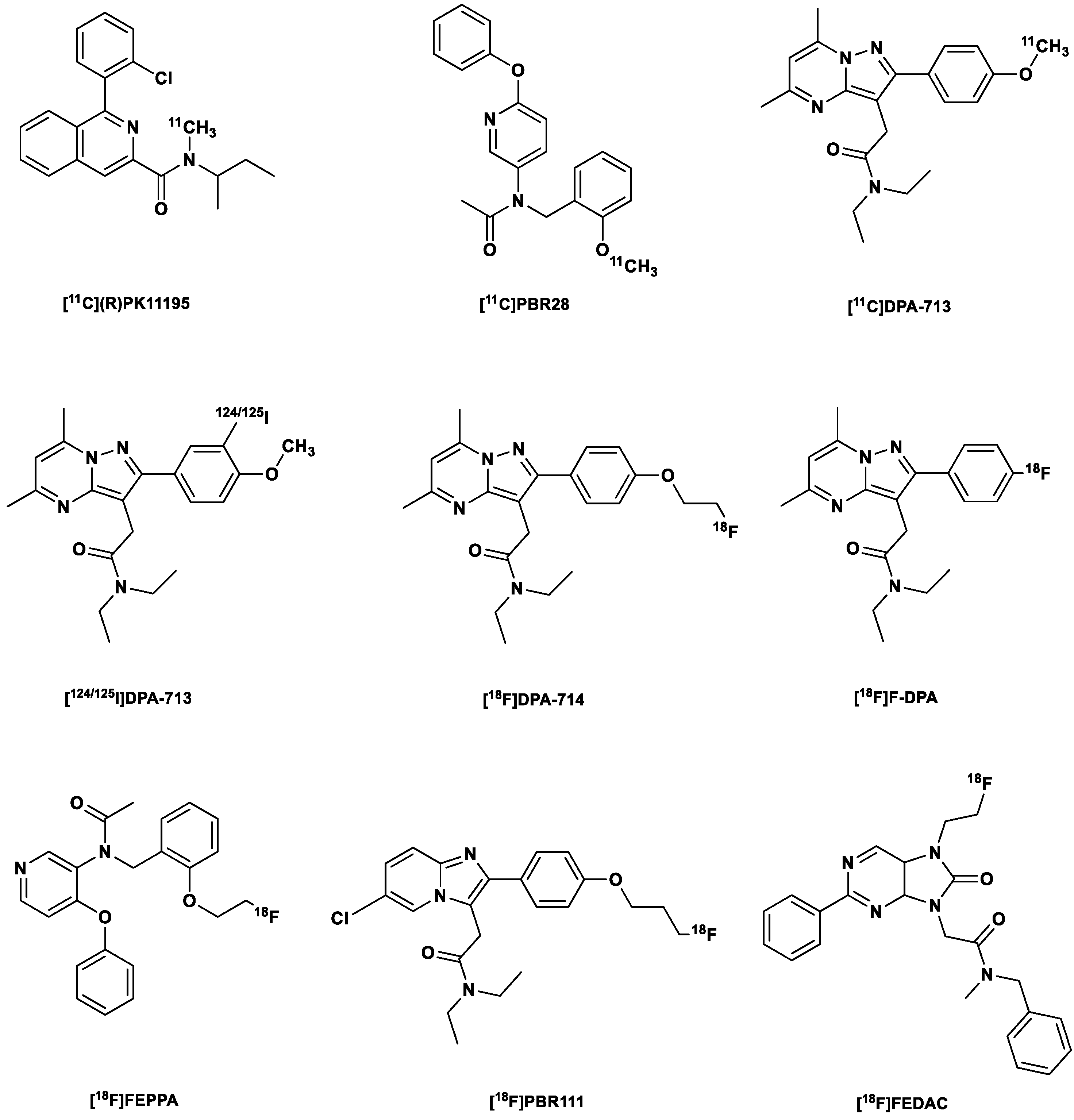
[11C](R)PK11195
2.3. Second-Generation TSPO Tracers
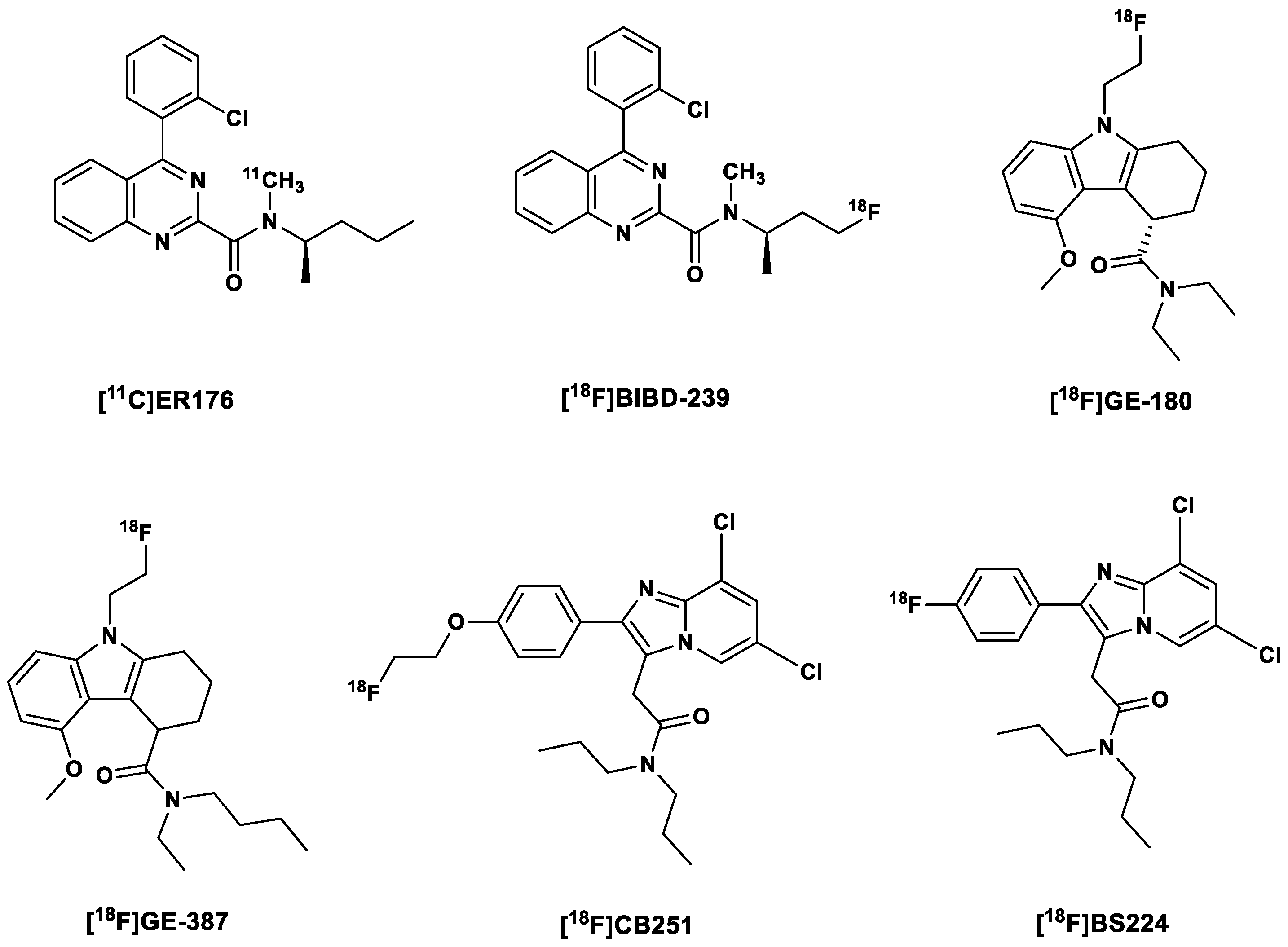
2.4. Third-Generation TSPO Tracers
3. The Cyclooxygenase-2 (COX-2) Enzyme
3.1. Introduction
3.2. PET COX-2 Radiotracers
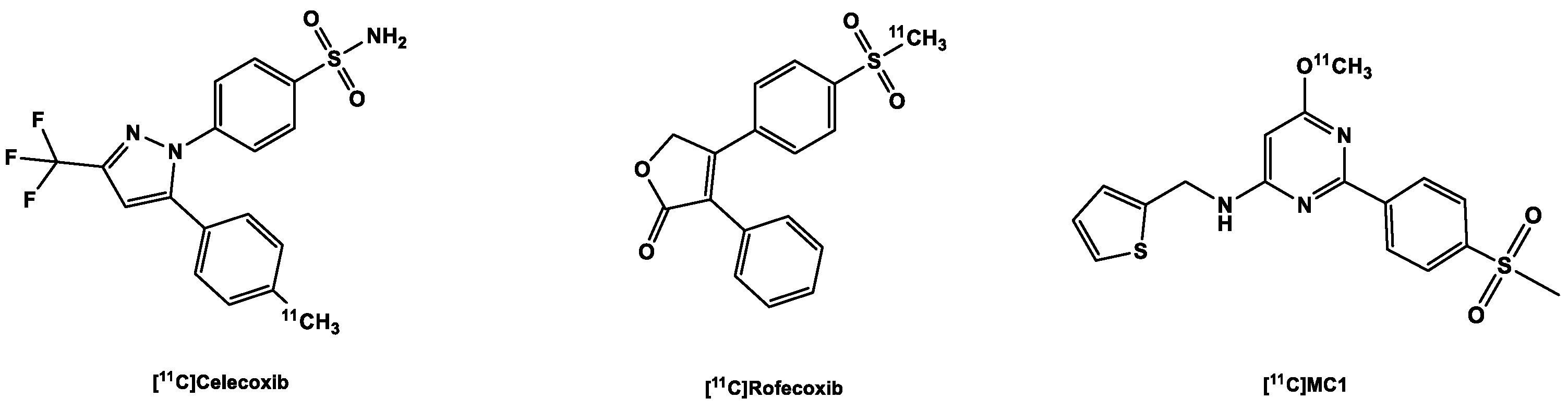
3.2.1. [11C]Carbon Labeled COX-2 Tracers

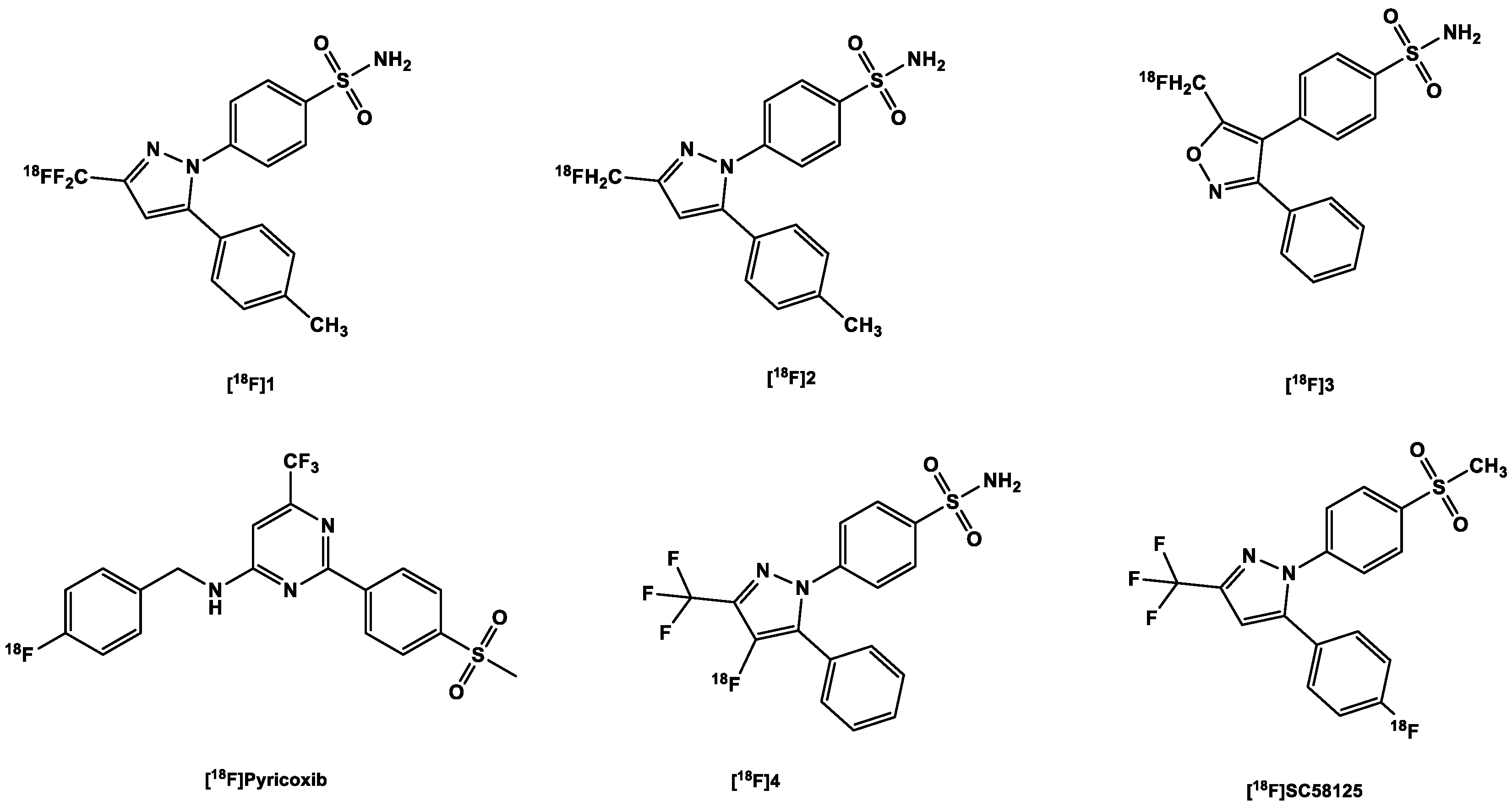
3.2.2. [18F]Fluorine-Labeled COX-2 Tracers
- A PET study comparing the uptake of the tracer in both inflamed (carrageenan-treated rat paws) and non-inflamed tissues (non-treated). It showed a 1.53-fold increase in the former over the non-treated paws [101];
- Pre-dosing with celecoxib (10 mg/kg), which significantly decreased tracer uptake in inflamed rat paws (there was only a 1.7-fold decrease in uptake [101];
- Experiments in COX-2 null mice further confirmed the specificity of the tracer; there was no increased tracer uptake in the inflamed carrageenan-treated paws of these mice compared to controls, i.e., non-inflamed carrageenan-treated paws of the same COX-2 knock-out mice with a ratio of 1.08. This contrasts significantly with the uptake in the inflamed paws of wild-type mice versus the control paws (1.48) [101];
- Results obtained using nude mice with both COX-2-positive 1483 HNSCC tumors and COX-2-negative HCT116 tumors suggest that the difference in the uptake in both tumor types correlates with the difference in their expression of COX-2 (3 times higher in the COX-2-positive tumor). The blocking of the COX-2 active site in the former prevented the binding of the tracer; compared to the control, tumor-to-muscle ratio was nearly three times lower [101].

3.3. SPECT COX-2 Radiotracers
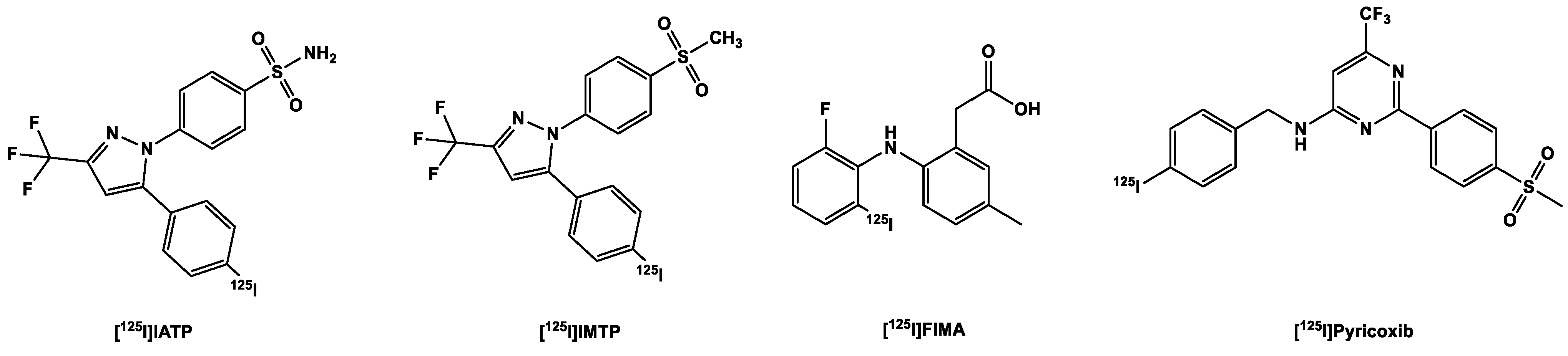
3.3.1. [123,125I]Iodine-Labeled COX-2 Tracers

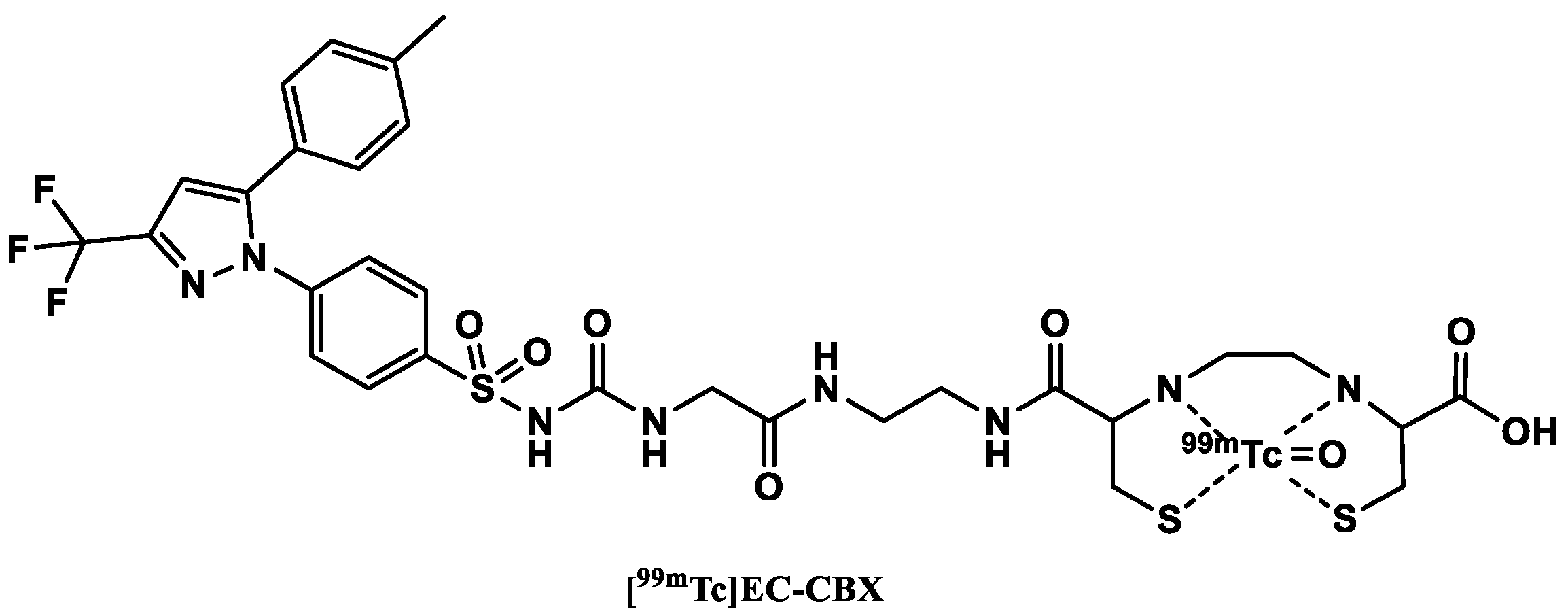
3.3.2. [99mTc]Technetium-Labeled COX-2 Tracers

4. PET and SPECT Imaging of TSPO and COX-2 in Non-Neuroinflammatory Diseases
4.1. Introduction
4.2. Pulmonary Inflammation
4.3. Autoimmune Diseases
4.4. Cardiovascular Pathology
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| %ID/g | % Percentage injected dose per gram |
| [18F]FDG | Fluorodeoxyglucose (18F) |
| A147T | Alanine is exchanged for threonine |
| ACPAs | Anti-citrullinated protein antibodies |
| AD | Alzheimer’s disease |
| Ala147 | Alanine-147 |
| ALS | Amyotrophic lateral sclerosis |
| Am | Molar activity |
| ANKA | ANKA strain of Plasmodium berghei |
| ApoE | Apolipoprotein E |
| ApoE−/− mice | Atherosclerosis-prone apolipoprotein E-deficient (ApoE−/−) mice |
| Aβ | Beta-amyloid plaque |
| BBB | Blood brain barrier |
| BPND | Binding potential (non-displaceable) |
| C3HeB/FeJ mice | C3HeB/FeJ mice: mouse strains hyper-susceptible to M. tuberculosis infection compared to other conventional mouse strains |
| CBR | Central benzodiazepine receptor |
| CBX | Ethyl 2-isocyanatoacetate of celecoxib |
| CD4+ | Cluster of differentiation 4 positive |
| CD68+ | Cluster of differentiation 68 positive |
| ClogP | Calculated log P |
| CNS | Central nervous system |
| COPD | Chronic obstructive pulmonary disease |
| COX | Cyclooxygenase |
| CSF1R | Colony-stimulating factor 1 receptor |
| Csp2 | Sp2 hybridized carbon |
| Csp3 | Sp3 hybridized carbon |
| CT | Computed tomography |
| CTA | Computed tomography angiography |
| DLB | Dementia with Lewy bodies |
| DMH | 1,2-dimethylhydrazine |
| EA-CBX | Product of Ethylenediamine and CBX reaction |
| EC–CBX | Product of L,L-ethylenedicysteine and EA-CBX reaction |
| FC | Frontal cortex |
| FLS | Fibroblast-like synoviocytes |
| FTD | Frontotemporal dementia |
| GABA | Gamma-aminobutyric acid (γ-Aminobutyric acid) |
| GBM | Glioblastoma |
| GL261 | Murine glioma cells are widely used as a syngeneic animal model of glioma |
| HandE | Hematoxylin and eosin stain |
| HABs | High-affinity binders |
| HCA-7 | Human colon adenocarcinoma |
| HCT-116 | Human colorectal carcinoma |
| HD | Huntington’s disease |
| HSV-1 | Herpes simplex encephalitis virus-1 |
| ICR mouse model | Institute of Cancer Research (USA) (strain of albino mice) |
| IFN-ɣ | Interferon-ɣ |
| IHC | Immunohistochemistry |
| kDa | KiloDalton |
| LABs | Low-affinity binders |
| LBD | Lewy body dementia |
| log D | Logarithm of the distribution coefficient |
| log P | Logarithm of the partition coefficient |
| LPS | Lipopolysaccharide |
| LVV | Large vessel vasculitis |
| M0 | M0 Non-activated macrophages |
| M1 macrophage | Pro-inflammatory M1 phenotype |
| M2 macrophage | Anti-inflammatory M2 phenotype |
| MA-ARDS | Malaria-associated acute respiratory distress syndrome |
| MABs | Medium-affinity binders |
| MAO-B | Monoamine oxidase-B |
| MHC-II | Major histocompatibility complex class II |
| MI | Myocardial infarction |
| mRNA | Messenger ribonucleic acid |
| MW | Molar weight |
| MS | Multiple sclerosis |
| NDD | Neurodegenerative diseases |
| NIH-III nude mice | Nude mice developed at the National Institutes of Health (USA). It has in addition to the nude gene (no thymus and T cell function) two other mutations which affects the regulation of the immune system |
| NNK | Nicotine-derived nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone |
| NSR | Normalized standard uptake value ratio |
| p.i. | Post injection |
| P2X | Purinoceptor |
| PBR | Peripheral benzodiazepine receptor |
| PD | Parkinson’s disease |
| PET | Positron emission tomography |
| PTC | Parietotemporal cortex |
| PTGS | Prostaglandin-endoperoxide synthase |
| RA | Rheumatic arthritis |
| RCY | Radiochemical yield |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SCIDY | Spirocyclic iodonium ylide |
| SD rat | Sprague Dawley rat |
| SeV | Sendai virus |
| SNAr | Nucleophilic substitution reaction |
| SNP | Single nucleotide polymorphism |
| SPECT | Single-emission computed tomography |
| SSTR2 | Somatostatin type 2 receptors |
| SUV | Standard uptake value |
| T/B | Target-to-background |
| TB | Tuberculosis |
| Thr147 | Threonine-147 |
| TNF-α | Tumor necrosis factor-α |
| TSPO | Translocator protein |
| VT | Volume of distribution |
| WHO | World Health Organization |
References
- Owen, D.R.; Yeo, A.J.; Gunn, R.N.; Song, K.; Wadsworth, G.; Lewis, A.; Rhodes, C.; Pulford, D.J.; Bennacef, I.; Parker, C.A.; et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2012, 32, 1–5. [Google Scholar] [CrossRef]
- Masdeu, J.C.; Pascual, B.; Fujita, M. Imaging Neuroinflammation in Neurodegenerative Disorders. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2022, 63, 45S–52S. [Google Scholar] [CrossRef]
- Horti, A.G.; Naik, R.; Foss, C.A.; Minn, I.; Misheneva, V.; Du, Y.; Wang, Y.; Mathews, W.B.; Wu, Y.; Hall, A.; et al. PET imaging of microglia by targeting macrophage colony-stimulating factor 1 receptor (CSF1R). Proc. Natl. Acad. Sci. USA 2019, 116, 1686–1691. [Google Scholar] [CrossRef]
- Ekblom, J.; Jossan, S.S.; Bergström, M.; Oreland, L.; Walum, E.; Aquilonius, S.M. Monoamine oxidase-B in astrocytes. Glia 1993, 8, 122–132. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Biber, K. The microglial ATP-gated ion channel P2X7 as a CNS drug target. Glia 2016, 64, 1772–1787. [Google Scholar] [CrossRef]
- Cserép, C.; Pósfai, B.; Lénárt, N.; Fekete, R.; László, Z.I.; Lele, Z.; Orsolits, B.; Molnár, G.; Heindl, S.; Schwarcz, A.D.; et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 2020, 367, 528–537. [Google Scholar] [CrossRef]
- Tronel, C.; Largeau, B.; Santiago Ribeiro, M.J.; Guilloteau, D.; Dupont, A.-C.; Arlicot, N. Molecular Targets for PET Imaging of Activated Microglia: The Current Situation and Future Expectations. Int. J. Mol. Sci. 2017, 18, 802. [Google Scholar] [CrossRef]
- Janssen, B.; Vugts, D.J.; Windhorst, A.D.; Mach, R.H. PET Imaging of Microglial Activation-Beyond Targeting TSPO. Molecules 2018, 23, 607. [Google Scholar] [CrossRef]
- Qiao, L.; Fisher, E.; McMurray, L.; Milicevic Sephton, S.; Hird, M.; Kuzhuppilly-Ramakrishnan, N.; Williamson, D.J.; Zhou, X.; Werry, E.; Kassiou, M.; et al. Radiosynthesis of (R,S)-18 FGE387: A Potential PET Radiotracer for Imaging Translocator Protein 18 kDa (TSPO) with Low Binding Sensitivity to the Human Gene Polymorphism rs6971. ChemMedChem 2019, 14, 982–993. [Google Scholar] [CrossRef]
- Damont, A.; Hinnen, F.; Kuhnast, B.; Schöllhorn-Peyronneau, M.-A.; James, M.; Luus, C.; Tavitian, B.; Kassiou, M.; Dollé, F. Radiosynthesis of [18F]DPA-714, a selective radioligand for imaging the translocator protein (18 kDa) with PET. J. Label. Compd. Radiopharm. 2008, 51, 286–292. [Google Scholar] [CrossRef]
- Largeau, B.; Dupont, A.-C.; Guilloteau, D.; Santiago-Ribeiro, M.-J.; Arlicot, N. TSPO PET Imaging: From Microglial Activation to Peripheral Sterile Inflammatory Diseases? Contrast Media Mol. Imaging 2017, 2017, 6592139. [Google Scholar] [CrossRef]
- Zanotti-Fregonara, P.; Zhang, Y.; Jenko, K.J.; Gladding, R.L.; Zoghbi, S.S.; Fujita, M.; Sbardella, G.; Castellano, S.; Taliani, S.; Martini, C.; et al. Synthesis and evaluation of translocator 18 kDa protein (TSPO) positron emission tomography (PET) radioligands with low binding sensitivity to human single nucleotide polymorphism rs6971. ACS Chem. Neurosci. 2014, 5, 963–971. [Google Scholar] [CrossRef]
- Chandra, A.; Valkimadi, P.-E.; Pagano, G.; Cousins, O.; Dervenoulas, G.; Politis, M. Applications of amyloid, tau, and neuroinflammation PET imaging to Alzheimer’s disease and mild cognitive impairment. Hum. Brain Mapp. 2019, 40, 5424–5442. [Google Scholar] [CrossRef]
- Werry, E.L.; Bright, F.M.; Piguet, O.; Ittner, L.M.; Halliday, G.M.; Hodges, J.R.; Kiernan, M.C.; Loy, C.T.; Kril, J.J.; Kassiou, M. Recent Developments in TSPO PET Imaging as A Biomarker of Neuroinflammation in Neurodegenerative Disorders. Int. J. Mol. Sci. 2019, 20, 3161. [Google Scholar] [CrossRef]
- Kreisl, W.C.; Henter, I.D.; Innis, R.B. Imaging Translocator Protein as a Biomarker of Neuroinflammation in Dementia. Adv. Pharmacol. 2018, 82, 163–185. [Google Scholar] [CrossRef]
- Guilarte, T.R.; Rodichkin, A.N.; McGlothan, J.L.; La Acanda De Rocha, A.M.; Azzam, D.J. Imaging neuroinflammation with TSPO: A new perspective on the cellular sources and subcellular localization. Pharmacol. Ther. 2022, 234, 108048. [Google Scholar] [CrossRef]
- Notter, T.; Schalbetter, S.M.; Clifton, N.E.; Mattei, D.; Richetto, J.; Thomas, K.; Meyer, U.; Hall, J. Neuronal activity increases translocator protein (TSPO) levels. Mol. Psychiatry 2021, 26, 2025–2037. [Google Scholar] [CrossRef]
- Camsonne, R.; Crouzel, C.; Comar, D.; Mazière, M.; Prenant, C.; Sastre, J.; Moulin, M.; Syrota, A. Synthesis of N-(11C) methyl, N-(methyl-1 propyl), (chloro-2 phenyl)-1 isoquinoleine carboxamide-3 (PK 11195): A new ligand for peripheral benzodiazepine receptors. J. Label. Compd. Radiopharm. 1984, 21, 985–991. [Google Scholar] [CrossRef]
- Yuan, J.; Yao, J.-Q.; Fang, X.-X.; Dai, W.; Wang, Y.-H.; Zhang, L.-M.; Li, Y.-F. Involvement of regulation of the excitation:inhibition functional balance in the mPFC in the antidepressant-anxiolytic effect of YL-IPA08, a novel TSPO ligand. Metab. Brain Dis. 2022, 37, 2305–2314. [Google Scholar] [CrossRef]
- Jučaite, A.; Cselényi, Z.; Arvidsson, A.; Ahlberg, G.; Julin, P.; Varnäs, K.; Stenkrona, P.; Andersson, J.; Halldin, C.; Farde, L. Kinetic analysis and test-retest variability of the radioligand 11C(R)-PK11195 binding to TSPO in the human brain—A PET study in control subjects. EJNMMI Res. 2012, 2, 15. [Google Scholar] [CrossRef]
- James, M.L.; Fulton, R.R.; Henderson, D.J.; Eberl, S.; Meikle, S.R.; Thomson, S.; Allan, R.D.; Dolle, F.; Fulham, M.J.; Kassiou, M. Synthesis and in vivo evaluation of a novel peripheral benzodiazepine receptor PET radioligand. Bioorganic Med. Chem. 2005, 13, 6188–6194. [Google Scholar] [CrossRef]
- Kreisl, W.C.; Fujita, M.; Fujimura, Y.; Kimura, N.; Jenko, K.J.; Kannan, P.; Hong, J.; Morse, C.L.; Zoghbi, S.S.; Gladding, R.L.; et al. Comparison of (11)C-(R)-PK 11195 and (11)CPBR28, two radioligands for translocator protein (18 kDa) in human and monkey: Implications for positron emission tomographic imaging of this inflammation biomarker. NeuroImage 2010, 49, 2924–2932. [Google Scholar] [CrossRef]
- Kobayashi, M.; Jiang, T.; Telu, S.; Zoghbi, S.S.; Gunn, R.N.; Rabiner, E.A.; Owen, D.R.; Guo, Q.; Pike, V.W.; Innis, R.B.; et al. 11C-DPA-713 has much greater specific binding to translocator protein 18 kDa (TSPO) in human brain than 11C-(R)-PK11195. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2018, 38, 393–403. [Google Scholar] [CrossRef]
- Ching, A.S.C.; Kuhnast, B.; Damont, A.; Roeda, D.; Tavitian, B.; Dollé, F. Current paradigm of the 18-kDa translocator protein (TSPO) as a molecular target for PET imaging in neuroinflammation and neurodegenerative diseases. Insights Imaging 2012, 3, 111–119. [Google Scholar] [CrossRef]
- Cagnin, A.; Brooks, D.J.; Kennedy, A.M.; Gunn, R.N.; Myers, R.; Turkheimer, F.E.; Jones, T.; Banati, R.B. In-vivo measurement of activated microglia in dementia. Lancet 2001, 358, 461–467. [Google Scholar] [CrossRef]
- Corcia, P.; Tauber, C.; Vercoullie, J.; Arlicot, N.; Prunier, C.; Praline, J.; Nicolas, G.; Venel, Y.; Hommet, C.; Baulieu, J.-L.; et al. Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e52941. [Google Scholar] [CrossRef]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with 11C(R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Gulyás, B.; Tóth, M.; Schain, M.; Airaksinen, A.; Vas, A.; Kostulas, K.; Lindström, P.; Hillert, J.; Halldin, C. Evolution of microglial activation in ischaemic core and peri-infarct regions after stroke: A PET study with the TSPO molecular imaging biomarker ((11))Cvinpocetine. J. Neurol. Sci. 2012, 320, 110–117. [Google Scholar] [CrossRef]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef]
- Pavese, N.; Gerhard, A.; Tai, Y.F.; Ho, A.K.; Turkheimer, F.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation correlates with severity in Huntington disease: A clinical and PET study. Neurology 2006, 66, 1638–1643. [Google Scholar] [CrossRef]
- Politis, M.; Lahiri, N.; Niccolini, F.; Su, P.; Wu, K.; Giannetti, P.; Scahill, R.I.; Turkheimer, F.E.; Tabrizi, S.J.; Piccini, P. Increased central microglial activation associated with peripheral cytokine levels in premanifest Huntington’s disease gene carriers. Neurobiol. Dis. 2015, 83, 115–121. [Google Scholar] [CrossRef]
- Yasuno, F.; Kosaka, J.; Ota, M.; Higuchi, M.; Ito, H.; Fujimura, Y.; Nozaki, S.; Takahashi, S.; Mizukami, K.; Asada, T.; et al. Increased binding of peripheral benzodiazepine receptor in mild cognitive impairment-dementia converters measured by positron emission tomography with 11CDAA1106. Psychiatry Res. 2012, 203, 67–74. [Google Scholar] [CrossRef]
- Gerhard, A. TSPO imaging in parkinsonian disorders. Clin. Transl. Imaging 2016, 4, 183–190. [Google Scholar] [CrossRef]
- Belloli, S.; Morari, M.; Murtaj, V.; Valtorta, S.; Moresco, R.M.; Gilardi, M.C. Translation Imaging in Parkinson’s Disease: Focus on Neuroinflammation. Front. Aging Neurosci. 2020, 12, 152. [Google Scholar] [CrossRef]
- Iannaccone, S.; Cerami, C.; Alessio, M.; Garibotto, V.; Panzacchi, A.; Olivieri, S.; Gelsomino, G.; Moresco, R.M.; Perani, D. In vivo microglia activation in very early dementia with Lewy bodies, comparison with Parkinson’s disease. Park. Relat. Disord. 2013, 19, 47–52. [Google Scholar] [CrossRef]
- Dollé, F.; Luus, C.; Reynolds, A.; Kassiou, M. Radiolabelled molecules for imaging the translocator protein (18 kDa) using positron emission tomography. Curr. Med. Chem. 2009, 16, 2899–2923. [Google Scholar] [CrossRef]
- Owen, D.R.J.; Gunn, R.N.; Rabiner, E.A.; Bennacef, I.; Fujita, M.; Kreisl, W.C.; Innis, R.B.; Pike, V.W.; Reynolds, R.; Matthews, P.M.; et al. Mixed-Affinity Binding in Humans with 18-kDa Translocator Protein Ligands. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2011, 52, 24–32. [Google Scholar] [CrossRef]
- Owen, D.R.; Howell, O.W.; Tang, S.-P.; Wells, L.A.; Bennacef, I.; Bergstrom, M.; Gunn, R.N.; Rabiner, E.A.; Wilkins, M.R.; Reynolds, R.; et al. Two Binding Sites for [3H]PBR28 in Human Brain: Implications for TSPO PET Imaging of Neuroinflammation. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2010, 30, 1608–1618. [Google Scholar] [CrossRef]
- Chauveau, F.; van Camp, N.; Dollé, F.; Kuhnast, B.; Hinnen, F.; Damont, A.; Boutin, H.; James, M.; Kassiou, M.; Tavitian, B. Comparative evaluation of the translocator protein radioligands 11C-DPA-713, 18F-DPA-714, and 11C-PK11195 in a rat model of acute neuroinflammation. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2009, 50, 468–476. [Google Scholar] [CrossRef]
- Fujita, M.; Kobayashi, M.; Ikawa, M.; Gunn, R.N.; Rabiner, E.A.; Owen, D.R.; Zoghbi, S.S.; Haskali, M.B.; Telu, S.; Pike, V.W.; et al. Comparison of four 11C-labeled PET ligands to quantify translocator protein 18 kDa (TSPO) in human brain: (R)-PK11195, PBR28, DPA-713, and ER176-based on recent publications that measured specific-to-non-displaceable ratios. EJNMMI Res. 2017, 7, 84. [Google Scholar] [CrossRef]
- Terada, T.; Yokokura, M.; Yoshikawa, E.; Futatsubashi, M.; Kono, S.; Konishi, T.; Miyajima, H.; Hashizume, T.; Ouchi, Y. Extrastriatal spreading of microglial activation in Parkinson’s disease: A positron emission tomography study. Ann. Nucl. Med. 2016, 30, 579–587. [Google Scholar] [CrossRef]
- Doorduin, J.; Klein, H.C.; Dierckx, R.A.; James, M.; Kassiou, M.; de Vries, E.F.J. 11C-DPA-713 and 18F-DPA-714 as new PET tracers for TSPO: A comparison with 11C-(R)-PK11195 in a rat model of herpes encephalitis. Mol. Imaging Biol. 2009, 11, 386–398. [Google Scholar] [CrossRef]
- Selleri, S.; Bruni, F.; Costagli, C.; Costanzo, A.; Guerrini, G.; Ciciani, G.; Costa, B.; Martini, C. 2-Arylpyrazolo1,5-apyrimidin-3-yl acetamides. New potent and selective peripheral benzodiazepine receptor ligands. Bioorganic Med. Chem. 2001, 9, 2661–2671. [Google Scholar] [CrossRef]
- Keller, T.; Krzyczmonik, A.; Forsback, S.; Picón, F.R.L.; Kirjavainen, A.K.; Takkinen, J.; Rajander, J.; Cacheux, F.; Damont, A.; Dollé, F.; et al. Radiosynthesis and Preclinical Evaluation of 18FF-DPA, A Novel Pyrazolo1,5apyrimidine Acetamide TSPO Radioligand, in Healthy Sprague Dawley Rats. Mol. Imaging Biol. 2017, 19, 736–745. [Google Scholar] [CrossRef]
- Wang, L.; Cheng, R.; Fujinaga, M.; Yang, J.; Zhang, Y.; Hatori, A.; Kumata, K.; Yang, J.; Vasdev, N.; Du, Y.; et al. A Facile Radiolabeling of 18FFDPA via Spirocyclic Iodonium Ylides: Preliminary PET Imaging Studies in Preclinical Models of Neuroinflammation. J. Med. Chem. 2017, 60, 5222–5227. [Google Scholar] [CrossRef]
- Wang, L.; Yao, S.; Tang, R.; Zhu, H.; Zhang, L.; Gong, J.; Chen, Q.; Collier, T.L.; Xu, H.; Liang, S.H. A concisely automated synthesis of TSPO radiotracer 18 FFDPA based on spirocyclic iodonium ylide method and validation for human use. J. Label. Comp. Radiopharm. 2020, 63, 119–128. [Google Scholar] [CrossRef]
- Keller, T.; López-Picón, F.R.; Krzyczmonik, A.; Forsback, S.; Takkinen, J.S.; Rajander, J.; Teperi, S.; Dollé, F.; Rinne, J.O.; Haaparanta-Solin, M.; et al. Comparison of high and low molar activity TSPO tracer 18FF-DPA in a mouse model of Alzheimer’s disease. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2020, 40, 1012–1020. [Google Scholar] [CrossRef]
- López-Picón, F.R.; Keller, T.; Bocancea, D.; Helin, J.S.; Krzyczmonik, A.; Helin, S.; Damont, A.; Dollé, F.; Rinne, J.O.; Haaparanta-Solin, M.; et al. Direct Comparison of 18FF-DPA with 18FDPA-714 and 11CPBR28 for Neuroinflammation Imaging in the same Alzheimer’s Disease Model Mice and Healthy Controls. Mol. Imaging Biol. 2022, 24, 157–166. [Google Scholar] [CrossRef]
- Ikawa, M.; Lohith, T.G.; Shrestha, S.; Telu, S.; Zoghbi, S.S.; Castellano, S.; Taliani, S.; Da Settimo, F.; Fujita, M.; Pike, V.W.; et al. 11C-ER176, a Radioligand for 18-kDa Translocator Protein, Has Adequate Sensitivity to Robustly Image All Three Affinity Genotypes in Human Brain. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2017, 58, 320–325. [Google Scholar] [CrossRef]
- Columbia University. Imaging Inflammation in Alzheimer’s Disease with 11C-ER176: NCT03744312, AAAR6570. Available online: https://clinicaltrials.gov/ct2/show/NCT03744312 (accessed on 3 November 2022).
- Chen, H.; Jiang, Z.; Cheng, X.; Zheng, W.; Sun, Y.; Yu, Z.; Yang, T.; Zhang, L.; Yan, J.; Liu, Y.; et al. 18FBIBD-239: 18F-Labeled ER176, a Positron Emission Tomography Tracer Specific for the Translocator Protein. Mol. Pharm. 2022, 19, 2351–2366. [Google Scholar] [CrossRef]
- Wadsworth, H.; Jones, P.A.; Chau, W.-F.; Durrant, C.; Fouladi, N.; Passmore, J.; O’Shea, D.; Wynn, D.; Morisson-Iveson, V.; Ewan, A.; et al. 18FGE-180: A novel fluorine-18 labelled PET tracer for imaging Translocator protein 18 kDa (TSPO). Bioorganic Med. Chem. Lett. 2012, 22, 1308–1313. [Google Scholar] [CrossRef]
- Reynolds, A.; Hanani, R.; Hibbs, D.; Damont, A.; Da Pozzo, E.; Selleri, S.; Dollé, F.; Martini, C.; Kassiou, M. Pyrazolo1,5-apyrimidine acetamides: 4-Phenyl alkyl ether derivatives as potent ligands for the 18 kDa translocator protein (TSPO). Bioorganic Med. Chem. Lett. 2010, 20, 5799–5802. [Google Scholar] [CrossRef]
- Chaney, A.; Cropper, H.C.; Johnson, E.M.; Lechtenberg, K.J.; Peterson, T.C.; Stevens, M.Y.; Buckwalter, M.S.; James, M.L. 11C-DPA-713 Versus 18F-GE-180: A Preclinical Comparison of Translocator Protein 18 kDa PET Tracers to Visualize Acute and Chronic Neuroinflammation in a Mouse Model of Ischemic Stroke. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2019, 60, 122–128. [Google Scholar] [CrossRef]
- Feeney, C.; Scott, G.; Raffel, J.; Roberts, S.; Coello, C.; Jolly, A.; Searle, G.; Goldstone, A.P.; Brooks, D.J.; Nicholas, R.S.; et al. Kinetic analysis of the translocator protein positron emission tomography ligand 18FGE-180 in the human brain. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 2201–2210. [Google Scholar] [CrossRef]
- Zanotti-Fregonara, P.; Pascual, B.; Rizzo, G.; Yu, M.; Pal, N.; Beers, D.; Carter, R.; Appel, S.H.; Atassi, N.; Masdeu, J.C. Head-to-Head Comparison of 11C-PBR28 and 18F-GE180 for Quantification of the Translocator Protein in the Human Brain. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2018, 59, 1260–1266. [Google Scholar] [CrossRef]
- Kreisl, W.C.; Jenko, K.J.; Hines, C.S.; Lyoo, C.H.; Corona, W.; Morse, C.L.; Zoghbi, S.S.; Hyde, T.; Kleinman, J.E.; Pike, V.W.; et al. A genetic polymorphism for translocator protein 18 kDa affects both in vitro and in vivo radioligand binding in human brain to this putative biomarker of neuroinflammation. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2013, 33, 53–58. [Google Scholar] [CrossRef]
- Zanotti-Fregonara, P.; Veronese, M.; Pascual, B.; Rostomily, R.C.; Turkheimer, F.; Masdeu, J.C. The validity of 18F-GE180 as a TSPO imaging agent. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1205–1207. [Google Scholar] [CrossRef]
- Albert, N.L.; Unterrainer, M.; Brendel, M.; Kaiser, L.; Zweckstetter, M.; Cumming, P.; Bartenstein, P. In response to: The validity of 18F-GE180 as a TSPO imaging agent. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1208–1211. [Google Scholar] [CrossRef]
- Sridharan, S.; Lepelletier, F.-X.; Trigg, W.; Banister, S.; Reekie, T.; Kassiou, M.; Gerhard, A.; Hinz, R.; Boutin, H. Comparative Evaluation of Three TSPO PET Radiotracers in a LPS-Induced Model of Mild Neuroinflammation in Rats. Mol. Imaging Biol. 2017, 19, 77–89. [Google Scholar] [CrossRef]
- López-Picón, F.R.; Snellman, A.; Eskola, O.; Helin, S.; Solin, O.; Haaparanta-Solin, M.; Rinne, J.O. Neuroinflammation Appears Early on PET Imaging and Then Plateaus in a Mouse Model of Alzheimer Disease. J. Nucl. Med. 2018, 59, 509–515. [Google Scholar] [CrossRef]
- Holzgreve, A.; Pötter, D.; Brendel, M.; Orth, M.; Weidner, L.; Gold, L.; Kirchner, M.A.; Bartos, L.M.; Unterrainer, L.M.; Unterrainer, M.; et al. Longitudinal 18FGE-180 PET Imaging Facilitates In Vivo Monitoring of TSPO Expression in the GL261 Glioblastoma Mouse Model. Biomedicines 2022, 10, 738. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, N.K.; Hird, M.; Thompson, S.; Williamson, D.J.; Qiao, L.; Owen, D.R.; Brooks, A.F.; Scott, P.J.H.; Bacallado, S.; O’Brien, J.T.; et al. Preclinical evaluation of (S)-18FGE387, a novel 18-kDa translocator protein (TSPO) PET radioligand with low binding sensitivity to human polymorphism rs6971. Eur. J. Nucl. Med. Mol. Imaging 2021, 49, 125–136. [Google Scholar] [CrossRef]
- Hobson, B.A.; Rowland, D.J.; Sisó, S.; Guignet, M.A.; Harmany, Z.T.; Bandara, S.B.; Saito, N.; Harvey, D.J.; Bruun, D.A.; Garbow, J.R.; et al. TSPO PET Using 18FPBR111 Reveals Persistent Neuroinflammation Following Acute Diisopropylfluorophosphate Intoxication in the Rat. Toxicol. Sci. 2019, 170, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Ottoy, J.; de Picker, L.; Verhaeghe, J.; Deleye, S.; Wyffels, L.; Kosten, L.; Sabbe, B.; Coppens, V.; Timmers, M.; van Nueten, L.; et al. 18F-PBR111 PET Imaging in Healthy Controls and Schizophrenia: Test–Retest Reproducibility and Quantification of Neuroinflammation. J. Nucl. Med. 2018, 59, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Kim, H.; Bae, S.-H.; Lee, S.-Y.; Kim, Y.-H.; Na, J.; Lee, C.-H.; Lee, M.S.; Ko, G.B.; Kim, K.Y.; et al. 18FCB251 PET/MR imaging probe targeting translocator protein (TSPO) independent of its Polymorphism in a Neuroinflammation Model. Theranostics 2020, 10, 9315–9331. [Google Scholar] [CrossRef]
- Lee, S.H.; Denora, N.; Laquintana, V.; Mangiatordi, G.F.; Lopedota, A.; Lopalco, A.; Cutrignelli, A.; Franco, M.; Delre, P.; Song, I.H.; et al. Radiosynthesis and characterization of [18F]BS224: A next-generation TSPO PET ligand insensitive to the rs6971 polymorphism. Eur. J. Nucl. Med. Mol. Imaging 2021, 49, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, J.; Zheng, Y.; Garavito, R.M.; Ferguson-Miller, S. Protein structure. Crystal structures of translocator protein (TSPO) and mutant mimic of a human polymorphism. Science 2015, 347, 555–558. [Google Scholar] [CrossRef]
- Jaremko, M.; Jaremko, Ł.; Giller, K.; Becker, S.; Zweckstetter, M. Structural Integrity of the A147T Polymorph of Mammalian TSPO. ChemBioChem 2015, 16, 1483–1489. [Google Scholar] [CrossRef]
- Jaremko, M.; Jaremko, Ł.; Giller, K.; Becker, S.; Zweckstetter, M. Backbone and side-chain resonance assignment of the A147T polymorph of mouse TSPO in complex with a high-affinity radioligand. Biomol. NMR Assign. 2016, 10, 79–83. [Google Scholar] [CrossRef]
- Berroterán-Infante, N.; Tadić, M.; Hacker, M.; Wadsak, W.; Mitterhauser, M. Binding Affinity of Some Endogenous and Synthetic TSPO Ligands Regarding the rs6971 Polymorphism. Int. J. Mol. Sci. 2019, 20, 563. [Google Scholar] [CrossRef]
- Midzak, A.S.; Akula, N.; Rone, M.B.; Papadopoulos, V. Computational modeling and biological validation of novel non-steroidal ligands for the cholesterol recognition/interaction amino acid consensus (CRAC) motif of the mitochondrial translocator protein (TSPO). Pharmacol. Res. 2015, 99, 393–403. [Google Scholar] [CrossRef]
- Rojas, C.; Stathis, M.; Coughlin, J.M.; Pomper, M.; Slusher, B.S. The Low-Affinity Binding of Second Generation Radiotracers Targeting TSPO is Associated with a Unique Allosteric Binding Site. J. Neuroimmune Pharmacol. 2018, 13, 1–5. [Google Scholar] [CrossRef]
- ENZYME—1.14.99.1 Prostaglandin-Endoperoxide Synthase. Available online: https://enzyme.expasy.org/EC/1.14.99.1 (accessed on 1 January 2021).
- Yamagata, K.; Matsumura, K.; Inoue, W.; Shiraki, T.; Suzuki, K.; Yasuda, S.; Sugiura, H.; Cao, C.; Watanabe, Y.; Kobayashi, S. Coexpression of microsomal-type prostaglandin E synthase with cyclooxygenase-2 in brain endothelial cells of rats during endotoxin-induced fever. J. Neurosci. 2001, 21, 2669–2677. [Google Scholar] [CrossRef]
- Rummel, C.; Sachot, C.; Poole, S.; Luheshi, G.N. Circulating interleukin-6 induces fever through a STAT3-linked activation of COX-2 in the brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R1316–R1326. [Google Scholar] [CrossRef]
- Steiner, A.A.; Hunter, J.C.; Phipps, S.M.; Nucci, T.B.; Oliveira, D.L.; Roberts, J.L.; Scheck, A.C.; Simmons, D.L.; Romanovsky, A.A. Cyclooxygenase-1 or -2—Which one mediates lipopolysaccharide-induced hypothermia? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R485–R494. [Google Scholar] [CrossRef]
- Tietz, O.; Wuest, M.; Marshall, A.; Glubrecht, D.; Hamann, I.; Wang, M.; Bergman, C.; Way, J.D.; Wuest, F. PET imaging of cyclooxygenase-2 (COX-2) in a pre-clinical colorectal cancer model. EJNMMI Res. 2016, 6, 37. [Google Scholar] [CrossRef]
- Le Fur, G.; Perrier, M.L.; Vaucher, N.; Imbault, F.; Flamier, A.; Benavides, J.; Uzan, A.; Renault, C.; Dubroeucq, M.C.; Guérémy, C. Peripheral benzodiazepine binding sites: Effect of PK 11195, 1-(2-chlorophenyl)-n-methyl-n-(1-methylpropyl)-3-isoquinolinecarboxamide. Life Sci. 1983, 32, 1839–1847. [Google Scholar] [CrossRef]
- Shrestha, S.; Kim, M.-J.; Eldridge, M.; Lehmann, M.L.; Frankland, M.; Liow, J.-S.; Yu, Z.-X.; Cortes-Salva, M.; Telu, S.; Henter, I.D.; et al. PET measurement of cyclooxygenase-2 using a novel radioligand: Upregulation in primate neuroinflammation and first-in-human study. J. Neuroinflamm. 2020, 17, 140. [Google Scholar] [CrossRef]
- Goetz Moro, M.; Vargas Sanchez, P.K.; Lupepsa, A.C.; Baller, E.M.; Nobre Franco, G.C. Biología de la ciclooxigenasa en la función renal—Revisión de la literatura. Rev. Colomb. Nefrol. 2017, 4, 27. [Google Scholar] [CrossRef]
- Seibert, K.; Zhang, Y.; Leahy, K.; Hauser, S.; Masferrer, J.; Perkins, W.; Lee, L.; Isakson, P. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc. Natl. Acad. Sci. USA 1994, 91, 12013–12017. [Google Scholar] [CrossRef]
- Krüger, K.; Bredehöft, J.; Mooren, F.C.; Rummel, C. Different effects of strength and endurance exercise training on COX-2 and mPGES expression in mouse brain are independent of peripheral inflammation. J. Appl. Physiol. 2016, 121, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Cortes, M.; Singh, P.; Morse, C.; Shrestha, S.; Jenko, K.; Kowalski, A.; Zoghbi, S.; Fujita, M.; Innis, R.; Pike, V. Synthesis of PET radioligands as potential probes for imaging COX-2 in neuroinflammation. J. Nucl. Med. 2015, 56, 1092. [Google Scholar]
- Kim, M.-J.; Shrestha, S.; Eldridge, M.; Cortes, M.; Singh, P.; Liow, J.-S.; Gladding, R.; Zoghbi, S.; Fujita, M.; Pike, V.; et al. Novel PET radioligands show that, in rhesus monkeys, COX-1 is constitutively expressed and COX-2 is induced by inflammation. J. Nucl. Med. 2017, 58, 203. [Google Scholar]
- Sejdiu, B.I.; Tieleman, D.P. COX-1—Lipid interactions: Arachidonic acid, cholesterol, and phospholipid binding to the membrane binding domain of COX-1. bioRxiv 2020, 5, 109363. [Google Scholar]
- Lebedev, A.; Jiao, J.; Lee, J.; Yang, F.; Allison, N.; Herschman, H.; Sadeghi, S. Radiochemistry on electrodes: Synthesis of an 18F-labelled and in vivo stable COX-2 inhibitor. PLoS ONE 2017, 12, e0176606. [Google Scholar] [CrossRef]
- Patel, R.; Attur, M.G.; Dave, M.; Abramson, S.B.; Amin, A.R. Regulation of Cytosolic COX-2 and Prostaglandin E2 Production by Nitric Oxide in Activated Murine Macrophages. J. Immunol. 1999, 162, 4191–4197. [Google Scholar] [CrossRef]
- Tietz, O.; Marshall, A.; Wuest, M.; Wang, M.; Wuest, F. Radiotracers for molecular imaging of cyclooxygenase-2 (COX-2) enzyme. Curr. Med. Chem. 2013, 20, 4350–4369. [Google Scholar] [CrossRef]
- Tietz, O.; Dzandzi, J.; Bhardwaj, A.; Valliant, J.F.; Wuest, F. Design and synthesis of (125)IPyricoxib: A novel (125)I-labeled cyclooxygenase-2 (COX-2) inhibitors. Bioorganic Med. Chem. Lett. 2016, 26, 1516–1520. [Google Scholar] [CrossRef]
- Teismann, P.; Tieu, K.; Choi, D.-K.; Wu, D.-C.; Naini, A.; Hunot, S.; Vila, M.; Jackson-Lewis, V.; Przedborski, S. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 5473–5478. [Google Scholar] [CrossRef]
- New Scientist. Up to 140,000 Heart Attacks Linked to Vioxx. Available online: https://www.newscientist.com/article/dn6918-up-to-140000-heart-attacks-linked-to-vioxx/?ignored=irrelevant (accessed on 3 January 2021).
- FDA/CDER. Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/020998s050lbl.pdf (accessed on 12 October 2023).
- Prabhakaran, J.; Underwood, M.D.; Parsey, R.V.; Arango, V.; Majo, V.J.; Simpson, N.R.; van Heertum, R.; Mann, J.J.; Kumar, J.S.D. Synthesis and in vivo evaluation of 18F-4-5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-ylbenzenesulfonamide as a PET imaging probe for COX-2 expression. Bioorganic Med. Chem. 2007, 15, 1802–1807. [Google Scholar] [CrossRef]
- Takashima-Hirano, M.; Takashima, T.; Katayama, Y.; Wada, Y.; Sugiyama, Y.; Watanabe, Y.; Doi, H.; Suzuki, M. Efficient sequential synthesis of PET Probes of the COX-2 inhibitor 11Ccelecoxib and its major metabolite 11CSC-62807 and in vivo PET evaluation. Bioorganic Med. Chem. 2011, 19, 2997–3004. [Google Scholar] [CrossRef] [PubMed]
- Fujisaki, Y.; Kawamura, K.; Wang, W.-F.; Ishiwata, K.; Yamamoto, F.; Kuwano, T.; Ono, M.; Maeda, M. Radiosynthesis and in vivo evaluation of 11C-labeled 1,5-diarylpyrazole derivatives for mapping cyclooxygenases. Ann. Nucl. Med. 2005, 19, 617–625. [Google Scholar] [CrossRef]
- Gao, M.; Wang, M.; Miller, K.D.; Hutchins, G.D.; Zheng, Q.-H. Synthesis of carbon-11 labeled celecoxib derivatives as new candidate PET radioligands for imaging of inflammation. Appl. Radiat. Isot. 2009, 67, 2019–2024. [Google Scholar] [CrossRef]
- de Vries, E.F.J.; Doorduin, J.; Dierckx, R.A.; van Waarde, A. Evaluation of (11)Crofecoxib as PET tracer for cyclooxygenase 2 overexpression in rat models of inflammation. Nucl. Med. Biol. 2008, 35, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Search Results for 11C-MC1—Clinical Trials Registry—ICH GCP. Available online: https://ichgcp.net/clinical-trials-registry/research/list?intr=11C-MC1 (accessed on 8 January 2021).
- Barrio, J.R.; Satyamurthy, N.; Huang, S.C.; Keen, R.E.; Nissenson, C.H.; Hoffman, J.M.; Ackermann, R.F.; Bahn, M.M.; Mazziotta, J.C.; Phelps, M.E. 3-(2’-18Ffluoroethyl)spiperone: In vivo biochemical and kinetic characterization in rodents, nonhuman primates, and humans. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 1989, 9, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.J.; Crews, B.C.; Ghebreselasie, K.; Huda, I.; Kingsley, P.J.; Ansari, M.S.; Tantawy, M.N.; Reese, J.; Marnett, L.J. Fluorinated COX-2 inhibitors as agents in PET imaging of inflammation and cancer. Cancer Prev. Res. 2011, 4, 1536–1545. [Google Scholar] [CrossRef]
- Lemal, D.M. Perspective on fluorocarbon chemistry. J. Org. Chem. 2004, 69, 1–11. [Google Scholar] [CrossRef]
- Secrieru, A.; O’Neill, P.M.; Cristiano, M.L.S. Revisiting the Structure and Chemistry of 3(5)-Substituted Pyrazoles. Molecules 2019, 25, 42. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kumadaki, I. Reactions of aromatic trifluoromethyl compounds with nucleophilic reagents. Acc. Chem. Res. 1978, 11, 197–204. [Google Scholar] [CrossRef]
- Toyokuni, T.; Kumar, J.S.D.; Walsh, J.C.; Shapiro, A.; Talley, J.J.; Phelps, M.E.; Herschman, H.R.; Barrio, J.R.; Satyamurthy, N. Synthesis of 4-(5-18Ffluoromethyl-3-phenylisoxazol-4-yl)benzenesulfonamide, a new 18Ffluorinated analogue of valdecoxib, as a potential radiotracer for imaging cyclooxygenase-2 with positron emission tomography. Bioorganic Med. Chem. Lett. 2005, 15, 4699–4702. [Google Scholar] [CrossRef]
- Cheng, K.; Qi, J.; Ren, X.; Zhang, J.; Li, H.; Xiao, H.; Wang, R.; Liu, Z.; Meng, L.; Ma, N.; et al. Developing Isoxazole as a Native Photo-Cross-Linker for Photoaffinity Labeling and Chemoproteomics. Angew. Chem. Int. Ed. Engl. 2022, 61, e202209947. [Google Scholar] [CrossRef]
- Weber, A.; Casini, A.; Heine, A.; Kuhn, D.; Supuran, C.T.; Scozzafava, A.; Klebe, G. Unexpected nanomolar inhibition of carbonic anhydrase by COX-2-selective celecoxib: New pharmacological opportunities due to related binding site recognition. J. Med. Chem. 2004, 47, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, M.E.; Beswick, P.J.; Gleave, R.J.; Green, R.H.; Bingham, S.; Bountra, C.; Carter, M.C.; Chambers, L.J.; Chessell, I.P.; Clayton, N.M.; et al. Identification of 4-4-(methylsulfonyl)phenyl-6-(trifluoromethyl)-2-pyrimidinyl amines and ethers as potent and selective cyclooxygenase-2 inhibitors. Bioorganic Med. Chem. Lett. 2009, 19, 4504–4508. [Google Scholar] [CrossRef]
- Tietz, O.; Sharma, S.K.; Kaur, J.; Way, J.; Marshall, A.; Wuest, M.; Wuest, F. Synthesis of three 18F-labelled cyclooxygenase-2 (COX-2) inhibitors based on a pyrimidine scaffold. Org. Biomol. Chem. 2013, 11, 8052–8064. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, T.J.; Sheriff, A.U.; Graneto, M.J.; Talley, J.J.; Welch, M.J. Radiosynthesis, in vitro validation, and in vivo evaluation of 18F-labeled COX-1 and COX-2 inhibitors. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2002, 43, 117–124. [Google Scholar]
- Breder, C.D. Cyclooxygenase systems in the mammalian brain. Ann. N. Y. Acad. Sci. 1997, 813, 296–301. [Google Scholar] [CrossRef]
- Kaufmann, W.E.; Andreasson, K.I.; Isakson, P.C.; Worley, P.F. Cyclooxygenases and the central nervous system. Prostaglandins 1997, 54, 601–624. [Google Scholar] [CrossRef]
- Kuge, Y.; Katada, Y.; Shimonaka, S.; Temma, T.; Kimura, H.; Kiyono, Y.; Yokota, C.; Minematsu, K.; Seki, K.-i.; Tamaki, N.; et al. Synthesis and evaluation of radioiodinated cyclooxygenase-2 inhibitors as potential SPECT tracers for cyclooxygenase-2 expression. Nucl. Med. Biol. 2006, 33, 21–27. [Google Scholar] [CrossRef]
- Kabalka, G.W.; Mereddy, A.R.; Schuller, H.M. Synthesis of an iodine-123-labeled celecoxib analogue: A potential spect agent. J. Label. Compd. Radiopharm. 2005, 48, 295–300. [Google Scholar] [CrossRef]
- Schuller, H.M.; Kabalka, G.; Smith, G.; Mereddy, A.; Akula, M.; Cekanova, M. Detection of overexpressed COX-2 in precancerous lesions of hamster pancreas and lungs by molecular imaging: Implications for early diagnosis and prevention. ChemMedChem 2006, 1, 603–610. [Google Scholar] [CrossRef]
- Uddin, M.J.; Crews, B.C.; Ghebreselasie, K.; Tantawy, M.N.; Marnett, L.J. I-Celecoxib Analogues as SPECT Tracers of Cyclooxygenase-2 in Inflammation. ACS Med. Chem. Lett. 2011, 2, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Mathis, C.A.; Lopresti, B.J.; Ikonomovic, M.D.; Klunk, W.E. Small-molecule PET Tracers for Imaging Proteinopathies. Semin. Nucl. Med. 2017, 47, 553–575. [Google Scholar] [CrossRef]
- Kuge, Y.; Obokata, N.; Kimura, H.; Katada, Y.; Temma, T.; Sugimoto, Y.; Aita, K.; Seki, K.-I.; Tamaki, N.; Saji, H. Synthesis and evaluation of a radioiodinated lumiracoxib derivative for the imaging of cyclooxygenase-2 expression. Nucl. Med. Biol. 2009, 36, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.J.; Bryant, J.; Chang, J.Y.; Mendez, R.; Oh, C.-S.; Yu, D.-F.; Ito, M.; Azhdarinia, A.; Kohanim, S.; Edmund Kim, E.; et al. Assessment of cyclooxygense-2 expression with 99mTc-labeled celebrex. Anticancer Drugs 2004, 15, 255–263. [Google Scholar] [CrossRef]
- Méric, J.-B.; Rottey, S.; Olaussen, K.; Soria, J.-C.; Khayat, D.; Rixe, O.; Spano, J.-P. Cyclooxygenase-2 as a target for anticancer drug development. Crit. Rev. Oncol. Hematol. 2006, 59, 51–64. [Google Scholar] [CrossRef]
- Farouk, N.; El-Tawoosy, M.; Ayoub, S.; El-Bayoumy, A.S. Optimization of the reaction conditions for the preparation of 99mTc-celecoxib and its biological evaluation. J. Radioanal. Nucl. Chem. 2011, 290, 685–690. [Google Scholar] [CrossRef]
- Chadha, V.D.; Pearl laird; Jan, G.; Khan, A.A. Radiosynthesis, Biodistribution and Scintigraphic Imaging of 99mtc-Celecoxib in Experimental Rat Model of Colon Carcinogenesis. Available online: https://austinpublishinggroup.com/nuclear-medicine-radiotherapy/fulltext/ajnmr-v2-id1010.php (accessed on 28 February 2021).
- Martín, A.; Boisgard, R.; Thézé, B.; van Camp, N.; Kuhnast, B.; Damont, A.; Kassiou, M.; Dollé, F.; Tavitian, B. Evaluation of the PBR/TSPO radioligand (18)FDPA-714 in a rat model of focal cerebral ischemia. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2010, 30, 230–241. [Google Scholar] [CrossRef]
- Cao, C.; Matsumura, K.; Yamagata, K.; Watanabe, Y. Endothelial cells of the rat brain vasculature express cyclooxygenase-2 mRNA in response to systemic interleukin-1 beta: A possible site of prostaglandin synthesis responsible for fever. Brain Res. 1996, 733, 263–272. [Google Scholar] [CrossRef]
- Kang, Y.-J.; Mbonye, U.R.; DeLong, C.J.; Wada, M.; Smith, W.L. Regulation of intracellular cyclooxygenase levels by gene transcription and protein degradation. Prog. Lipid Res. 2007, 46, 108–125. [Google Scholar] [CrossRef]
- Uzuegbunam, B.C.; Librizzi, D.; Hooshyar Yousefi, B. PET Radiopharmaceuticals for Alzheimer’s Disease and Parkinson’s Disease Diagnosis, the Current and Future Landscape. Molecules 2020, 25, 977. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-X.; Zhou, M.; Ma, H.-L.; Qiao, Y.-B.; Li, Q.-S. The Role of Chronic Inflammation in Various Diseases and Anti-inflammatory Therapies Containing Natural Products. ChemMedChem 2021, 16, 1576–1592. [Google Scholar] [CrossRef] [PubMed]
- Peters, E.; Del Rey, A.; Krüger, K.; Rummel, C. 2nd European Psychoneuroimmunology Network Autumn School: The Skin-Brain Axis and the Breaking of Barriers. Neuroimmunomodulation 2023, 30 (Suppl. S1), 3–7. [Google Scholar] [CrossRef]
- Bajinka, O.; Simbilyabo, L.; Tan, Y.; Jabang, J.; Saleem, S.A. Lung-brain axis. Crit. Rev. Microbiol. 2022, 48, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Rummel, C.; Del, R.A.; Bähr, L.; Krüger, K.; Peters, E. 1st European Psychoneuroimmunology Network (EPN) Autumn School: Lung-Brain Axis in Health and Disease. Neuroimmunomodulation 2022, 29 (Suppl. S2), 3–8. [Google Scholar] [CrossRef]
- Hosang, L.; Canals, R.C.; van der Flier, F.J.; Hollensteiner, J.; Daniel, R.; Flügel, A.; Odoardi, F. The lung microbiome regulates brain autoimmunity. Nature 2022, 603, 138–144. [Google Scholar] [CrossRef]
- Li, C.; Chen, W.; Lin, F.; Li, W.; Wang, P.; Liao, G.; Zhang, L. Functional Two-Way Crosstalk Between Brain and Lung: The Brain-Lung Axis. Cell. Mol. Neurobiol. 2022, 43, 991–1003. [Google Scholar] [CrossRef]
- Alzghool, O.M.; van Dongen, G.; van de Giessen, E.; Schoonmade, L.; Beaino, W. α-Synuclein Radiotracer Development and In Vivo Imaging: Recent Advancements and New Perspectives. Mov. Disord. 2022, 37, 936–948. [Google Scholar] [CrossRef]
- Hernandez, J.; Schäffer, J.; Herden, C.; Pflieger, F.J.; Reiche, S.; Körber, S.; Kitagawa, H.; Welter, J.; Michels, S.; Culmsee, C.; et al. n-3 Polyunsaturated Fatty Acids Modulate LPS-Induced ARDS and the Lung-Brain Axis of Communication in Wild-Type versus Fat-1 Mice Genetically Modified for Leukotriene B4 Receptor 1 or Chemerin Receptor 23 Knockout. Int. J. Mol. Sci. 2023, 24, 13524. [Google Scholar] [CrossRef]
- Goggi, J.L.; Claser, C.; Hartimath, S.V.; Hor, P.X.; Tan, P.W.; Ramasamy, B.; Abdul Rahman, H.; Cheng, P.; Chang, Z.W.; Nguee, S.Y.T.; et al. PET Imaging of Translocator Protein as a Marker of Malaria-Associated Lung Inflammation. Infect. Immun. 2021, 89, 10. [Google Scholar] [CrossRef]
- Chen, D.L.; Agapov, E.; Wu, K.; Engle, J.T.; Solingapuram Sai, K.K.; Arentson, E.; Spayd, K.J.; Moreland, K.T.; Toth, K.; Byers, D.E.; et al. Selective Imaging of Lung Macrophages Using 11CPBR28-Based Positron Emission Tomography. Mol. Imaging Biol. 2021, 23, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Narayan, N.; Mandhair, H.; Smyth, E.; Dakin, S.G.; Kiriakidis, S.; Wells, L.; Owen, D.; Sabokbar, A.; Taylor, P. The macrophage marker translocator protein (TSPO) is down-regulated on pro-inflammatory ‘M1’ human macrophages. PLoS ONE 2017, 12, e0185767. [Google Scholar] [CrossRef] [PubMed]
- Hatori, A.; Yui, J.; Yamasaki, T.; Xie, L.; Kumata, K.; Fujinaga, M.; Yoshida, Y.; Ogawa, M.; Nengaki, N.; Kawamura, K.; et al. PET imaging of lung inflammation with 18FFEDAC, a radioligand for translocator protein (18 kDa). PLoS ONE 2012, 7, e45065. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.A.; Valind, S.O.; Clark, I.C.; Bolden, G.E.; Krausz, T.; Schofield, J.B.; Boobis, A.R.; Haslett, C. Kinetics of lung macrophages monitored in vivo following particulate challenge in rabbits. Toxicol. Appl. Pharmacol. 2002, 183, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Sinharay, S.; Patel, R.; Solomon, J.; Lee, J.H.; Schreiber-Stainthorp, W.; Basuli, F.; Zhang, X.; Hagen, K.R.; Reeder, R.; et al. PET imaging of TSPO expression in immune cells can assess organ-level pathophysiology in high-consequence viral infections. Proc. Natl. Acad. Sci. USA 2022, 119, e2110846119. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, A.A.; Pokkali, S.; DeMarco, V.P.; Klunk, M.; Mease, R.C.; Foss, C.A.; Pomper, M.G.; Jain, S.K. Radioiodinated DPA-713 imaging correlates with bactericidal activity of tuberculosis treatments in mice. Antimicrob. Agents Chemother. 2015, 59, 642–649. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ruiz-Bedoya, C.A.; Mota, F.; Ordonez, A.A.; Foss, C.A.; Singh, A.K.; Praharaj, M.; Mahmud, F.J.; Ghayoor, A.; Flavahan, K.; de Jesus, P.; et al. 124I-Iodo-DPA-713 Positron Emission Tomography in a Hamster Model of SARS-CoV-2 Infection. Mol. Imaging Biol. 2022, 24, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Van der Krogt, J.M.A.; van Binsbergen, W.H.; van der Laken, C.J.; Tas, S.W. Novel positron emission tomography tracers for imaging of rheumatoid arthritis. Autoimmun. Rev. 2021, 20, 102764. [Google Scholar] [CrossRef]
- Verstappen, M.; van Steenbergen, H.W.; de Jong, P.H.P.; van der Helm-van Mil, A.H.M. Unraveling heterogeneity within ACPA-negative rheumatoid arthritis: The subgroup of patients with a strong clinical and serological response to initiation of DMARD treatment favor disease resolution. Arthritis Res. Ther. 2022, 24, 4. [Google Scholar] [CrossRef]
- Novella-Navarro, M.; Plasencia, C.; Tornero, C.; Navarro-Compán, V.; Cabrera-Alarcón, J.L.; Peiteado-López, D.; Nuño, L.; Monjo-Henry, I.; Franco-Gómez, K.; Villalba, A.; et al. Clinical predictors of multiple failure to biological therapy in patients with rheumatoid arthritis. Arthritis Res. Ther. 2020, 22, 284. [Google Scholar] [CrossRef]
- Wang, Z.; Huang, J.; Xie, D.; He, D.; Lu, A.; Liang, C. Toward Overcoming Treatment Failure in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 755844. [Google Scholar] [CrossRef] [PubMed]
- Bruijnen, S.T.G.; Verweij, N.J.F.; Gent, Y.Y.J.; Huisman, M.C.; Windhorst, A.D.; Kassiou, M.; van de Ven, P.M.; Lammertsma, A.A.; Hoekstra, O.S.; Voskuyl, A.E.; et al. Imaging disease activity of rheumatoid arthritis by macrophage targeting using second generation translocator protein positron emission tomography tracers. PLoS ONE 2019, 14, e0222844. [Google Scholar] [CrossRef] [PubMed]
- Narayan, N.; Owen, D.R.; Mandhair, H.; Smyth, E.; Carlucci, F.; Saleem, A.; Gunn, R.N.; Rabiner, E.A.; Wells, L.; Dakin, S.G.; et al. Translocator Protein as an Imaging Marker of Macrophage and Stromal Activation in Rheumatoid Arthritis Pannus. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2018, 59, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Gent, Y.Y.J.; Voskuyl, A.E.; Kloet, R.W.; van Schaardenburg, D.; Hoekstra, O.S.; Dijkmans, B.A.C.; Lammertsma, A.A.; van der Laken, C.J. Macrophage positron emission tomography imaging as a biomarker for preclinical rheumatoid arthritis: Findings of a prospective pilot study. Arthritis Rheum. 2012, 64, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Gent, Y.Y.; Ahmadi, N.; Voskuyl, A.E.; Hoetjes, N.; van Kuijk, C.; Britsemmer, K.; Turkstra, F.; Boers, M.; Hoekstra, O.S.; van der Laken, C.J. Detection of subclinical synovitis with macrophage targeting and positron emission tomography in patients with rheumatoid arthritis without clinical arthritis. J. Rheumatol. 2014, 41, 2145–2152. [Google Scholar] [CrossRef] [PubMed]
- Gent, Y.Y.J.; Ter Wee, M.M.; Voskuyl, A.E.; den Uyl, D.; Ahmadi, N.; Dowling, C.; van Kuijk, C.; Hoekstra, O.S.; Boers, M.; Lems, W.F.; et al. Subclinical synovitis detected by macrophage PET, but not MRI, is related to short-term flare of clinical disease activity in early RA patients: An exploratory study. Arthritis Res. Ther. 2015, 17, 266. [Google Scholar] [CrossRef]
- Pugh, D.; Karabayas, M.; Basu, N.; Cid, M.C.; Goel, R.; Goodyear, C.S.; Grayson, P.C.; McAdoo, S.P.; Mason, J.C.; Owen, C.; et al. Large-vessel vasculitis. Nat. Rev. Dis. Primers 2022, 7, 93. [Google Scholar] [CrossRef]
- Weyand, C.M.; Goronzy, J.J. Immune mechanisms in medium and large-vessel vasculitis. Nat. Rev. Rheumatol. 2013, 9, 731–740. [Google Scholar] [CrossRef]
- Qi, X.; Xu, J.; Wang, F.; Xiao, J. Translocator protein (18 kDa): A promising therapeutic target and diagnostic tool for cardiovascular diseases. Oxidative Med. Cell. Longev. 2012, 2012, 162934. [Google Scholar] [CrossRef]
- Lamare, F.; Hinz, R.; Gaemperli, O.; Pugliese, F.; Mason, J.C.; Spinks, T.; Camici, P.G.; Rimoldi, O.E. Detection and quantification of large-vessel inflammation with 11C-(R)-PK11195 PET/CT. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2011, 52, 33–39. [Google Scholar] [CrossRef]
- Pugliese, F.; Gaemperli, O.; Kinderlerer, A.R.; Lamare, F.; Shalhoub, J.; Davies, A.H.; Rimoldi, O.E.; Mason, J.C.; Camici, P.G. Imaging of vascular inflammation with 11C-PK11195 and positron emission tomography/computed tomography angiography. J. Am. Coll. Cardiol. 2010, 56, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xiao, J.; Liang, D.; Zhang, H.; Zhang, G.; Liu, Y.; Zhang, Y.; Liu, Y.; Yu, Z.; Yan, B.; et al. Inhibition of mitochondrial translocator protein prevents atrial fibrillation. Eur. J. Pharmacol. 2010, 632, 60–64. [Google Scholar] [CrossRef]
- Hellberg, S.; Silvola, J.M.U.; Kiugel, M.; Liljenbäck, H.; Savisto, N.; Li, X.-G.; Thiele, A.; Lehmann, L.; Heinrich, T.; Vollmer, S.; et al. 18-kDa translocator protein ligand 18F-FEMPA: Biodistribution and uptake into atherosclerotic plaques in mice. J. Nucl. Cardiol. 2017, 24, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Maulik, S.K.; Kumar, S. Oxidative stress and cardiac hypertrophy: A review. Toxicol. Mech. Methods 2012, 22, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Liang, D.; Zhang, H.; Liu, Y.; Li, F.; Chen, Y.-H. 4′-Chlorodiazepam, a translocator protein (18 kDa) antagonist, improves cardiac functional recovery during postischemia reperfusion in rats. Exp. Biol. Med. 2010, 235, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Mou, T.; Tian, J.; Tian, Y.; Yun, M.; Li, J.; Dong, W.; Lu, X.; Zhu, Z.; Mi, H.; Zhang, X.; et al. Automated synthesis and preliminary evaluation of 18FFDPA for cardiac inflammation imaging in rats after myocardial infarction. Sci. Rep. 2020, 10, 18685. [Google Scholar] [CrossRef]
- Thackeray, J.T.; Bengel, F.M. Molecular Imaging of Myocardial Inflammation With Positron Emission Tomography Post-Ischemia: A Determinant of Subsequent Remodeling or Recovery. JACC Cardiovasc. Imaging 2018, 11, 1340–1355. [Google Scholar] [CrossRef]
- Thackeray, J.T.; Hupe, H.C.; Wang, Y.; Bankstahl, J.P.; Berding, G.; Ross, T.L.; Bauersachs, J.; Wollert, K.C.; Bengel, F.M. Myocardial Inflammation Predicts Remodeling and Neuroinflammation After Myocardial Infarction. J. Am. Coll. Cardiol. 2018, 71, 263–275. [Google Scholar] [CrossRef]
- Meissner, A.; Visanji, N.P.; Momen, M.A.; Feng, R.; Francis, B.M.; Bolz, S.-S.; Hazrati, L.-N. Tumor Necrosis Factor-α Underlies Loss of Cortical Dendritic Spine Density in a Mouse Model of Congestive Heart Failure. J. Am. Heart Assoc. 2015, 4, e001920. [Google Scholar] [CrossRef]
- Hellberg, S.; Liljenbäck, H.; Eskola, O.; Morisson-Iveson, V.; Morrison, M.; Trigg, W.; Saukko, P.; Ylä-Herttuala, S.; Knuuti, J.; Saraste, A.; et al. Positron Emission Tomography Imaging of Macrophages in Atherosclerosis with 18F-GE-180, a Radiotracer for Translocator Protein (TSPO). Contrast Media Mol. Imaging 2018, 2018, 9186902. [Google Scholar] [CrossRef]
- Malmberg, C.; Ripa, R.S.; Johnbeck, C.B.; Knigge, U.; Langer, S.W.; Mortensen, J.; Oturai, P.; Loft, A.; Hag, A.M.; Kjær, A. 64Cu-DOTATATE for Noninvasive Assessment of Atherosclerosis in Large Arteries and Its Correlation with Risk Factors: Head-to-Head Comparison with 68Ga-DOTATOC in 60 Patients. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2015, 56, 1895–1900. [Google Scholar] [CrossRef] [PubMed]
- Tarkin, J.M.; Joshi, F.R.; Evans, N.R.; Chowdhury, M.M.; Figg, N.L.; Shah, A.V.; Starks, L.T.; Martin-Garrido, A.; Manavaki, R.; Yu, E.; et al. Detection of Atherosclerotic Inflammation by 68Ga-DOTATATE PET Compared to 18FFDG PET Imaging. J. Am. Coll. Cardiol. 2017, 69, 1774–1791. [Google Scholar] [CrossRef] [PubMed]
- Toner, Y.C.; Ghotbi, A.A.; Naidu, S.; Sakurai, K.; van Leent, M.M.T.; Jordan, S.; Ordikhani, F.; Amadori, L.; Sofias, A.M.; Fisher, E.L.; et al. Systematically evaluating DOTATATE and FDG as PET immuno-imaging tracers of cardiovascular inflammation. Sci. Rep. 2022, 12, 6185. [Google Scholar] [CrossRef] [PubMed]
- Foss, C.A.; Bedja, D.; Mease, R.C.; Wang, H.; Kass, D.A.; Chatterjee, S.; Pomper, M.G. Molecular imaging of inflammation in the ApoE−/− mouse model of atherosclerosis with IodoDPA. Biochem. Biophys. Res. Commun. 2015, 461, 70–75. [Google Scholar] [CrossRef]
- Kopecky, C.; Pandzic, E.; Parmar, A.; Szajer, J.; Lee, V.; Dupuy, A.; Arthur, A.; Fok, S.; Whan, R.; Ryder, W.J.; et al. Translocator protein localises to CD11b+ macrophages in atherosclerosis. Atherosclerosis 2019, 284, 153–159. [Google Scholar] [CrossRef]
- Gaemperli, O.; Shalhoub, J.; Owen, D.R.J.; Lamare, F.; Johansson, S.; Fouladi, N.; Davies, A.H.; Rimoldi, O.E.; Camici, P.G. Imaging intraplaque inflammation in carotid atherosclerosis with 11C-PK11195 positron emission tomography/computed tomography. Eur. Heart J. 2012, 33, 1902–1910. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| № | Generation | Radiotracer |
|---|---|---|
| 1. | First | [11C](R)PK11195 |
| 2. | Second | [11C]PBR28 |
| 3. | [11C]DPA-713 | |
| 4. | [123I]/[124I]/[125I]DPA-713 | |
| 5. | [18F]DPA-714 (PBR099) | |
| 6. | [18F]F-DPA | |
| 7. | [18F]FEPPA | |
| 8. | [18F]PBR111 | |
| 9. | [18F]FEDAC | |
| 10. | Third | [11C]ER176 |
| 11. | [18F]BIBD-239 | |
| 12. | [18F]GE-180 (Flutriciclamide) | |
| 13. | [18F]GE-387 | |
| 14. | [18F]CB251 | |
| 15. | [18F]BS224 |
| № | Imaging Modality | Radiotracer |
|---|---|---|
| 1. | PET | [11C]Celecoxib |
| 2. | [11C]Rofexcoxib | |
| 3. | [11C]MC1 | |
| 4. | [18F]1 (Celecoxib derivative) | |
| 5. | [18F]2 (Celecoxib derivative) | |
| 6. | [18F]3 (Valdecoxib derivative) | |
| 7. | [18F]4 (Celecoxib derivative) | |
| 8. | [18F]Pyricoxib | |
| 9. | SPECT | [125I]IATP |
| 10. | [125I]IMTP | |
| 11. | [125I]FIMA (Lumiracoxib derivative) | |
| 12. | [125I]Pyricoxib | |
| 13. | [99mTc]Celecoxib |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uzuegbunam, B.C.; Rummel, C.; Librizzi, D.; Culmsee, C.; Hooshyar Yousefi, B. Radiotracers for Imaging of Inflammatory Biomarkers TSPO and COX-2 in the Brain and in the Periphery. Int. J. Mol. Sci. 2023, 24, 17419. https://doi.org/10.3390/ijms242417419
Uzuegbunam BC, Rummel C, Librizzi D, Culmsee C, Hooshyar Yousefi B. Radiotracers for Imaging of Inflammatory Biomarkers TSPO and COX-2 in the Brain and in the Periphery. International Journal of Molecular Sciences. 2023; 24(24):17419. https://doi.org/10.3390/ijms242417419
Chicago/Turabian StyleUzuegbunam, Bright Chukwunwike, Christoph Rummel, Damiano Librizzi, Carsten Culmsee, and Behrooz Hooshyar Yousefi. 2023. "Radiotracers for Imaging of Inflammatory Biomarkers TSPO and COX-2 in the Brain and in the Periphery" International Journal of Molecular Sciences 24, no. 24: 17419. https://doi.org/10.3390/ijms242417419
APA StyleUzuegbunam, B. C., Rummel, C., Librizzi, D., Culmsee, C., & Hooshyar Yousefi, B. (2023). Radiotracers for Imaging of Inflammatory Biomarkers TSPO and COX-2 in the Brain and in the Periphery. International Journal of Molecular Sciences, 24(24), 17419. https://doi.org/10.3390/ijms242417419