1. Introduction
Molecular structures are highly dynamic. The same also holds for biological macromolecules governing the complex vital processes in living organisms. From the helical structures of nucleic acids to the intricate folds of proteins, the three-dimensional architectures of biomolecules are intertwined with the microscopic origins of life. A plethora of both experimental and theoretical studies have consistently demonstrated that biological macromolecules exist as a large ensemble of conformations, rather than adhering to a single static structure [
1,
2,
3]. Small potential barriers between conformations result in continuous thermal fluctuations across various structures, imparting flexibility to biological macromolecules [
4,
5]. For proteins, this dynamic nature gives rise to the concept ‘protein dynamics’, a topic that has become a notable agenda in the field of structural biology [
4,
6]. Flexibility in biological macromolecules grants a physiological adaptability essential to the function of biological macromolecules [
7,
8,
9,
10,
11,
12,
13]. Representatively, flexible structures facilitate the interactions between different molecules that are essential for processes such as molecular recognition [
4,
11]. For instance, structural flexibility accounts for the folding of ribonucleic acids (RNAs) [
10] or the protein–nucleic acid recognition [
7,
8]. Furthermore, the capacity of proteins to adapt to the external environment stems from structural fluctuations [
13]. Accordingly, a comprehensive understanding of these dynamic features in biological macromolecules is essential for unraveling the complex molecular interplay of life.
Biological macromolecules can adopt multiple structural conformations, each of which may potentially contribute to the physiological functions in vivo. Therefore, to scrutinize the intricate relationship between the physiological function of a molecule and its dynamic structure, it is largely desirable to capture all conformations. On the other hand, traditional structure determination techniques, such as X-ray diffraction (XRD) [
12,
14,
15,
16], small-angle X-ray scattering (SAXS) [
17,
18,
19,
20], and nuclear magnetic resonance (NMR) [
21,
22], predominantly capture time-averaged molecular conformations. In these techniques, the structural information is derived from a huge ensemble of biological molecules, each existing in a distinct conformation. If the molecular structure is rigid, meaning that all molecules within the ensemble adopt a single, identical conformation, then these techniques can be employed to retrieve the structure with high spatial resolutions. However, when dealing with biological macromolecules that exhibit structural flexibility, the observables measured from the ensemble average may not accurately represent those of the individual molecules within the ensemble. Thus, directly retrieving the full spectrum of possible structural conformations of the molecule is of paramount importance, yet it presents significant challenges.
To overcome these limitations, a myriad of cutting-edge techniques has been proposed in the fields of XRD [
23,
24,
25], cryo-electron microscopy (cryo-EM) [
26,
27,
28,
29,
30,
31,
32], NMR [
10,
22,
33,
34], and optical spectroscopy [
35,
36,
37,
38,
39]. Notably, single-molecule spectroscopy and nanoparticle-assisted cryo-electron microscopy sampling (NACS) [
27,
32] have emerged as viable strategies for capturing single-molecule structures, each presenting distinct benefits and limitations. Single-molecule spectroscopy is invaluable for capturing molecular behaviors and interactions at the individual molecule level, thus shedding light on the complex and dynamic processes of biomolecules. Techniques such as single-molecule FRET (smFRET) [
36,
37,
38] can elucidate dynamic protein structural ensembles in solution by measuring distances between fluorophores. However, the distance limitation of less than 10 nm for typical fluorophores and the influence of fluorophore orientation can complicate interpretations. NACS has been developed as a complementary approach. NACS utilizes gold nanoparticles (AuNPs) to enhance the visualization of protein conformational distributions. By labeling two distinct sites on a target sample, NACS measures distance distributions, leveraging the principles of cryo-EM. However, it is important to note that cryo-EM involves flash freezing molecules, and the freezing process typically takes several nanoseconds to tens of nanoseconds, during which time molecules may undergo structural rearrangements. This raises questions about the ability of cryo-EM, and by extension NACS, to accurately capture the “instantaneous” conformation of molecules, particularly those that are highly unstable and rarely populated. By contrast, our study employs a novel experimental scheme, called single-object scattering sampling (SOSS) [
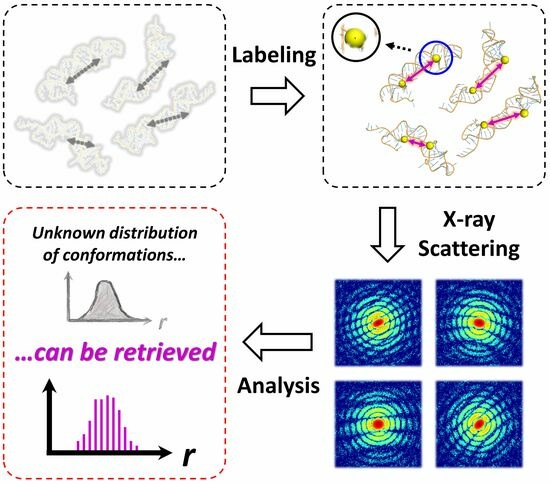
25], which is based on single-molecule X-ray diffraction (
Figure 1a). This technique is tailored to capture and identify individual structural conformations of dynamic biological macromolecules. The SOSS procedure involves irradiating an isolated molecule with a coherent, ultrashort, and high-intensity X-ray pulse. The duration of this X-ray pulse is significantly shorter than the time scale of the typical molecular structural fluctuations, allowing the pulse to capture the conformation of the molecule at a specific instant, as if “freezing” the molecule in time. A repeated measurement of the instantaneous structures allows for sampling all possible structural conformations of the molecule. The relative abundance of each conformation, as determined by repetitive sampling, is then used to reconstruct the population distribution of the structural conformations. In light of these capabilities, SOSS emerges as a promising technique, holding significant potential for investigating the dynamic features of the biological macromolecules.
Nevertheless, two challenges remain in implementing SOSS on biomolecules. First, one should obtain the single molecule scattering snapshot with sufficiently high signal-to-noise ratios in order to retrieve the structural information from the snapshot. However, it is challenging to obtain a scattering image of a large biomolecule that is strong enough to provide three-dimensional structure information with the flux of incident photons from currently available X-ray sources, including the state-of-the-art X-ray free-electron lasers (XFELs) [
24,
40,
41,
42]. In addition, for macromolecules such as proteins and nucleic acids, their complex three-dimensional structures, which are composed of more than thousands of atoms, make it practically impossible to extract the structure from a single two-dimensional scattering image regardless of the strength of the scattering signal. One compromise solution to overcome these two challenging obstacles would be the site-directed labeling of biological macromolecules using metal nanoparticles [
43,
44]. Utilizing nanoparticles composed of heavy elements facilitates the acquisition of clear single-shot single-molecule scattering snapshots [
45]. This is particularly useful when two nanoparticles are labeled at distinct sites on a biomolecule, as the distance between these two labeled sites can be routinely determined from the strong interference pattern observed in the scattering snapshot (
Figure 1b). Consequently, the site-directed labeling of two nanoparticles enables the extraction of a crucial instantaneous structural parameter—the distance between two labeled sites—from a single scattering snapshot. In this work, we perform proof-of-concept simulations of the SOSS method on nanoparticle-labeled biological macromolecules, a technique denoted as Bio-SOSS. Through these simulations, we demonstrate the feasibility and the potential of Bio-SOSS as a novel approach to probing the dynamic nature of biological molecules.
2. Results and Discussion
We present a proof-of-concept of a novel algorithm designed to accurately retrieve the distribution of three-dimensional (3D) interatomic distances within a fluctuating single-stranded ribonucleic acid (ssRNA) chain comprising 2255 atoms (
Figure 2a). Each nucleotide in the ssRNA is composed of a ribose sugar, a phosphate group, and one of four bases: adenine (A), cytosine (C), guanine (G), and uracil (U). Owing to its dynamic nature, the structure of the ssRNA incessantly fluctuates, resulting in time-dependent variations in the distances between any two nucleosides of interest. These structural fluctuations were sampled in a total of 100 snapshots generated through molecular dynamics (MD) simulations. For each snapshot, we performed labeling of two selected nucleotides on the RNA with gold nanoparticles (AuNPs). The two sites for labeling were carefully selected to prevent any potential collisions with other atoms in the RNA chain. The details of this selection process are comprehensively described in the
Section 3. Consequently, we conducted simulations for a total of 18 pairs of labeling sites. For each snapshot, we generated the single-shot single-molecule X-ray scattering pattern for the RNA labeled with AuNPs. To generate the patterns, we assumed a hypothetical X-ray scattering setup with a two-dimensional (2D) area detector. We then simulated the mock experimental single-object X-ray scattering pattern, S
exp, as detected by the area detector (
Figure 2). To check the dependence of S
exp and the efficiency of retrieving the inter-site distances on various experimental parameters, S
exp was simulated with varying experimental parameters including the size of the AuNPs, the energy of the X-ray photons, the focal size of the X-ray pulse, and the intensity of the X-ray pulse. By analyzing each scattering pattern, we extracted the 3D distance information (r) between the AuNPs. We assumed that this value corresponds to the distance between the two labeled nucleotides on the RNA, denoted as r
i,j. Repeating this process, we were able to construct a histogram of the distances between the two nucleotides.
An experimental single-object X-ray scattering pattern should contain the interference from all pairs of atoms in the ssRNA as well as AuNPs. In addition, the experimental data contain noise. Considering this, S
exp was generated by adding the simulated single-object X-ray scattered image from the AuNP-labeled ssRNA, S
AuNP–RNA, and the sum of Poisson random noise and uniform random noise at each detector pixel, S
noise (
Figure 2b). The goal is to extract the distance between two AuNPs from the S
exp, while the instant structure of ssRNA at the moment of X-ray scattering is not known. The X-ray scattering intensity from a molecule depends on the form factor of constituent atoms, as well as the effects of molecular structures such as interatomic distances. In the case of the former, the atomic form factor increases with an increase in the number of electrons, or the atomic number. Given that the ssRNA chain is composed exclusively of lighter atoms—phosphorus, oxygen, nitrogen, carbon, and hydrogen—the scattering pattern originating solely from the RNA is weaker in comparison to those inclusive of the considerably heavier gold (Z = 79) nanoparticles, as depicted in
Figure 2c. Hence, the incorporation of AuNP labels significantly amplifies the scattering signal, thereby facilitating the extraction of precise structural information.
More specifically, the signal S
AuNP–RNA can be depicted as follows:
where S
AuNP represents the scattering signal from AuNPs, S
RNA denotes the scattering signal from the RNA, and S
AuNP,1 and S
AuNP,2 correspond to the scattering signals from each of the two individual AuNPs. S
AuNP*RNA represents the scattering cross-term between the AuNPs and the RNA, and S
AuNP, 1*2 denotes the scattering cross-term between one AuNP and the other.
fAuNP,1 and
fAuNP,2 correspond to the scattering amplitude of each of the two individual AuNPs, and
fRNA denotes the scattering amplitude of the RNA. Assuming that the two AuNPs are Identical in terms of their composition and shape, |
fAuNP,1|
2 = |
fAuNP,2|
2. When the scattering signal from the AuNPs is significantly stronger than that from the RNA, that is, |
fAuNP,1| >> |
fRNA|, Equation (1) can be approximated as follows:
Our strategy involves the assumption that |fAuNP,1| >> |fRNA|. Consequently, Equation (2) holds true, allowing for accurately approximating the single-object scattering pattern from an AuNP-labeled ssRNA with that from two AuNPs alone.
To test this idea, we first simulated the single-object X-ray scattering images, S
theo, using only the scattering signals from the two AuNPs, S
AuNP, while neglecting the contribution from the fluctuating RNA backbone (
Figure 2c). The resulting S
theo was then compared to S
exp, which is the sum of S
AuNP–RNA and S
noise, with the primary contribution coming from S
AuNP–RNA. The qualitative comparison, as illustrated in
Figure 2b,c, confirms that S
AuNP–RNA can indeed be accurately approximated by S
AuNP, given that the principal oscillating features in these patterns are nearly identical in shapes. To quantitatively validate this observation, we conducted further analysis. In this process, the distance r was iteratively optimized to minimize the sum-of-squares of residual at each detector pixel intensity, |S
exp − S
AuNP|
2, scaled by the error at each pixel. To confirm the validity of our approach, we compared the optimized distance, r
fit, with the actual distance between the two labeled AuNPs, denoted as r
AuNP. Our simulation results validate that the optimized interparticle distances, r
fit, closely mirror the actual distances between the AuNPs, r
AuNP. Consequently, the reconstructed distribution of r
fit, denoted as p(r
fit), obtained through Bio-SOSS, accurately mirrors the true distribution, p(r
AuNP) (
Figure 3). The consistency was observed regardless of the labeling sites.
We quantitatively evaluated the accuracy of the distance retrieval method by calculating the discrepancies between r
fit and r
AuNP at each snapshot (
Figure 4). For this analysis, we gathered 1800 pairs of r
fit and r
AuNP values corresponding to all 18 labeling-site pairs, as analyzing only 100 pairs specific to a particular nucleotide pair would result in insufficient data. The resulting distributions of the 1800 r
fit and r
AuNP values, denoted as p(r
fit) and p(r
AuNP), are plotted and compared in
Figure 4a. A satisfactory agreement between p(r
fit) and p(r
AuNP) can be observed. By examining the distribution of discrepancies between r
fit and r
AuNP, denoted as p(r
fit − r
AuNP), we found that the errors in r-values, Δr
AuNP, follow a Gaussian distribution centered at zero (
Figure 4b). This distribution has a standard deviation of approximately 0.23 Å, demonstrating that the structural retrieval error of Bio-SOSS is within this range.
To provide a more comprehensive analysis, we decomposed both r
fit and r
AuNP into their constituent components, x
fit, y
fit, and z
fit, and x
AuNP, y
AuNP, and z
AuNP, respectively. These components represent the 3D projections of the vector pointing from one AuNP to the other. The comparisons of x
fit and x
AuNP, y
fit and y
AuNP, and z
fit and z
AuNP are depicted in
Figure 4c. Subsequently, we calculated the discrepancies between x
fit and x
AuNP, y
fit and y
AuNP, and z
fit and z
AuNP to obtain Δx, Δy, and Δz. Decomposing the 3D distance r into its x, y, and z directional components elucidates the key factor that governs the accuracy of distance retrieval.
Figure 4d illustrates the distinct widths of the distributions of Δx, Δy, and Δz. It is noteworthy that the width of Δz is comparable to that of Δr
AuNP, indicating that the accuracy in determining the r value is primarily influenced by the accuracy achieved in quantifying the distance along the z-direction. This can be rationalized by considering the richness of information available in the x and y directions, which lie perpendicular to the propagation direction of the X-ray pulse, in comparison to the z-direction. In terms of the momentum transfer vector q, the maximum magnitudes of q in the x- and y-directions, denoted as q
x and q
y, covered by a detector are substantially larger than the component in the z-direction, denoted as q
z. Taking the Fourier-transform relationship between r-space and q-space into account, as the maximum achievable magnitude q in q-space increases, the minimum resolvable r in the r-space decreases. Therefore, due to the limitations in q
z compared to q
x and q
y, the precision in resolving distances in the z-direction is compromised relative to the x and y directions.
The distributions of Δx, Δy, and Δz were quantitatively evaluated, revealing a marked discrepancy in the breadth of Δz compared to Δx and Δy. For this analysis, we employed Gaussian functions to fit the respective distributions and then extracted the center and width (standard deviation) of the optimum Gaussian fits. Consequently, we found that the center values for Δx and Δy were −4.6 × 10
−3 and −6.0 × 10
−5 Å, respectively, which are practically zero. The standard deviations for Δx and Δy were calculated to be 5.3 × 10
−2 and 5.0 × 10
−2 Å, respectively, both residing within the atomic scale. Conversely, the center value and standard deviation for Δz, at 1.8 × 10
−2 and 0.37 Å, respectively, were considerably higher compared to the other two axes. Nevertheless, these values remain smaller relative to the typical pair distances in a macromolecule, which are depicted for the RNA on the horizontal axis of the distribution diagrams for r in
Figure 3 and
Figure 4a. Such findings underscore the potential of Bio-SOSS in accurately delineating the local structural configurations inherent to biological macromolecules.
To comprehensively evaluate the versatility of Bio-SOSS on a wider spectrum of biomolecules, we conducted a quantitative investigation of its accuracy using ten distinct biomolecules (
Figure S1), comprising four RNAs, five proteins, and an RNA–protein complex. Our results demonstrate a notable agreement between the actual distances (r
AuNP) and those retrieved by Bio-SOSS (r
fit), as depicted in
Figures S18–S21. This level of agreement is comparable to that observed for the MD snapshots of the target RNA (PDB ID: 1KXK). Employing the same experimental parameters as those used to obtain the simulation results shown in
Figure 4, the standard deviation of the distance differences, σ(Δr), for these ten biomolecules ranged from 0.097 Å to 0.33 Å (
Tables S1 and S2). In addition, the results suggest a positive correlation between the σ(Δr) value and the size of the biomolecules, which can be characterized either by the molecular weight or the number of residues (
Figure S6). A more detailed discussion on this topic is provided in the “Bio-SOSS on various biomolecules” section of the
Supporting Information.
To determine the optimal conditions for the retrieval of interatomic distances using the Bio-SOSS method, we quantitatively assessed the impact of the experimental parameters on the statistical distributions of ΔrAuNP. Six conditions were varied in the simulations: the size of the AuNPs (R), the wavelength of the X-ray photons (λ), the focal size of the X-ray pulse (f), the intensity of the X-ray pulse (I), the sample-to-detector distance (l), and the number of pixels in the detector (p). It should be emphasized that f and I of the X-ray pulse are interrelated, as they collectively define a key independent variable—the number of X-ray photons per unit area. Despite this interconnection, it is essential to recognize the capacity to independently adjust these parameters during the experimental procedure. In light of this, we investigated the effects of varying the two parameters to comprehensively understand their impact on our results.
Scattering images were generated under a variety of experimental conditions, and from these images, the 3D distances, r
fit, were subsequently extracted. To isolate the influence of each parameter, we systematically varied one parameter at a time while keeping the other parameters constant. The fitted distance, r
fit, for each image was compared to the actual distance, r
AuNP. Subsequently, the distributions of discrepancies between r
fit and r
AuNP, Δr
AuNP = r
fit − r
AuNP, were generated and fitted with a Gaussian function (
Figures S7–S17). The standard deviations of the Gaussian functions obtained from the fitting results, plotted according to conditions, are presented in
Figure 5 and
Figure 6. We decomposed both r
fit and r
AuNP into their respective x, y, and z directional components and performed a comparative analysis. The deviations in these components, denoted as Δx, Δy, and Δz, were quantitatively assessed by fitting their respective distributions to Gaussian functions and subsequently determining the center and standard deviation of the best-fit Gaussian functions. The standard deviation values of Δx, Δy, and Δz, along with the standard deviation values of Δr
AuNP, are collectively displayed in
Figure 5 and
Figure 6.
One of the most crucial parameters governing the accuracy of Bio-SOSS is the size of the AuNPs used for labeling, denoted as
R. To quantify the effect of
R, we traced the dependence of the standard deviation of the difference distribution, σ(Δr), at five distinct
Rs, namely 6, 7.5, 9, 10.5, and 12 Å. We found a monotonic decrease in σ(Δr), or an enhancement in the Bio-SOSS accuracy, as
R increased (
Figure 5a). These results can be attributed to the positive dependence of the AuNP form factor,
fAuNP, on
R (
Figure 5b), which qualitatively accords with the tendency in
Figure 5a. As
R increased, the relative contribution of X-ray scatterings from AuNPs became more significant compared to those from the fluctuating RNA backbone. On the other hand, it is noteworthy that when
R became large, a disparity between the two AuNPs and the actual distance between the two labeled sites on the biomolecule also arose. This potentially poses negative impacts on the accuracy of Bio-SOSS. Furthermore, for the practical implementation of Bio-SOSS, it is essential to consider both chemical and physical interactions between the AuNPs and the target biomolecule to be labeled. Larger nanoparticles, particularly those with substantial surface charges, are prone to stronger interactions with the target biomolecule, potentially leading to distortions in its native conformation [
46]. Since the primary objective of the Bio-SOSS method is to retrieve the native conformation of the biomolecule, rather than the altered conformation due to AuNPs, it is generally less preferable to use nanoparticles of large sizes during the experimental labeling process. In this regard, the choice of metal for nanoparticle labeling can also be of critical importance. For example, AuNPs offer several advantages: (1) they can be synthesized in small sizes, (2) their surface charges can be tailored to desired levels, and (3) they exhibit weak oxidative properties. Collectively, these characteristics make AuNPs superior candidates for Bio-SOSS applications. It is important to note that our simulated labeling process and the results presented in
Figure 5 do not take these chemical and physical interactions into account. Instead, our primary aim is to demonstrate the Bio-SOSS method’s capability to retrieve distances in biomolecules, regardless of whether the labeled molecule undergoes denaturation. In practice, the successful application of Bio-SOSS relies on selecting an optimal AuNP size that strikes a balance between the effects of scattering interactions, as discussed in this paragraph (where a larger
R is advantageous due to the increased scattering contribution from the AuNPs), and the effects of chemical and physical interactions (where a smaller
R is preferable to minimize distortions from the native conformation).
The X-ray wavelength, or energy, used for the scattering experiment directly determines the maximum range of q (q
max) that encrypts the structural information of a molecule. Since the reciprocal (q) and radial (r) spaces are interrelated by Fourier transformation, the scattering data with larger q improve the accuracy of structural retrieval at the entire radial space. Therefore, it is commonly assumed that a higher X-ray energy is more beneficial in X-ray scattering experiments. However, our observation in
Figure 6a counters this intuition, as the standard deviation of Δr, σ(Δr), increases as the X-ray energy increases. In other words, using an X-ray at higher energy is proven to be disadvantageous for the accuracy of the Bio-SOSS. To assess these results, we calculated the radial resolution limit r
lim at each X-ray condition by implementing the well-known relationship r
lim = 2π/q
max, and obtained: 6.7, 4.4, 3.3, 2.2, 1.7, 1.3, and 1.1 Å (at 3, 4.5, 6, 9, 12, 15, and 18 keV, respectively). All of these values are notably larger than the extracted standard deviations of Δr, implying that the additional information obtained from the lengthened q range is not crucial to the functioning of the Bio-SOSS structure. On the other hand, when we computed the dq value corresponding to a single pixel at the center of the detector, we obtained the values 0.009, 0.013, 0018, 0.027, 0.036, 0.044, and 0.053 Å
−1 at 3, 4.5, 6, 9, 12, 15, and 18 keV, respectively, which are increasing according to the X-ray energy. Therefore, we conclude that the primary factor determining the statistical accuracy of Bio-SOSS is not the maximum range of q-space but rather the q-space resolution corresponding to a single pixel in the detector. It is also expected from the form factors of an AuNP (
Figure 5b) where
fAuNP(q) at smaller qs are notably greater by several orders in a logarithmic scale than those in larger qs. To systematically look into the impacts of the two countering effects, we investigated the statistical dependence of the Bio-SOSS accuracy on the size of a 2D area detector. Considering that the pixel size is kept at a constant value of 234 μm, a larger detector increases the maximum available q range while maintaining the single-pixel dq as constant.
Figure 6b confirms that the standard deviation of Δr remained almost constant as the number of detector pixels increases. This observation coincides with our hypothesis that the single-pixel dq, rather than the maximum q value, primarily decides the accuracy of Bio-SOSS.
We also observed how σ(Δr) depends on
I (
Figure 6c). A straightforward tendency was observed up to 10
16 photons, where brighter X-ray reduced σ(Δr), improving the accuracy of the Bio-SOSS. On the other hand, when we used an ultra-intense X-ray with a 10
17 photon count, the accuracy of the structure retrieval between the two AuNPs significantly worsened. This is because the scattering image from these ultra-intense X-ray sources is sensitive not only to the distance between two AuNPs but also to the delicate structures of RNA. Thus, at these high fluences, we expect that the structural resolution of the RNA backbone becomes high enough to resolve the light atoms at that cost of reducing the distance retrieval accuracy between the two AuNPs. Nevertheless, considering that a typical X-ray photon flux for the X-ray scattering experiments at X-ray linear free electron lasers (XFELs) lies between 10
12 and 10
13, we can assume that the accuracy of Bio-SOSS will increase at stronger fluences within the experimental conditions available in near future.
The
f at the point of interaction also plays a critical role in the spatial resolution of the reconstructed molecular structure. If the X-ray pulses are spatially broad, the intensity per unit area diminishes and the overall scattering intensity decreases. On the other hand, if the X-ray pulses are too tightly focused, the scattering contribution from the RNA backbone and other minor distances becomes non-negligible. In this case, the X-ray scattering images contain mixed structural information not only from the two AuNPs but also from the RNA backbone, as in the case where we use the X-ray pulses with 10
17 photon flux. The standard deviation of Δr as a function of the X-ray focal size (
Figure 6d,e) portrays the combined effect of these two contradicting dependencies, leading to a U-shaped optimal range of focal sizes from 3.2 to 100 nm. Finally, the trend of σ(Δr) with respect to
l (
Figure 6f) shows a combined effect of (1) maximum q, (2) single-pixel dq, and (3) X-ray intensity per unit area. As
l lengthens, all of these three dependent variables decrease. Here, the increase in single-pixel dq resolution accounts for the initial enhancement in σ(Δr) from 20 to 40 mm while the reduction in unit X-ray intensity predominantly contributes to the decay of the Bio-SOSS accuracy from 40 mm to 80 mm.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





