
Recent Updates on the Therapeutic Prospects of Reversion-Inducing Cysteine-Rich Protein with Kazal Motifs (RECK) in Liver Injuries

 ,
,  ,
,  , and
, and 
Abstract
:1. Introduction
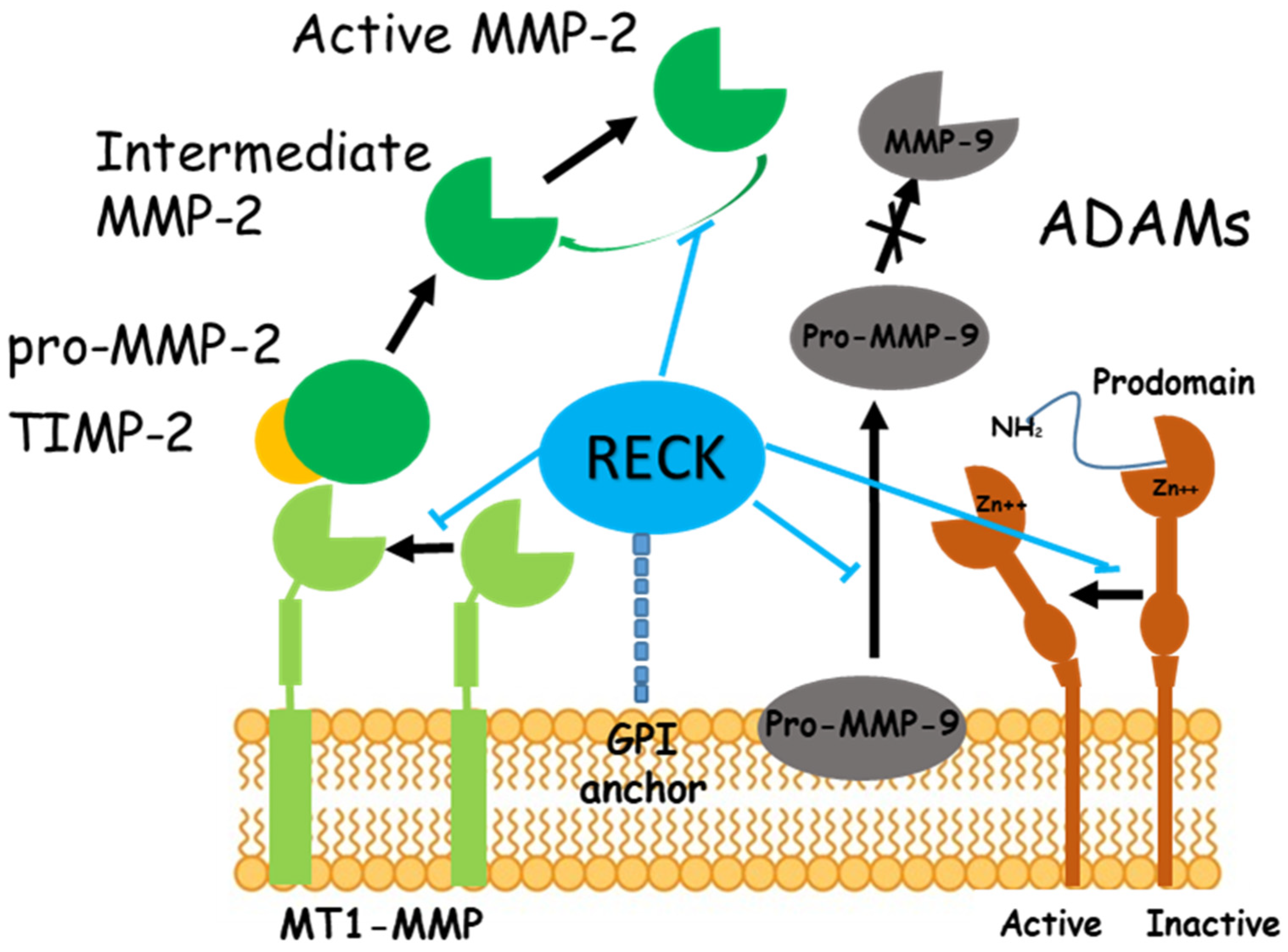
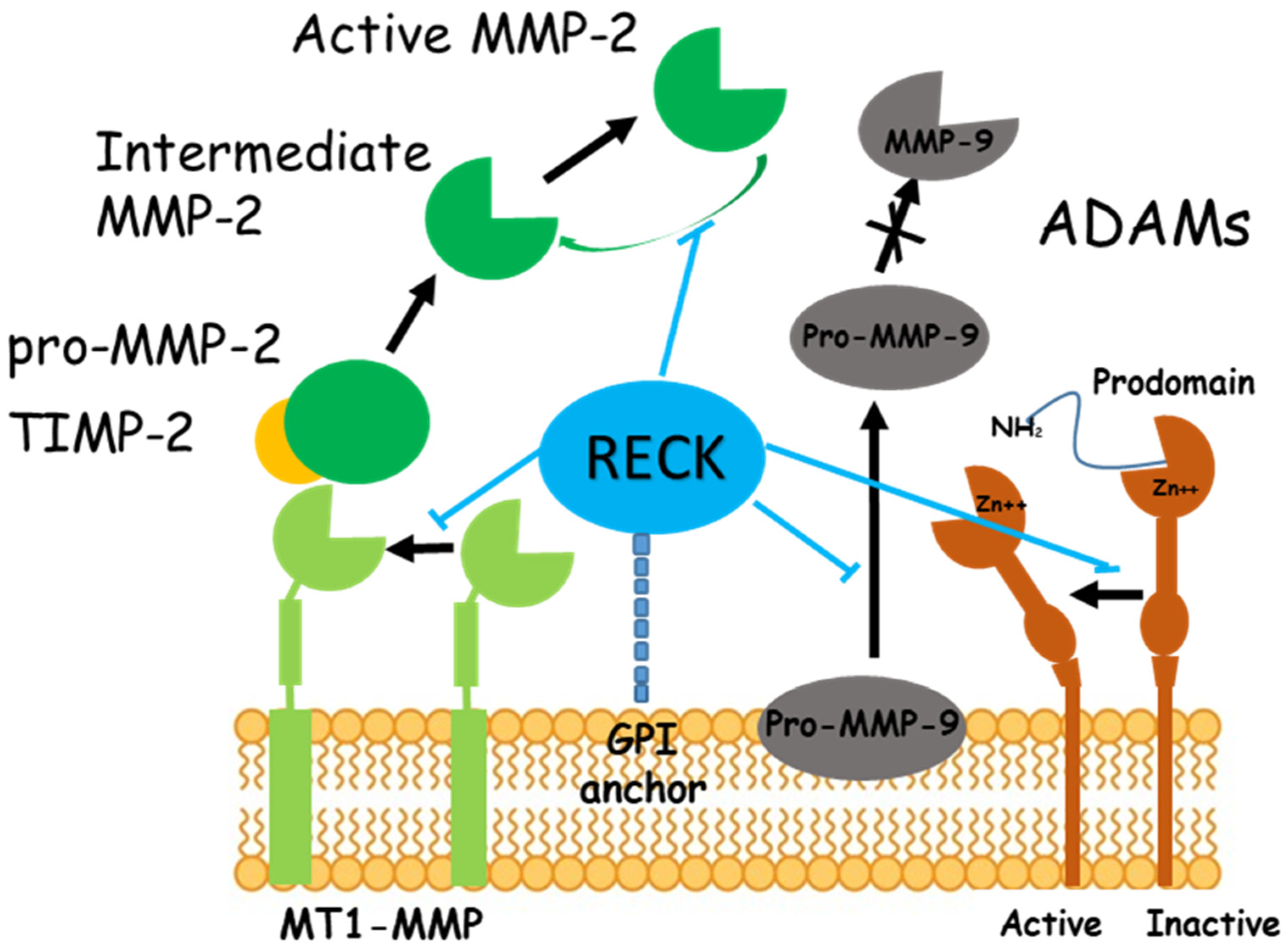
2. Ischemia/Reperfusion (I/R) Damage and RECK
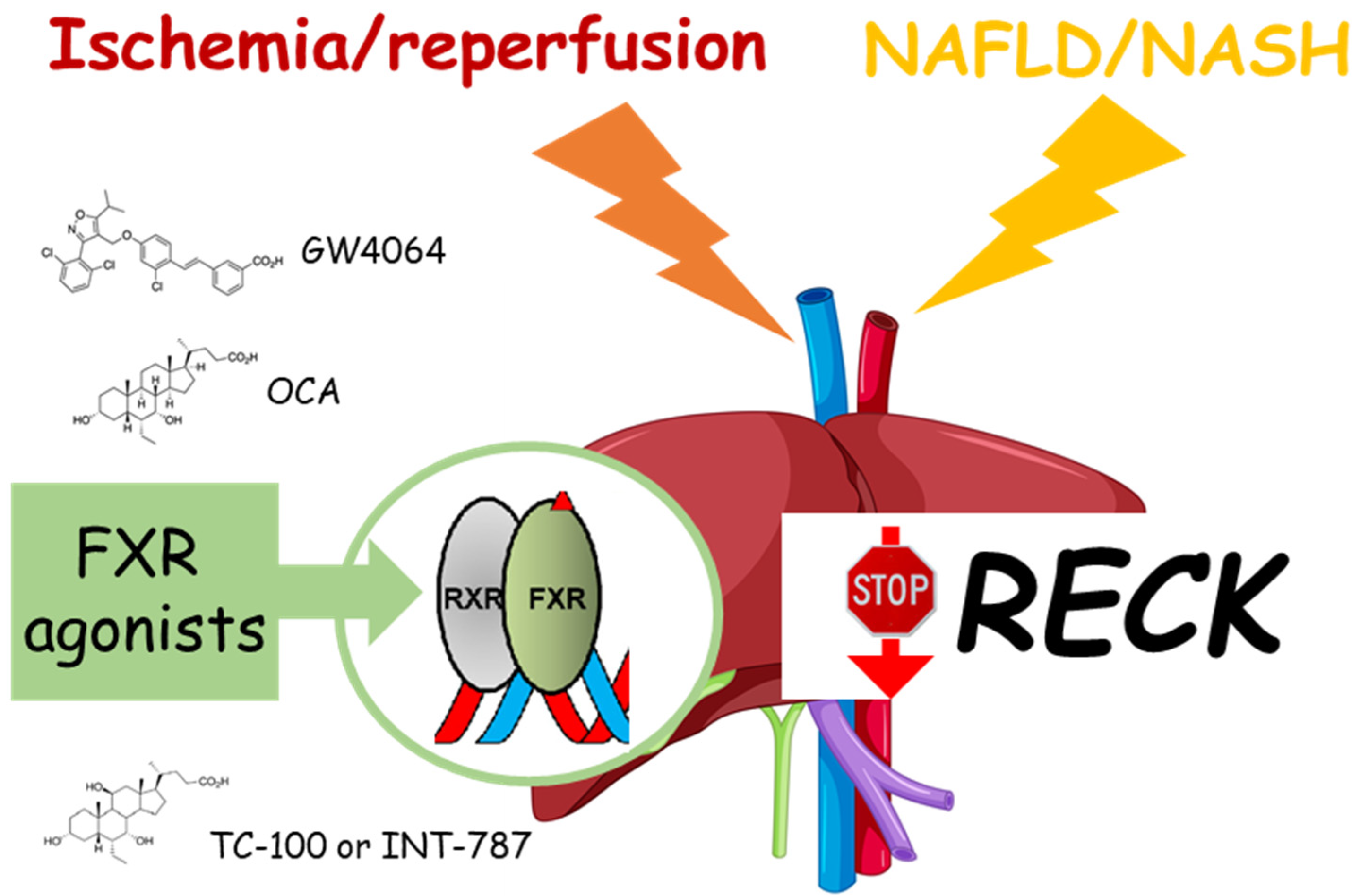
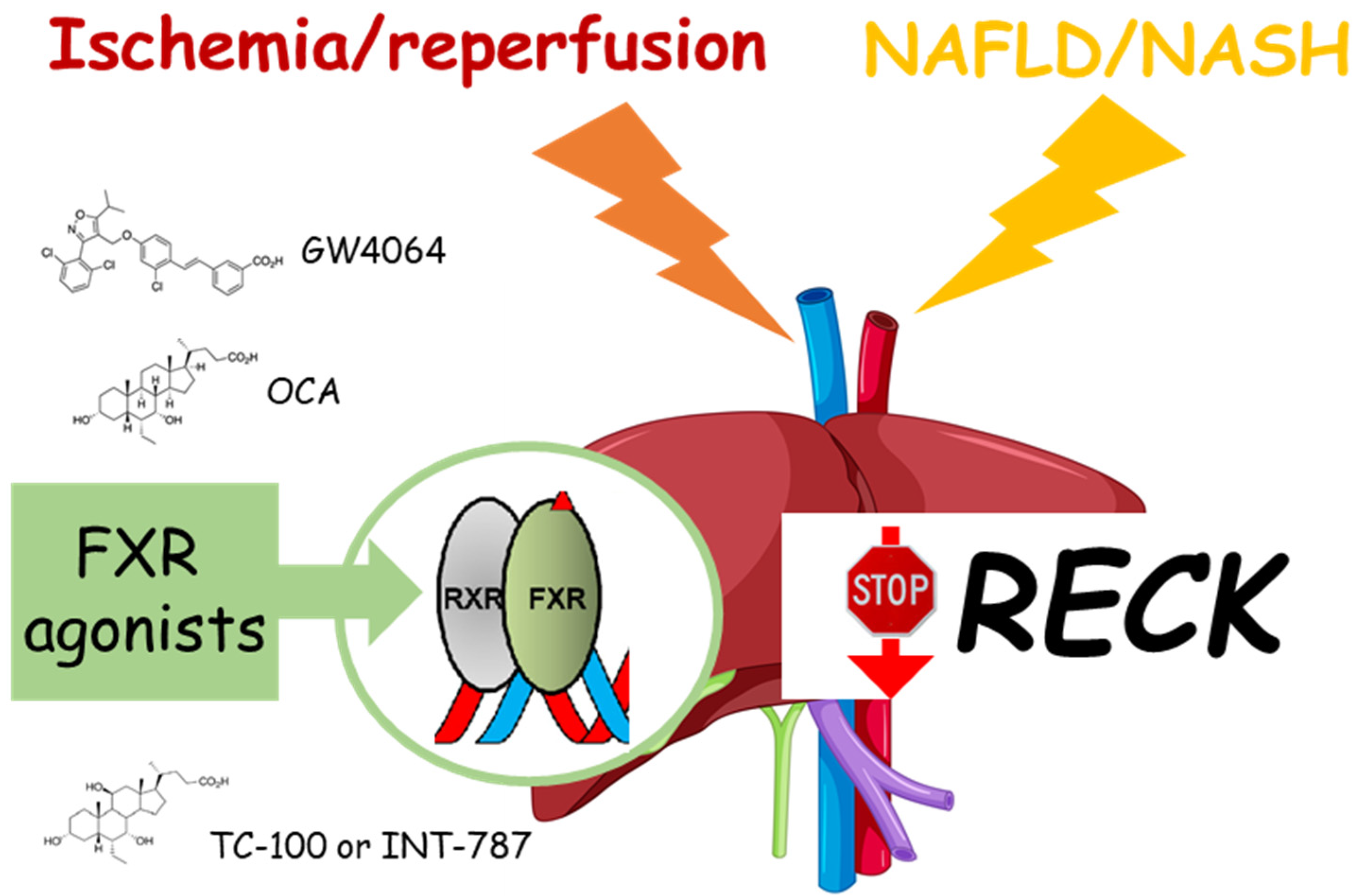
3. NAFLD and RECK
4. Cholangiocarcinoma (CCA) and RECK
5. Hepatocellular Carcinoma (HCC) and RECK
6. Viral Hepatitis and RECK
7. Future Prospective
8. Conclusions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Oh, J.; Takahashi, R.; Kondo, S.; Mizoguchi, A.; Adachi, E.; Sasahara, R.M.; Nishimura, S.; Imamura, Y.; Kitayama, H.; Alexander, D.B.; et al. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell 2001, 107, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, C.; Akiyama, N.; Matsuzaki, T.; Takai, S.; Kitayama, H.; Noda, M. Characterization of a human MSX-2 cDNA and its fragment isolated as a transformation suppressor gene against v-Ki-ras oncogene. Oncogene 1996, 12, 2137–2146. [Google Scholar] [PubMed]
- Omura, A.; Matsuzaki, T.; Mio, K.; Ogura, T.; Yamamoto, M.; Fujita, A.; Okawa, K.; Kitayama, H.; Takahashi, C.; Sato, C.; et al. RECK forms cowbell-shaped dimers and inhibits matrix metalloproteinase-catalyzed cleavage of fibronectin. J. Biol. Chem. 2009, 284, 3461–3469. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Sáez, E.; Moracho, N.; Learte, A.I.R.; Collignon, A.; Arroyo, A.G.; Noel, A.; Sounni, N.E.; Sánchez-Camacho, C. Molecular Mechanisms Driven by MT4-MMP in Cancer Progression. Int. J. Mol. Sci. 2023, 24, 9944. [Google Scholar] [CrossRef]
- Xu, D.; Dai, R.; Chi, H.; Ge, W.; Rong, J. Long Non-Coding RNA MEG8 Suppresses Hypoxia-Induced Excessive Proliferation, Migration and Inflammation of Vascular Smooth Muscle Cells by Regulation of the miR-195-5p/RECK Axis. Front. Mol. Biosci. 2021, 8, 697273. [Google Scholar] [CrossRef]
- Noda, M.; Takahashi, C. Recklessness as a hallmark of aggressive cancer. Cancer Sci. 2007, 98, 1659–1665. [Google Scholar] [CrossRef]
- Matsuzaki, T.; Keene, D.R.; Nishimoto, E.; Noda, M. Reversion-inducing cysteine-rich protein with Kazal motifs and MT1-MMP promote the formation of robust fibrillin fibers. J. Cell. Physiol. 2021, 236, 1980–1995. [Google Scholar] [CrossRef]
- Kurzepa, J.; Mdro, A.; Czechowska, G.; Kurzepa, J.; Celiński, K.; Kazmierak, W.; Slstrokomka, M. Role of MMP-2 and MMP-9 and their natural inhibitors in liver fibrosis, chronic pancreatitis and non-specific inflammatory bowel diseases. Hepatobiliary Pancreat. Dis. Int. 2014, 13, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Mendes, S.R.; del Amo-Maestro, L.; Marino-Puertas, L.; de Diego, I.; Goulas, T.; Gomis-Rüth, F.X. Analysis of the inhibiting activity of reversion-inducing cysteine-rich protein with Kazal motifs (RECK) on matrix metalloproteinases. Sci. Rep. 2020, 10, 6317. [Google Scholar] [CrossRef]
- Di Pasqua, L.G.; Cagna, M.; Palladini, G.; Croce, A.C.; Cadamuro, M.; Fabris, L.; Perlini, S.; Adorini, L.; Ferrigno, A.; Vairetti, M. FXR agonists INT-787 and OCA increase RECK and inhibit liver steatosis and inflammation in diet-induced ob/ob mouse model of NASH. Liver Int. 2023. [Google Scholar] [CrossRef]
- Hong, K.J.; Wu, D.C.; Cheng, K.H.; Chen, L.T.; Hung, W.C. RECK inhibits stemness gene expression and tumorigenicity of gastric cancer cells by suppressing ADAM-mediated Notch1 activation. J. Cell. Physiol. 2014, 229, 191–201. [Google Scholar] [CrossRef]
- Saad, M.I.; Jenkins, B.J. The protease ADAM17 at the crossroads of disease: Revisiting its significance in inflammation, cancer, and beyond. FEBS J. 2023. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Lin, Y.; Liu, L.; Yang, S.; Lin, Y.Y.; He, J.; Shao, Y. ADAM10-a “multitasker” in sepsis: Focus on its posttranslational target. Inflamm. Res. 2023, 72, 395–423. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Kim, K.J.; Wang, X.; Bartolome, A.; Salomao, M.; Dongiovanni, P.; Meroni, M.; Graham, M.J.; Yates, K.P.; Diehl, A.M.; et al. Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med. 2018, 10, eaat0344. [Google Scholar] [CrossRef]
- D’Souza, B.; Miyamoto, A.; Weinmaster, G. The many facets of Notch ligands. Oncogene 2008, 27, 5148–5167. [Google Scholar] [CrossRef] [PubMed]
- Zolkiewska, A. ADAM proteases: Ligand processing and modulation of the Notch pathway. Cell. Mol. Life Sci. 2008, 65, 2056–2068. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, B.; Banerjee, S.; Paranjpe, S.; Koral, K.; Mars, W.M.; Stoops, J.W.; Orr, A.; Bowen, W.C.; Locker, J.; Michalopoulos, G.K. Pharmacologic Inhibition of Epidermal Growth Factor Receptor Suppresses Nonalcoholic Fatty Liver Disease in a Murine Fast-Food Diet Model. Hepatology 2019, 70, 1546–1563. [Google Scholar] [CrossRef]
- Liang, D.; Chen, H.; Zhao, L.; Zhang, W.; Hu, J.; Liu, Z.; Zhong, P.; Wang, W.; Wang, J.; Liang, G. Inhibition of EGFR attenuates fibrosis and stellate cell activation in diet-induced model of nonalcoholic fatty liver disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 133–142. [Google Scholar] [CrossRef]
- Robert, S.; Gicquel, T.; Bodin, A.; Lagente, V.; Boichot, E. Characterization of the MMP/TIMP imbalance and collagen production induced by IL-1 β or TNF-α release from human hepatic stellate cells. PLoS ONE 2016, 11, e0153118. [Google Scholar] [CrossRef]
- Russell, J.J.; Grisanti, L.A.; Brown, S.M.; Bailey, C.A.; Bender, S.B.; Chandrasekar, B. Reversion inducing cysteine rich protein with Kazal motifs and cardiovascular diseases: The RECKlessness of adverse remodeling. Cell. Signal. 2021, 83, 106–115. [Google Scholar] [CrossRef]
- Clark, J.C.M.; Thomas, D.M.; Choong, P.F.M.; Dass, C.R. RECK—A newly discovered inhibitor of metastasis with prognostic significance in multiple forms of cancer. Cancer Metastasis Rev. 2007, 26, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Palladini, G.; Ferrigno, A.; Rizzo, V.; Boncompagni, E.; Richelmi, P.; Freitas, I.; Perlini, S.; Vairetti, M. Lobe-specific heterogeneity and matrix metalloproteinase activation after ischemia/reperfusion injury in rat livers. Toxicol. Pathol. 2012, 40, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Dejonckheere, E.; Vandenbroucke, R.E.; Libert, C. Matrix metalloproteinases as drug targets in ischemia/reperfusion injury. Drug Discov. Today 2011, 16, 762–778. [Google Scholar] [CrossRef]
- Wang, H.; Imamura, Y.; Ishibashi, R.; Chandana, E.P.S.; Yamamoto, M.; Noda, M. The Reck tumor suppressor protein alleviates tissue damage and promotes functional recovery after transient cerebral ischemia in mice. J. Neurochem. 2010, 115, 385–398. [Google Scholar] [CrossRef]
- Ferrigno, A.; Di Pasqua, L.G.; Palladini, G.; Berardo, C.; Verta, R.; Richelmi, P.; Perlini, S.; Collotta, D.; Collino, M.; Vairetti, M. Transient expression of reck under hepatic ischemia/reperfusion conditions is associated with mapk signaling pathways. Biomolecules 2020, 10, 747. [Google Scholar] [CrossRef]
- Jeon, H.W.; Lee, K.-J.; Lee, S.H.; Kim, W.-H.; Lee, Y.M. Attenuated expression and function of the RECK tumor suppressor under hypoxic conditions is mediated by the MAPK signaling pathways. Arch. Pharm. Res. 2011, 34, 137–145. [Google Scholar] [CrossRef]
- Tüfek, A.; Tokgöz, O.; Aliosmanoglu, I.; Alabalik, U.; Evliyaoglu, O.; Çiftçi, T.; Güzel, A.; Yildirim, Z.B. The protective effects of dexmedetomidine on the liver and remote organs against hepatic ischemia reperfusion injury in rats. Int. J. Surg. 2013, 11, 96–100. [Google Scholar] [CrossRef]
- Palladini, G.; Cagna, M.; Di Pasqua, L.G.; Adorini, L.; Croce, A.C.; Perlini, S.; Ferrigno, A.; Berardo, C.; Vairetti, M. Obeticholic Acid Reduces Kidney Matrix Metalloproteinase Activation Following Partial Hepatic Ischemia/Reperfusion Injury in Rats. Pharmaceuticals 2022, 15, 524. [Google Scholar] [CrossRef]
- Peng, X.; Wu, W.; Zhu, B.; Sun, Z.; Ji, L.; Ruan, Y.; Zhou, M.; Zhou, L.; Gu, J. Activation of farnesoid X receptor induces RECK expression in mouse liver. Biochem. Biophys. Res. Commun. 2014, 443, 211–216. [Google Scholar] [CrossRef]
- Wang, Y.D.; Chen, W.D.; Moore, D.D.; Huang, W. FXR: A metabolic regulator and cell protector. Cell Res. 2008, 18, 1087–1095. [Google Scholar] [CrossRef]
- Ferrigno, A.; Palladini, G.; Di Pasqua, L.G.; Berardo, C.; Richelmi, P.; Cadamuro, M.; Fabris, L.; Perlini, S.; Adorini, L.; Vairetti, M. Obeticholic acid reduces biliary and hepatic matrix metalloproteinases activity in rat hepatic ischemia/reperfusion injury. PLoS ONE 2020, 15, e0238543. [Google Scholar] [CrossRef] [PubMed]
- Gofton, C.; Upendran, Y.; Zheng, M.H.; George, J. MAFLD: How is it different from NAFLD? Clin. Mol. Hepatol. 2023, 29, S17–S31. [Google Scholar] [CrossRef] [PubMed]
- Pipitone, R.M.; Ciccioli, C.; Infantino, G.; La Mantia, C.; Parisi, S.; Tulone, A.; Pennisi, G.; Grimaudo, S.; Petta, S. MAFLD: A multisystem disease. Ther. Adv. Endocrinol. Metab. 2023, 14, 20420188221145549. [Google Scholar] [CrossRef] [PubMed]
- Song, B.G.; Choi, S.C.; Goh, M.J.; Kang, W.; Sinn, D.H.; Gwak, G.Y.; Paik, Y.H.; Choi, M.S.; Lee, J.H.; Paik, S.W. Metabolic dysfunction-associated fatty liver disease and the risk of hepatocellular carcinoma. JHEP Rep. Innov. Hepatol. 2023, 5, 100810. [Google Scholar] [CrossRef]
- Cunningham, R.; Sheldon, R.; Meers, G.; Kandikattu, H.K.; Chandrasekar, B.; Rector, R.S. Western diet feeding downregulates hepatic RECK expression and induces NASH with fibrosis. FASEB J. 2017, 31, 885–887. [Google Scholar] [CrossRef]
- Cunningham, R.P.; Moore, M.P.; Nguyen, N.; Dashek, R.; Boccardi, L.; Jepkemoi, V.; Meers, G.M.; Chandrasekar, B.; Rector, R.S. Hepatic Knockdown of RECK Increases NASH Susceptibility. FASEB J. 2019, 33, 582–585. [Google Scholar] [CrossRef]
- Palladini, G.; Di Pasqua, L.G.; Cagna, M.; Croce, A.C.; Perlini, S.; Mannucci, B.; Profumo, A.; Ferrigno, A.; Vairetti, M. MCD Diet Rat Model Induces Alterations in Zinc and Iron during NAFLD Progression from Steatosis to Steatohepatitis. Int. J. Mol. Sci. 2022, 23, 6817. [Google Scholar] [CrossRef]
- Dashek, R.J.; Diaz, C.J.; Chandrasekar, B.; Rector, R.S. A Mechanistic Role for RECK in the Regulation of Hepatocellular Inflammation. FASEB J. 2022, 36. [Google Scholar] [CrossRef]
- Jalil, A.T.; Abdulhadi, M.A.; Al-Ameer, L.R.; Khaleel, L.A.; Abdulameer, S.J.; Hadi, A.M.; Merza, M.S.; Zabibah, R.S.; Ali, A. Small but mighty: How microRNAs drive the deadly progression of cholangiocarcinoma. Pathol. Res. Pract. 2023, 247, 154565. [Google Scholar] [CrossRef]
- Namwat, N.; Puetkasichonpasutha, J.; Loilome, W.; Yongvanit, P.; Techasen, A.; Puapairoj, A.; Sripa, B.; Tassaneeyakul, W.; Khuntikeo, N.; Wongkham, S. Downregulation of reversion-inducing-cysteine-rich protein with Kazal motifs (RECK) is associated with enhanced expression of matrix metalloproteinases and cholangiocarcinoma metastases. J. Gastroenterol. 2011, 46, 664–675. [Google Scholar] [CrossRef]
- Hirashita, T.; Iwashita, Y.; Ohta, M.; Komori, Y.; Eguchi, H.; Yada, K.; Kitano, S. Expression of Matrix Metalloproteinase-7 is an Unfavorable Prognostic Factor in Intrahepatic Cholangiocarcinoma. J. Gastrointest. Surg. 2012, 16, 842–848. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Zheng, Q. Expression of RECK gene and MMP-9 in hilar cholangiocarcinoma and its clinical significance. J. Huazhong Univ. Sci. Technol. Med. Sci. 2005, 25, 552–554. [Google Scholar] [CrossRef]
- Liu, L.T.; Chang, H.C.; Chiang, L.C.; Hung, W.C. Induction of RECK by nonsteroidal anti-inflammatory drugs in lung cancer cells. Oncogene 2002, 21, 8347–8350. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.C.; Chang, H.C.; Hung, W.C. HER-2/neu represses the metastasis suppressor RECK via ERK and Sp transcription factors to promote cell invasion. J. Biol. Chem. 2006, 281, 4718–4725. [Google Scholar] [CrossRef] [PubMed]
- Loayza-Puch, F.; Yoshida, Y.; Matsuzaki, T.; Takahashi, C.; Kitayama, H.; Noda, M. Hypoxia and RAS-signaling pathways converge on, and cooperatively downregulate, the RECK tumor-suppressor protein through microRNAs. Oncogene 2010, 29, 2638–2648. [Google Scholar] [CrossRef] [PubMed]
- Namwat, N.; Chusorn, P.; Loilome, W.; Techasen, A.; Puetkasichonpasutha, J.; Pairojkul, C.; Khuntikeo, N.; Yongvanit, P. Expression profiles of oncomir miR-21 and tumor suppressor let-7a in the progression of Opisthorchiasis-associated cholangiocarcinoma. Asian Pac. J. Cancer Prev. 2012, 13, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Liu, L.; Liu, C.H.; You, H.; Shao, F.; Xie, F.; Lin, X.S.; Hu, S.Y.; Zhang, C.H. MicroRNA-21 regulates the invasion and metastasis in cholangiocarcinoma and may be a potential biomarker for cancer prognosis. Asian Pac. J. Cancer Prev. 2013, 14, 829–834. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, Y.; Liu, K.; Tan, L. Tumor Cell-Derived Extracellular Vesicles Promote the Growth, Metastasis and Chemoresistance in Cholangiocarcinoma by Delivering microRNA-210 to Downregulate RECK. Mol. Biotechnol. 2023, 65, 1151–1164. [Google Scholar] [CrossRef]
- Zhu, H.; Mi, Y.; Jiang, X.; Zhou, X.; Li, R.; Wei, Z.; Jiang, H.; Lu, J.; Sun, X. Hepatocyte nuclear factor 6 inhibits the growth and metastasis of cholangiocarcinoma cells by regulating miR-122. J. Cancer Res. Clin. Oncol. 2016, 142, 969–980. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Quigley, J.P. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006, 25, 9–34. [Google Scholar] [CrossRef]
- Furumoto, K.; Arii, S.; Mori, A.; Furuyama, H.; Rivas, M.J.G.; Nakao, T.; Isobe, N.; Murata, T.; Takahashi, C.; Noda, M.; et al. RECK gene expression in hepatocellular carcinoma: Correlation with invasion-related clinicopathological factors and its clinical significance. Hepatology 2001, 33, 189–195. [Google Scholar] [CrossRef]
- Zhang, C.; Ling, Y.; Zhang, C.; Xu, Y.; Gao, L.; Li, R.; Zhu, J.; Fan, L.; Wei, L. The silencing of RECK gene is associated with promoter hypermethylation and poor survival in hepatocellular carcinoma. Int. J. Biol. Sci. 2012, 8, 451–458. [Google Scholar] [CrossRef]
- Dong, Z.R.; Chen, Z.Q.; Yang, X.Y.; Ding, Z.N.; Liu, K.X.; Yan, L.J.; Meng, G.X.; Yang, Y.F.; Yan, Y.C.; Yao, S.Y.; et al. RECK expression is associated with angiogenesis and immunogenic Tumor Microenvironment in Hepatocellular Carcinoma, and is a prognostic factor for better survival. J. Cancer 2021, 12, 3827–3840. [Google Scholar] [CrossRef]
- Said, E.M.; Salem, A.A.; Shousha, H.I.; Ahmad, E.S.; Alazzouny, M.A.; Ahmed, I.A.; Elfeky, H.M.; Abdelsalam, F.M. RECK gene polymorphisms in hepatitis B-related hepatocellular carcinoma: A case-control study. Arab. J. Gastroenterol. 2022, 23, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Fakhry, A.B.; Ahmed, A.I.; AbdelAlim, M.A.; Ramadan, D.I. RECK gene promoter rs10814325 polymorphism in Egyptian patients with hepatocellular carcinoma on top of chronic hepatitis c viral infection. Asian Pac. J. Cancer Prev. 2016, 17, 2383–2388. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Lin, Y.; Niu, J.; Cai, L. Association between polymorphisms in tumor suppressor genes and oncogenes and risk of hepatocellular carcinoma: A case-control study in an HCC epidemic area within the Han Chinese population. Med. Oncol. 2014, 31, 356. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.T.; Yeh, C.B.; Li, Y.C.; Su, S.C.; Chien, M.H.; Yang, S.F.; Hsieh, Y.H. Effect of RECK gene polymorphisms on hepatocellular carcinoma susceptibility and clinicopathologic features. PLoS ONE 2012, 7, e33517. [Google Scholar] [CrossRef]
- Nassara, A.K.; El-Toukhya, N.E.-T.R.; Mokhlesa, M.A.; Mohamedb, A.A.; Siddika, R.I.; El-Hanafic, H.; Abd-Elsalamd, S. RECK gene polymorphism in patients with hepatocellular carcinomaNo Title. Meta Gene 2019, 19, 149–154. [Google Scholar] [CrossRef]
- Bahgat, D.M.R.; Shahin, R.M.H.; Makar, N.N.; Aziz, A.O.A.; Hunter, S.S. Reversion-Inducing-Cysteine-Rich Protein with Kazal Motifs (RECK) Gene Single Nucleotide Polymorphism with Hepatocellular Carcinoma: A Case-Control Study. J. Clin. Lab. Anal. 2016, 30, 36–40. [Google Scholar] [CrossRef]
- Abd-Elfatah, G.; Gad-Allah, A.N.A.A. RASSF1A, RECK genotypes and haplotypes in Egyptian population with Hepatocellular carcinoma. Immunol. Lett. 2016, 173, 36–41. [Google Scholar] [CrossRef]
- Bayram, S. RASSF1A Ala133Ser polymorphism is associated with increased susceptibility to hepatocellular carcinoma in a Turkish population. Gene 2012, 498, 264–269. [Google Scholar] [CrossRef]
- Li, Y.; Xu, D.; Bao, C.; Zhang, Y.; Chen, D.; Zhao, F.; Ding, J.; Liang, L.; Wang, Q.; Liu, L.; et al. MicroRNA-135b, a HSF1 target, promotes tumor invasion and metastasis by regulating RECK and EVI5 in hepatocellular carcinoma. Oncotarget 2015, 6, 2421–2433. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yu, J.; Yu, S.; Lavker, R.M.; Cai, L.; Liu, W.; Yang, K.; He, X.; Chen, S. MicroRNA-21 acts as an oncomir through multiple targets in human hepatocellular carcinoma. J. Hepatol. 2010, 53, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, X.; Wei, F.; Zhang, X.; Yu, J.; Zhao, H.; Sun, Q.; Yan, F.; Yan, C.; Li, H.; et al. Diagnostic and prognostic value of circulating miR-21 for cancer: A systematic review and meta-analysis. Gene 2014, 533, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Yang, Z.X.; Song, W.J.; Li, Q.J.; Yang, F.; Wang, D.S.; Zhang, N.; Dou, K.F. MicroRNA-21 regulates the migration and invasion of a stem-like population in hepatocellular carcinoma. Int. J. Oncol. 2013, 43, 661–669. [Google Scholar] [CrossRef]
- Chen, M.; Liu, Y.; Varley, P.; Chang, Y.; He, X.X.; Huang, H.; Tang, D.; Lotze, M.T.; Lin, J.; Tsung, A. High-Mobility Group Box 1 Promotes Hepatocellular Carcinoma Progression through miR-21-Mediated Matrix Metalloproteinase Activity. Cancer Res. 2015, 75, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Löffler, D.; Brocke-Heidrich, K.; Pfeifer, G.; Stocsits, C.; Hackermüller, J.; Kretzschmar, A.K.; Burger, R.; Gramatzki, M.; Blumert, C.; Bauer, K.; et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 2007, 110, 1330–1333. [Google Scholar] [CrossRef]
- Zhang, N.; Duan, W.D.; Leng, J.J.; Zhou, L.; Wang, X.; Xu, Y.Z.; Wang, X.D.; Zhang, A.Q.; Dong, J.H. STAT3 regulates the migration and invasion of a stem-like subpopulation through microRNA-21 and multiple targets in hepatocellular carcinoma. Oncol. Rep. 2015, 33, 1493–1498. [Google Scholar] [CrossRef]
- Zhou, R.; Chen, K.K.; Zhang, J.; Xiao, B.; Huang, Z.; Ju, C.; Sun, J.; Zhang, F.; Lv, X.B.; Huang, G. The decade of exosomal long RNA species: An emerging cancer antagonist. Mol. Cancer 2018, 17, 75. [Google Scholar] [CrossRef]
- Lin, C.; Yang, L. Long Noncoding RNA in Cancer: Wiring Signaling Circuitry. Trends Cell Biol. 2018, 28, 287. [Google Scholar] [CrossRef]
- Zhang, G.; Chen, X.; Ma, L.; Ding, R.; Zhao, L.; Ma, F.; Deng, X. LINC01419 facilitates hepatocellular carcinoma growth and metastasis through targeting EZH2-regulated RECK. Aging 2020, 12, 11071–11084. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Jiang, J. GAS5 Regulates RECK Expression and Inhibits Invasion Potential of HCC Cells by Sponging miR-135b. BioMed Res. Int. 2019, 2019, 2973289. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.; Hu, C.; Yang, B.; Xiao, W.; Zhou, Q.; Li, Y.; Li, Z. Salvianolic acid B targets mortalin and inhibits the migration and invasion of hepatocellular carcinoma via the RECK/STAT3 pathway. Cancer Cell Int. 2021, 21, 654. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Guo, Z.Y.; Chen, Z.T.; Zhi, X.T.; Li, D.K.; Dong, Z.R.; Chen, Z.Q.; Hu, S.Y.; Li, T. TMPRSS4 facilitates epithelial-mesenchymal transition of hepatocellular carcinoma and is a predictive marker for poor prognosis of patients after curative resection. Sci. Rep. 2015, 5, 12366. [Google Scholar] [CrossRef] [PubMed]
- Murugan, R.S.; Uchida, K.; Hara, Y.; Nagini, S. Black tea polyphenols modulate xenobiotic-metabolizing enzymes, oxidative stress and adduct formation in a rat hepatocarcinogenesis model. Free Radic. Res. 2008, 42, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Huang, L.R.; Wohlleber, D.; Reisinger, F.; Jenne, C.N.; Cheng, R.L.; Abdullah, Z.; Schildberg, F.A.; Odenthal, M.; Dienes, H.P.; Van Rooijen, N.; et al. Intrahepatic myeloid-cell aggregates enable local proliferation of CD8 + T cells and successful immunotherapy against chronic viral liver infection. Nat. Immunol. 2013, 14, 574–583. [Google Scholar] [CrossRef]
- Gao, Y.; Nepal, N.; Jin, S.Z. Toll-like receptors and hepatitis C virus infection. Hepatobiliary Pancreat. Dis. Int. 2021, 20, 521–529. [Google Scholar] [CrossRef]
- Peroval, M.Y.; Boyd, A.C.; Young, J.R.; Smith, A.L. A Critical Role for MAPK Signalling Pathways in the Transcriptional Regulation of Toll Like Receptors. PLoS ONE 2013, 8, e51243. [Google Scholar] [CrossRef]
- Panteva, M.; Korkaya, H.; Jameel, S. Hepatitis viruses and the MAPK pathway: Is this a survival strategy? Virus Res. 2003, 92, 131–140. [Google Scholar] [CrossRef]
- Cheung, C.; Luo, H.; Yanagawa, B.; Hon, S.L.; Samarasekera, D.; Lai, J.C.K.; Suarez, A.; Zhang, J.; McManus, B.M. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in coxsackievirus-induced myocarditis. Cardiovasc. Pathol. 2006, 15, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.N.; Deng, Y.F.; Li, R.H.; Yin, P.; Ye, C.S. Concurrent alterations of RAGE, RECK, and MMP9 protein expression are relevant to Epstein-Barr virus infection, metastasis, and survival in nasopharyngeal carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 3245–3254. [Google Scholar] [PubMed]
- Abo El-khair, S.M.; Elalfy, H.; Diasty, M.; Ebrahim, E.E.; Elsamanoudy, A.Z. Methylation degree of metalloproteinase inhibitor RECK gene: Links to RECK protein level and hepatocellular carcinoma in chronic HCV infection patients. J. Biochem. Mol. Toxicol. 2021, 35, e22886. [Google Scholar] [CrossRef] [PubMed]
- Song, S.Y.; Son, H.J.; Nam, E.; Rhee, J.C.; Park, C. Expression of reversion-inducing-cysteine-rich protein with Kazal motifs (RECK) as a prognostic indicator in gastric cancer. Eur. J. Cancer 2006, 42, 101–108. [Google Scholar] [CrossRef]
- van der Jagt, M.F.P.; Sweep, F.C.G.J.; Waas, E.T.; Hendriks, T.; Ruers, T.J.M.; Merry, A.H.H.; Wobbes, T.; Span, P.N. Correlation of reversion-inducing cysteine-rich protein with kazal motifs (RECK) and extracellular matrix metalloproteinase inducer (EMMPRIN), with MMP-2, MMP-9, and survival in colorectal cancer. Cancer Lett. 2006, 237, 289–297. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Compound | Mechanism | Disease |
|---|---|---|
| FXR agonist | Decreased MMPs and ADAMs | I/R, NAFLD/NASH |
| NSAIDs (aspirin) | Inhibited MMP activity | CCA |
| Salvianolic acid B | Inhibition STAT3 pathway; downregulation of MMP-2 and MMP-9 | HCC |
| Polyphenon-B | Decrease MMPs, TIMPs, and angiogenesis | HCC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palladini, G.; Di Pasqua, L.G.; Croce, A.C.; Ferrigno, A.; Vairetti, M. Recent Updates on the Therapeutic Prospects of Reversion-Inducing Cysteine-Rich Protein with Kazal Motifs (RECK) in Liver Injuries. Int. J. Mol. Sci. 2023, 24, 17407. https://doi.org/10.3390/ijms242417407
Palladini G, Di Pasqua LG, Croce AC, Ferrigno A, Vairetti M. Recent Updates on the Therapeutic Prospects of Reversion-Inducing Cysteine-Rich Protein with Kazal Motifs (RECK) in Liver Injuries. International Journal of Molecular Sciences. 2023; 24(24):17407. https://doi.org/10.3390/ijms242417407
Chicago/Turabian StylePalladini, Giuseppina, Laura Giuseppina Di Pasqua, Anna Cleta Croce, Andrea Ferrigno, and Mariapia Vairetti. 2023. "Recent Updates on the Therapeutic Prospects of Reversion-Inducing Cysteine-Rich Protein with Kazal Motifs (RECK) in Liver Injuries" International Journal of Molecular Sciences 24, no. 24: 17407. https://doi.org/10.3390/ijms242417407
APA StylePalladini, G., Di Pasqua, L. G., Croce, A. C., Ferrigno, A., & Vairetti, M. (2023). Recent Updates on the Therapeutic Prospects of Reversion-Inducing Cysteine-Rich Protein with Kazal Motifs (RECK) in Liver Injuries. International Journal of Molecular Sciences, 24(24), 17407. https://doi.org/10.3390/ijms242417407