
Transcriptomic and Metabolomic Analyses Reveal the Importance of Lipid Metabolism and Photosynthesis Regulation in High Salinity Tolerance in Barley (Hordeum vulgare L.) Leaves Derived from Mutagenesis Combined with Microspore Culture

 ,
,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
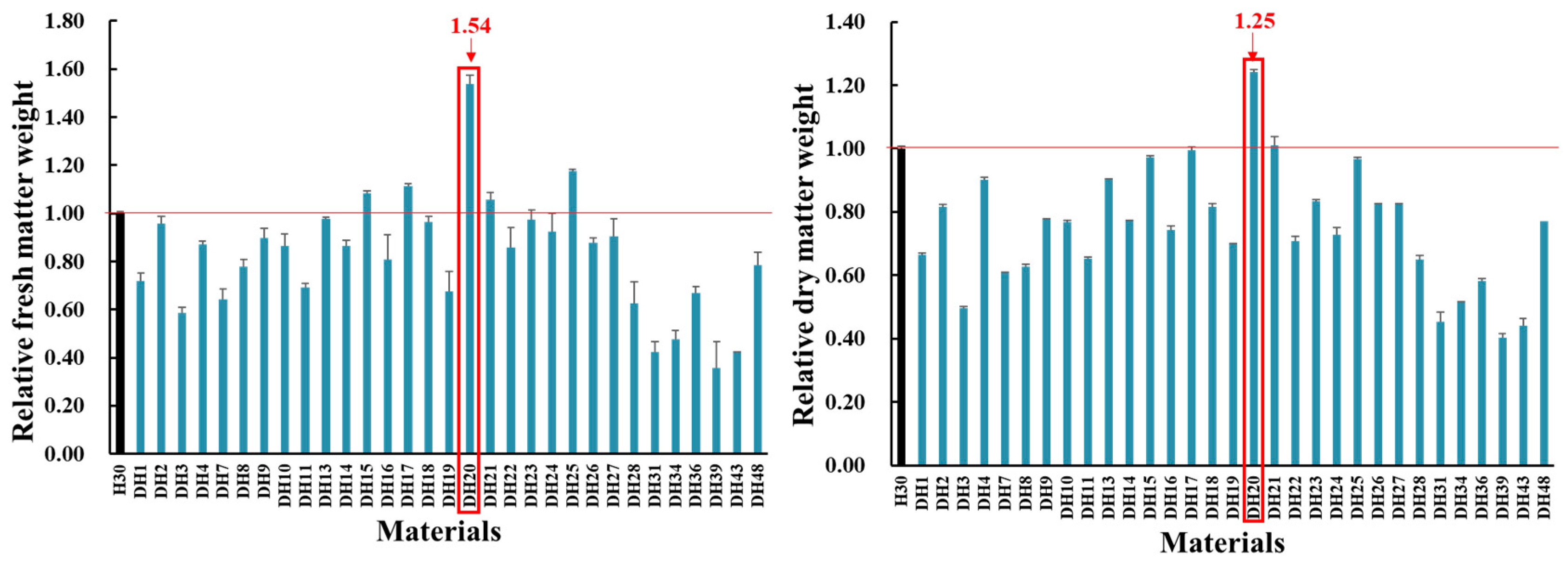
2.1. Identification of DH20 with Higher Salt Tolerance than H30 by Screening DH Lines Produced by Mutagenesis Combined with Isolated Microspore Culture
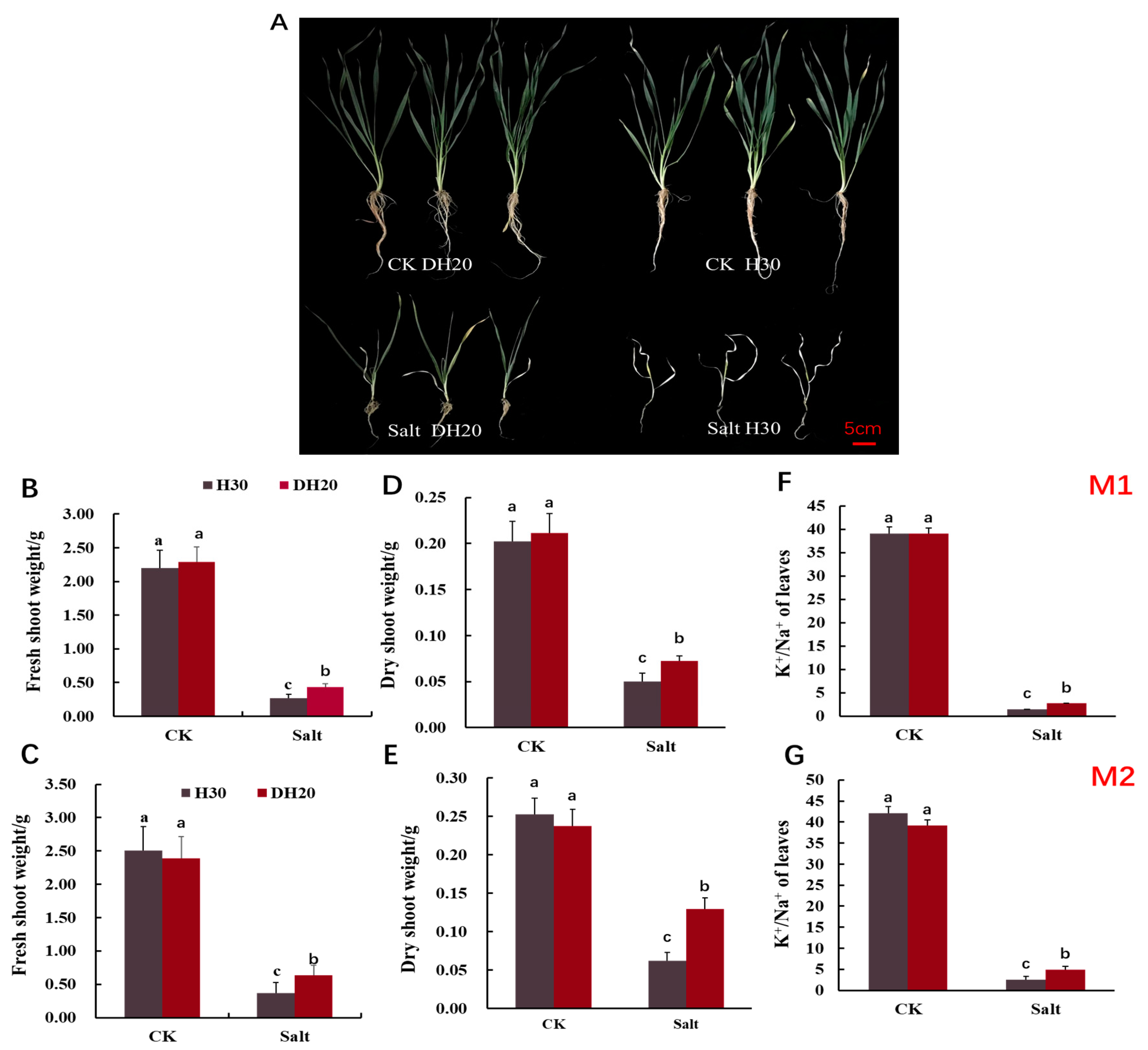
2.2. Comparison of Phenotype and K+/Na+ Ratio between H30 and DH20 under Control and Salt Conditions
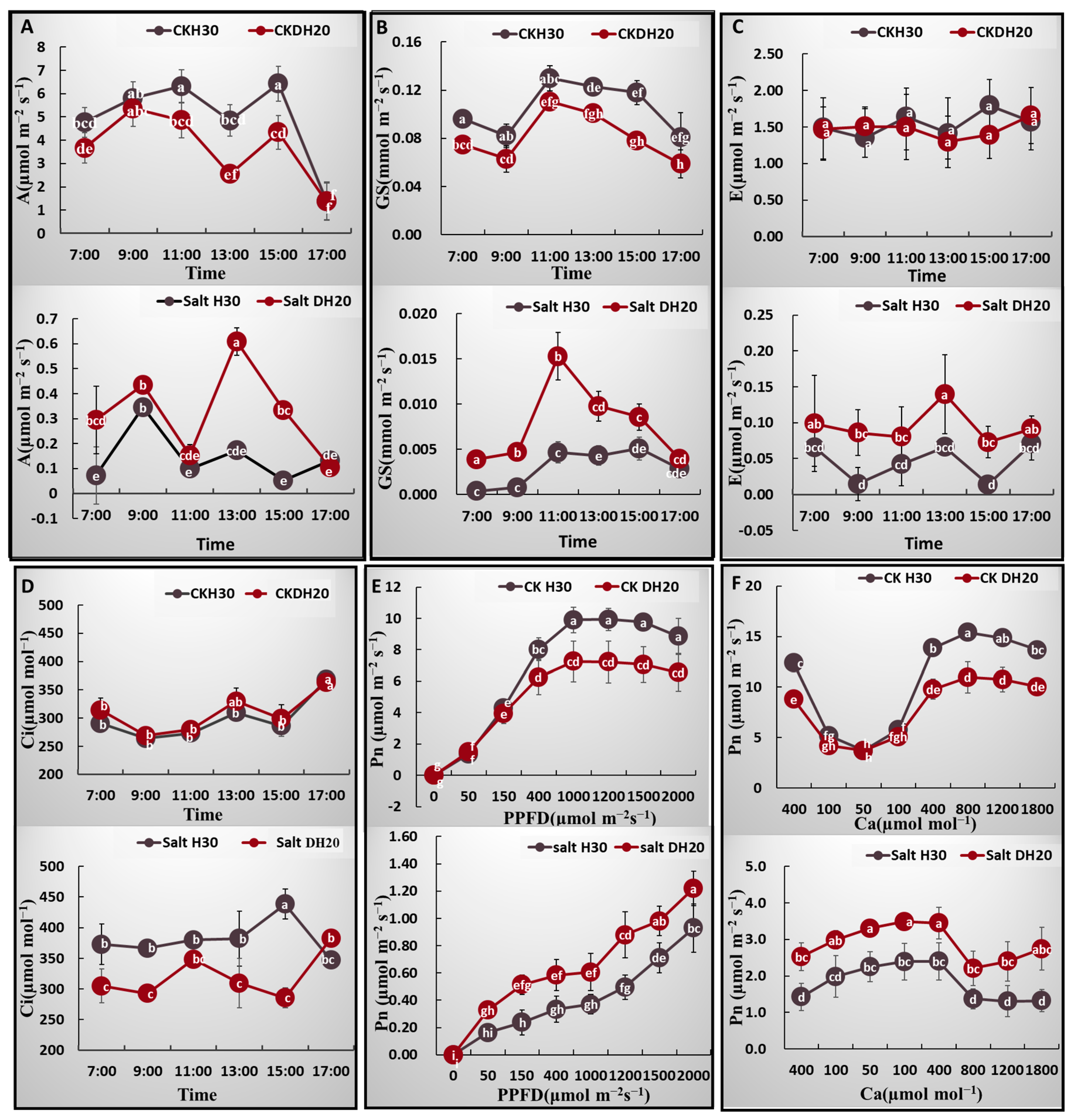
2.3. Comparison of Photosynthetic Characteristics between H30 and DH20 under Control and Salt Conditions
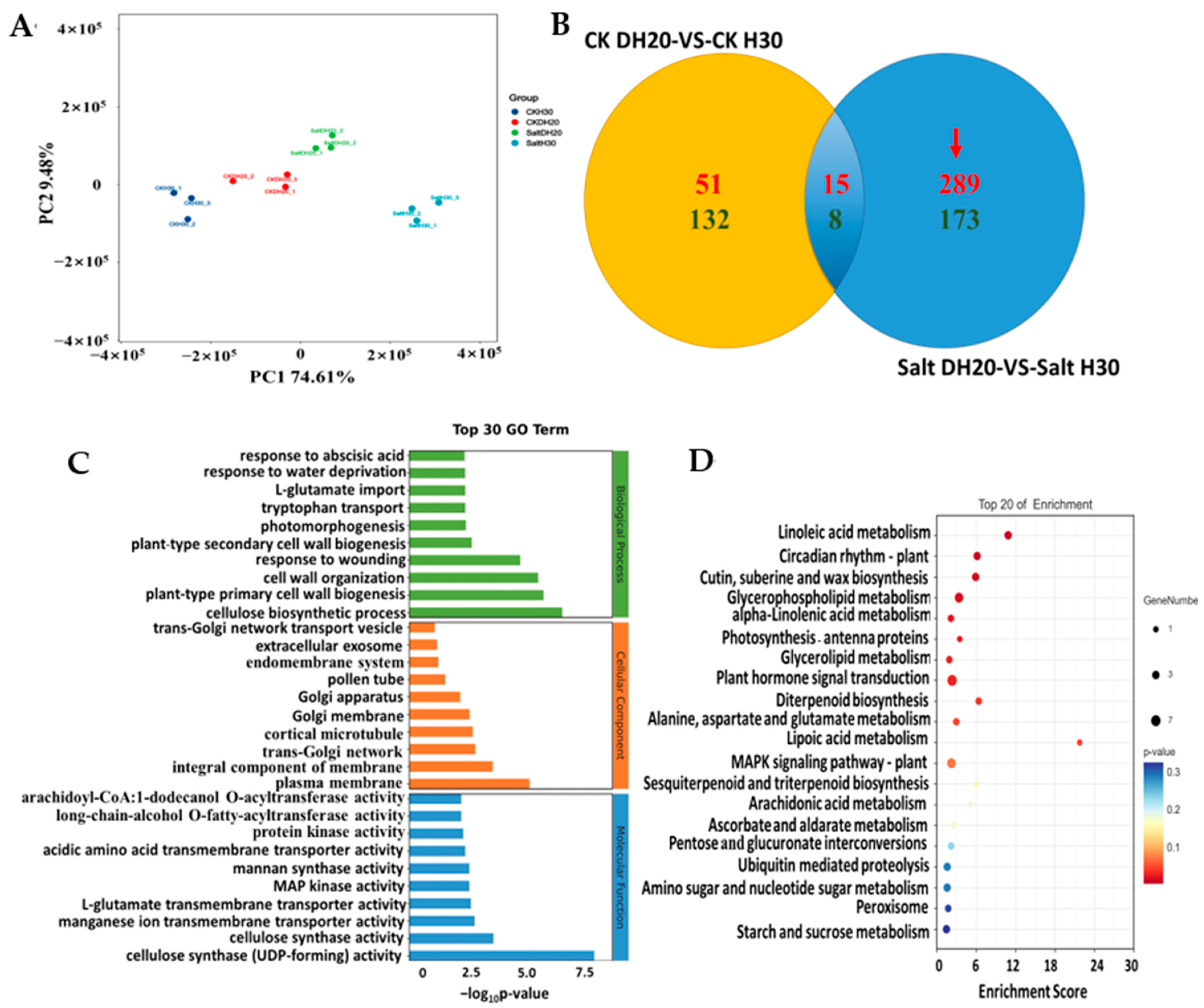
2.4. Differentially Expressed Genes of DH20 in Response to Salt Stress
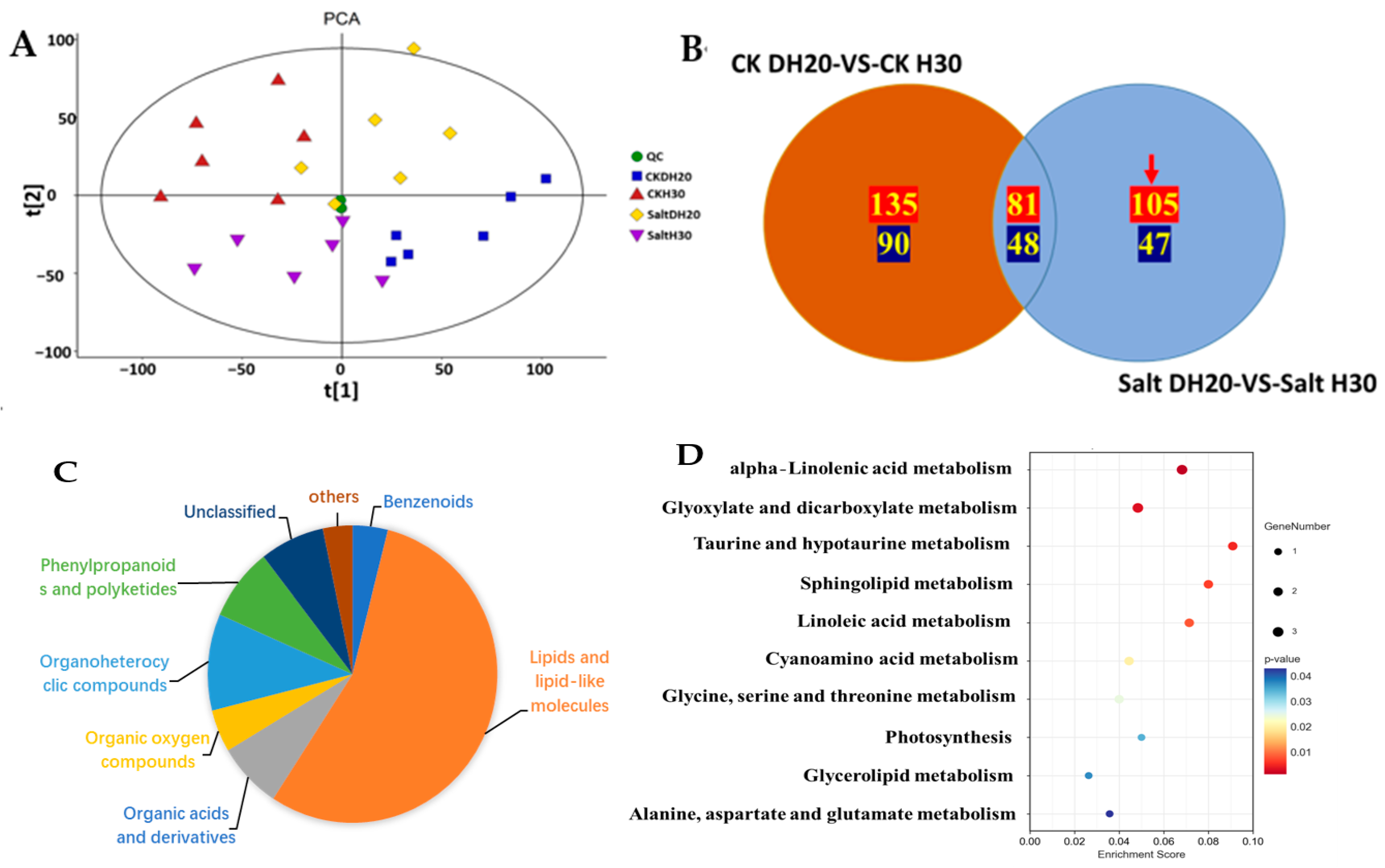
2.5. Differentially Abundant Metabolites of DH20 in Response to Salt Stress
2.6. Association Analysis of DEGs and DAMs in Response to Salt Stress
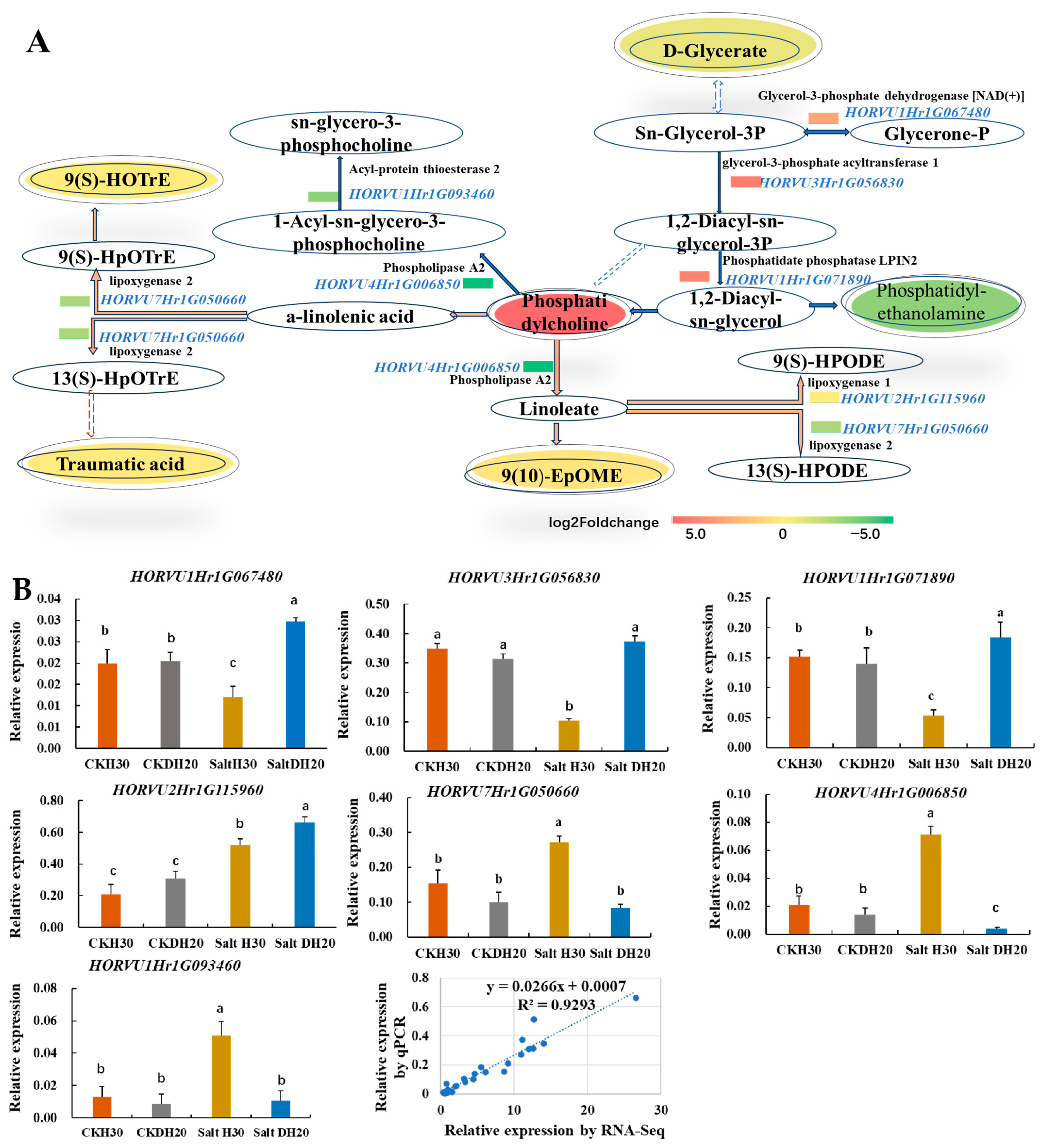
2.7. Improving Phosphatidylcholine in the Lipid Metabolism of DH20 in Response to Salt Stress
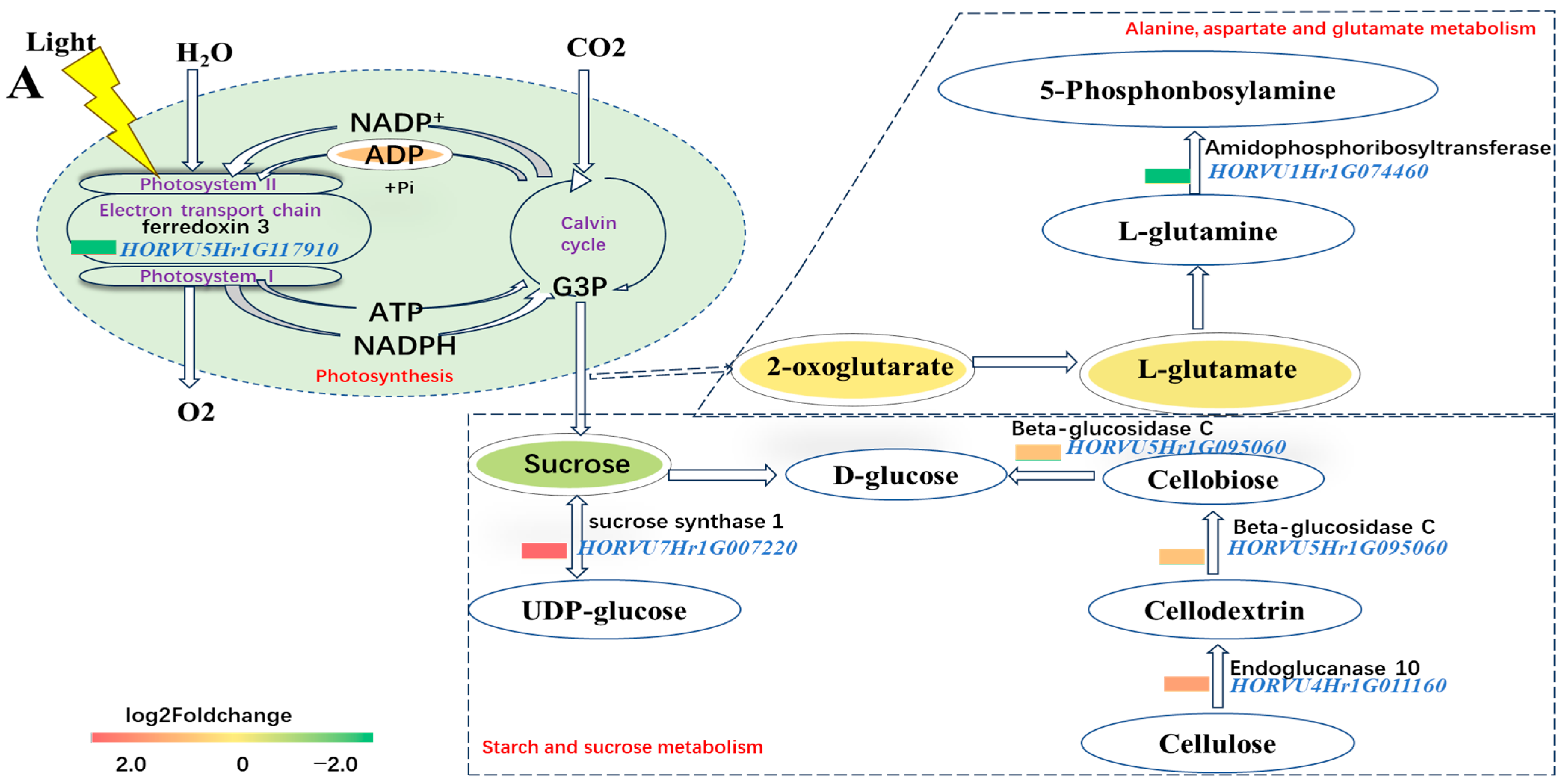
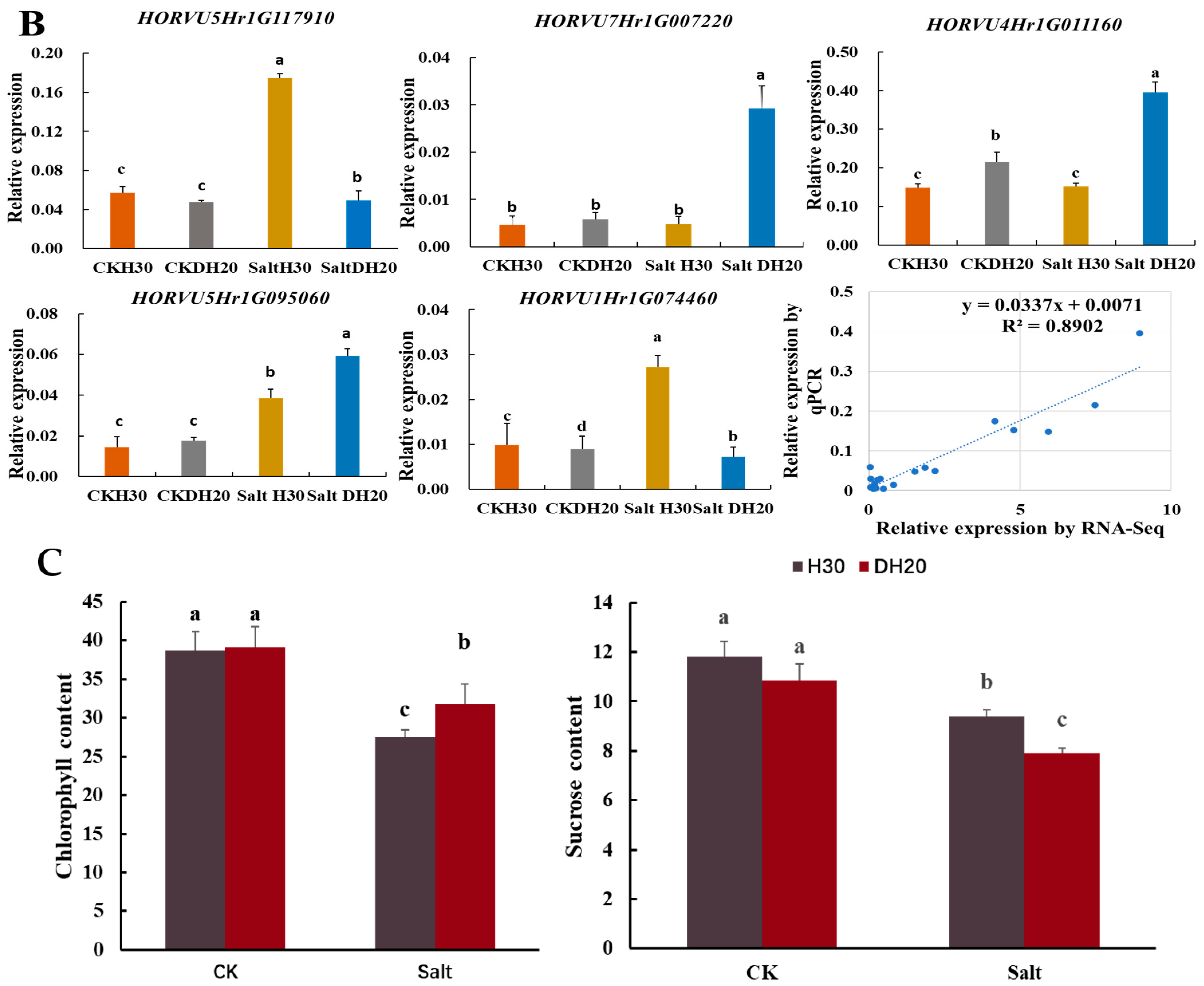
2.8. Maintaining ADP and Decreasing L-glutamate of DH20 in Response to Salt Stress
3. Discussion
3.1. Rapid Production of DH Lines for Salt Tolerance by Mutagenesis Combined with Microspore Culture Technology
3.2. Lipid Metabolism Underlying the DH20 Response to Salt Stress
3.3. Adaptive Strategies of Photosynthesis Underlying the DH20 Response to Salt Stress
3.4. Molecular Biomarkers Underlying the DH20 Response to Salt Stress
4. Materials and Methods
4.1. Plant Materials and Isolated Microspore Culture
4.2. Hydroponic Culture Screening
4.3. Determination of Shoot Fresh Weight, Shoot Dry Weight, Shoot K+/Na+ Content and Leaf Photosynthetic Parameters
4.4. RNA Extraction, Library Construction, Illumina Sequencing and Transcriptome Analysis
4.5. Metabolite Profiling and Data Analysis
4.6. Correlation Analysis of Transcript and Metabolite Profiles
4.7. Quantitative Real-Time PCR Analysis
4.8. Chlorophyll and Sucrose Content
4.9. Statistical Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Munns, R.; Day, D.A.; Fricke, W.; Watt, M.; Arsova, B.; Barkla, B.J.; Bose, J.; Byrt, C.S.; Chen, Z.; Foster, K.J.; et al. Energy costs of salt tolerance in crop plants. New Phytol. 2019, 225, 1072–1090. [Google Scholar] [CrossRef]
- Li, L.; Qi, Q.; Zhang, H.; Dong, Q.; Iqbal, A.; Gui, H.; Kayoumu, M.; Song, M.; Zhang, X.; Wang, X. Ameliorative effects of silicon against salt stress in Gossypium hirsutum L. Antioxidants 2022, 11, 1520. [Google Scholar] [CrossRef]
- Food and Agriculture Organization of the United Nations. FAO Extent of Salt-Affected Soils. 2020. Available online: http://www.fao.org/soils-portal/soil-management/management-of-some-problem-soils/salt-affected-soils/more-information-on-salt-affected-soils/en/ (accessed on 13 May 2020).
- Zhang, Y.; Wang, Y.; Xing, J.; Wan, J.; Wang, X.; Zhang, J.; Wang, X.; Li, Z.; Zhang, M. Copalyl diphosphate synthase mutation improved salt tolerance in maize (Zea mays. L.) via enhancing vacuolar Na+ sequestration and maintaining ROS homeostasis. Front. Plant Sci. 2020, 11, 457. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, J.; Shan, Q.; Chen, G.; Wang, Y.; Shen, L.; Pan, C.; Wu, H.; Abarquez, A. Soil salinization and ecological remediation by planting trees in China. In Proceedings of the 2010 International Conference on Mechanic Automation and Control Engineering, Wuhan, China, 26–28 June 2010; pp. 1349–1352. [Google Scholar]
- Wang, N.; Long, T.; Yao, W.; Xiong, L.; Zhang, Q.; Wu, C. Mutant resources for the functional analysis of the rice genome. Mol. Plant 2013, 6, 596–604. [Google Scholar] [CrossRef]
- Ren, J.; Wu, P.; Trampe, B.; Tian, X.; Chen, S. Novel technologies in doubled haploid line development. Plant Biotechnol. J. 2017, 15, 1361–1370. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Li, G.; Wurschum, T.; Zhang, Y.; Zheng, D.; Yang, X.; Li, J.; Liu, W.; Yan, J.; Chen, S. Genome-wide association study of haploid male fertility in maize (Zea Mays L.). Front. Plant Sci. 2018, 9, 974. [Google Scholar] [CrossRef]
- Dwivedi, S.L.; Britt, A.B.; Tripathi, L.; Sharma, S.; Upadhyaya, H.D.; Ortiz, R. Haploids: Constraints and opportunities in plant breeding. Biotechnol. Adv. 2015, 33, 812–829. [Google Scholar] [CrossRef] [PubMed]
- Chaikam, V.; Nair, S.K.; Martinez, L.; Lopez, L.A.; Utz, H.F.; Melchinger, A.E.; Boddupalli, P.M. Marker-assisted breeding of improved maternal haploid inducers in maize for the Tropical/Subtropical Regions. Front. Plant Sci. 2018, 9, 1527. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Chen, Z.; Gao, R.; He, T.; Wang, Y.; Xu, H.; Guo, G.; Liu, C.; Huang, J. Genotypes-independent optimization of nitrogen supply for isolated microspore cultures in barley. Biomed. Res. Int. 2016, 2016, 1801646. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Guo, G.; Fang, C.; Huang, S.; Chen, J.; Lu, R.; Huang, J.; Fan, X.; Liu, C. Rapid Generation of Barley Mutant Lines with high nitrogen uptake efficiency by microspore mutagenesis and field screening. Front. Plant Sci. 2018, 9, 450. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Liu, Z.; Li, D.; Yao, R.; Meng, Q.; Feng, H. Screening of chinese cabbage mutants produced by 60Co γ-ray mutagenesis of isolated microspore cultures. Plant Breed. 2014, 133, 480–488. [Google Scholar] [CrossRef]
- Beaith, M.E.; Fletcher, R.S.; Kott, L.S. Reduction of saturated fats by mutagenesis and heat selection in Brassica napus L. Euphytica 2005, 144, 1–9. [Google Scholar] [CrossRef]
- Zhu, J.; Fan, Y.; Shabala, S.; Li, C.; Lv, C.; Guo, B.; Xu, R.; Zhou, M. Understanding mechanisms of salinity tolerance in barley by proteomic and biochemical analysis of near-isogenic lines. Int. J. Mol. Sci. 2020, 21, 1516. [Google Scholar] [CrossRef]
- Fu, L.; Shen, Q.; Kuang, L.; Yu, J.; Wu, D.; Zhang, G. Metabolite profiling and gene expression of Na/K transporter analyses reveal mechanisms of the difference in salt tolerance between barley and rice. Plant Physiol. Bioch 2018, 130, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Gharaghanipor, N.; Arzani, A.; Rahimmalek, M.; Ravash, R. Physiological and transcriptome indicators of salt tolerance in wild and cultivated barley. Front. Plant Sci. 2022, 13, 819282. [Google Scholar] [CrossRef]
- Henderson, C.A.P.; Pauls, K.P. The use of haploidy to develop plants that express several recessive traits using light seeded canola (Brassica napus) as an example. TAG 1992, 83, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, Q.; Li, X.; Zheng, S.; Wang, S.; Guo, L.; Zhang, L.; Custers, J.B.M. Haploid plant production in Zantedeschia aethiopica ‘Hong Gan’ using anther culture. Sci. Hortic. 2011, 129, 335–342. [Google Scholar] [CrossRef]
- Liu, C.; Lu, R.; Guo, G.; He, T.; Li, Y.; Xu, H.; Gao, R.; Chen, Z.; Huang, J. Transcriptome analysis reveals translational regulation in barley microspore-derived embryogenic callus under salt stress. Plant Cell Rep. 2016, 35, 1719–1728. [Google Scholar] [CrossRef]
- Colmer, T.D.; Flowers, T.J.; Munns, R. Use of wild relatives to improve salt tolerance in wheat. J. Exp. Bot. 2006, 57, 1059–1078. [Google Scholar] [CrossRef]
- Yousefirad, S.; Soltanloo, H.; Ramezanpour, S.S.; Nezhad, K.Z.; Shariati, V. The RNA-seq transcriptomic analysis reveals genes mediating salt tolerance through rapid triggering of ion transporters in a mutant barley. PLoS ONE 2020, 15, e0229513. [Google Scholar] [CrossRef]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef]
- Yousfi, S.; Rabhi, M.; Hessini, K.; Abdelly, C.; Gharsalli, M. Differences in efficient metabolite management and nutrient metabolic regulation between wild and cultivated barley grown at high salinity. Plant Biology 2010, 12, 650–658. [Google Scholar] [CrossRef]
- Xu, H.; Li, Y.; Gao, R.; Xu, R.; Guo, G.; Lu, R.; Halford, N.G.; Chen, Z.; Liu, C. Rapid Generation and Analysis of a Barley Doubled Haploid Line with Higher Nitrogen Use Efficiency Than Parental Lines by F1 Microspore Embryogenesis. Plants 2021, 10, 1588. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Ufer, G.; Bartels, D. Lipid signaling in plant responses to abiotic stress. Plant Cell Environ. 2016, 39, 1029–1048. [Google Scholar] [CrossRef]
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Dennis, E.A. A comprehensive classification system for lipids. JLR 2005, 46, 839–862. [Google Scholar]
- Vu, H.S.; Tamura, P.; Galeva, N.A.; Chaturvedi, R.; Roth, M.R.; Williams, T.D.; Wang, X.; Shah, J.; Welti, R. Direct infusion mass spectrometry of oxylipin-containing Arabidopsis membrane lipids reveals varied patterns in different stress responses. Plant Physiol. 2012, 158, 324–339. [Google Scholar] [CrossRef] [PubMed]
- Mène-Saffrané, L.; Dubugnon, L.; Chételat, A.; Stolz, S.; Gouhier-Darimont, C.; Farmer, E.E. Nonenzymatic oxidation of trienoic fatty acids contributes to reactive oxygen species management in Arabidopsis. J. Biol. Chem. 2009, 284, 1702–1708. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Liang, J.; Liu, D.; Liu, Y.; Liu, G.; Wei, S. Melatonin-induced physiology and transcriptome changes in banana seedlings under salt stress conditions. Front. Plant Sci. 2022, 13, 938262. [Google Scholar] [CrossRef]
- Xie, Z.; Wang, J.; Wang, W.; Wang, Y.; Xu, J.; Li, Z.; Zhao, X.; Fu, B. Integrated analysis of the transcriptome and metabolome revealed the molecular mechanisms underlying the enhanced salt tolerance of rice due to the application of exogenous melatonin. Front. Plant Sci. 2021, 11, 618680. [Google Scholar] [CrossRef]
- Ji, C.; Liang, Z.; Cao, H.; Chen, Z.; Kong, X.; Xin, Z.; He, M.; Wang, J.; Wei, Z.; Xing, J.; et al. Transcriptome-based analysis of the effects of compound microbial agents on gene expression in wheat roots and leaves under salt stress. Front. Plant Sci. 2023, 14, 1109077. [Google Scholar] [CrossRef]
- Natera, S.H.A.; Hill, C.B.; Rupasinghe, T.W.T.; Roessner, U. Salt-stress induced alterations in the root lipidome of two barley genotypes with contrasting responses to salinity. Funct. Plant Biol. 2016, 43, 207–219. [Google Scholar] [CrossRef]
- Sarabia, L.D.; Boughton, B.A.; Rupasinghe, T.; VandeMeene, A.M.L.; Callahan, D.L.; Hill, C.B.; Roessner, U. High-mass-resolution MALDI mass spectrometry imaging reveals detailed spatial distribution of metabolites and lipids in roots of barley seedlings in response to salinity stress. Metabolomics 2018, 14, 63. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Liu, X.; Gao, H.; Zhang, B.; Peng, F.; Xiao, Y. Phosphatidylcholine enhances homeostasis in peach seedling cell membrane and increases its salt stress tolerance by phosphatidic acid. Int. J. Mol. Sci. 2022, 23, 2585. [Google Scholar] [CrossRef] [PubMed]
- Tasseva, G.; Richard, L.; Zachowski, A. Regulation of phosphatidylcholine biosynthesis under salt stress involves choline kinases in Arabidopsis thaliana. FEBS Lett. 2004, 566, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Chalbi, N.; Hessini, K.; Gandour, M.; Mohamed, S.N.; Smaoui, A.; Abdelly, C.; Ben Youssef, N. Are changes in membrane lipids and fatty acid composition related to salt-stress resistance in wild and cultivated barley? J. Plant Nutr. Soil. Sci. 2013, 176, 138–147. [Google Scholar] [CrossRef]
- Parida, A.K.; Das, A.B. Salt tolerance and salinity effects on plants: A review. Ecotoxicol. Environ. Saf. 2005, 60, 324–349. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Boughton, B.A.; Hill, C.B.; Feussner, I.; Roessner, U.; Rupasinghe, T.W.T. Insights into oxidized lipid modification in barley roots as an adaptation mechanism to salinity stress. Front. Plant Sci. 2020, 11, 1. [Google Scholar] [CrossRef]
- Chaves, M.M.; Flexas, J.; Pinheiro, C. Photosynthesis under drought and salt stress: Regulation mechanisms from whole plant to cell. Ann. Bot. 2009, 103, 551–560. [Google Scholar] [CrossRef]
- Shi, C.; Yang, F.; Liu, Z.; Li, Y.; Di, X.; Wang, J.; Lin, J. Uniform water potential induced by salt, alkali, and drought stresses has different impacts on the seedling of hordeum jubatum: From growth, photosynthesis, and chlorophyll fluorescence. Front. Plant Sci. 2021, 12, 733236. [Google Scholar] [CrossRef]
- Patsikka, E.; Kairavuo, M.; Sersen, F.; Aro, E.-M.; Tyystjarvi, E. Excess copper predisposes photosystem II to photoinhibition in vivo by outcompeting iron and causing decrease in leaf chlorophyll. Plant Physiol. 2002, 129, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Srivastava, S.; Lokhande, V.H.; D’Souza, S.F.; Suprasanna, P. Salt stress reveals differential antioxidant and energetics responses in glycophyte (Brassica juncea L.) and halophyte (Sesuvium portulacastrum L.). Front. Environ. 2015, 3, 19. [Google Scholar] [CrossRef]
- Wu, D.; Cai, S.; Chen, M.; Ye, L.; Chen, Z.; Zhang, H.; Dai, F.; Wu, F.; Zhang, G. Tissue metabolic responses to salt stress in wild and cultivated barley. PLoS ONE 2013, 8, e55431. [Google Scholar] [CrossRef] [PubMed]
- Chaves, M.M.; Oliveira, M.M. Mechanisms underlying plant resilience to water deficits: Prospects for water-saving agriculture. J. Exp. Bot. 2004, 55, 2365–2384. [Google Scholar] [CrossRef] [PubMed]
- Mathan, J.; Singh, A.; Ranjan, A. Sucrose transport in response to drought and salt stress involves ABA-mediated induction of OsSWEET13 and OsSWEET15 in rice. Physiol. Plant 2021, 171, 620–637. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, X.; Lu, X.; Zhao, B.; Yang, Y.; Liu, J. Integrative analysis of transcriptome and metabolome reveal mechanism of tolerance to salt stress in oat (Avena sativa L.). Plant Physiol. Bioch. 2021, 160, 315–328. [Google Scholar] [CrossRef]
- Cuin, T.A.; Shabala, S. Amino acids regulate salinity-induced potassium efflux in barley root epidermis. Planta 2007, 225, 753–761. [Google Scholar] [CrossRef]
- Wang, F.; Deng, X.W. Plant ubiquitin-proteasome pathway and its role in gibberellin signaling. Cell Res. 2011, 21, 1286–1294. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, X.; Zhao, C.; Fang, Z.; Jiao, P. Transcriptomic and metabolomic analysis reveals the role of CoA in the salt tolerance of Zygophyllum spp. BMC Plant Biol. 2020, 20, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qin, H.; Zhou, S.; Wei, P.; Zhang, H.; Zhou, Y.; Miao, Y.; Huang, R. The ubiquitin-binding protein OsDSK2a mediates seedling growth and salt responses by regulating gibberellin metabolism in rice. Plant Cell 2020, 32, 414–428. [Google Scholar] [CrossRef]
- Guo, J.; Chen, J.; Yang, J.; Yu, Y.; Yang, Y.; Wang, W. Identification, characterization and expression analysis of the VQ motif-containing gene family in tea plant (Camellia sinensis). BMC Genom. 2018, 19, 710. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, L.; Wang, H.; Zhang, L.; Wang, F.; Yu, D. Arabidopsis transcription factor WRKY8 functions antagonistically with its interacting partner VQ9 to modulate salinity stress tolerance. Plant J. 2013, 74, 730–745. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zou, Y.; Hu, J.; Ding, Y. Genome-wide analysis of the rice PPR gene family and their expression profiles under different stress treatments. BMC Genom. 2018, 19, 720. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, J.; Lu, P.; Chen, M.; Guo, C.; Xu, Z.; Ma, Y. The E-Subgroup pentatricopeptide repeat protein family in Arabidopsis thaliana and confirmation of the responsiveness PPR96 to abiotic stresses. Front. Plant Sci. 2016, 7, 1825. [Google Scholar] [CrossRef]
- Nijhawan, A.; Jain, M.; Tyagi, A.K.; Khurana, J.P. Genomic survey and gene expression analysis of the basic leucine zipper transcription factor family in rice. Plant Physiol. 2008, 146, 333–350. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.X.; Srivastava, R.; Che, P.; Howell, S.H. An endoplasmic reticulum stress response in Arabidopsis is mediated by proteolytic processing and nuclear relocation of a membrane-associated transcription factor, bZIP28. Plant Cell 2007, 19, 4111–4119. [Google Scholar] [CrossRef]
- Stankovi’c, B.; Vian, A.; Henry-Vian, C.; Davies, E. Molecular cloning and characterization of a tomato cDNA encoding a systemically wound-inducible bZIP DNA-binding protein. Planta 2000, 212, 60–66. [Google Scholar]
- Liu, H.; Tang, X.; Zhang, N.; Li, S.; Si, H. Role of bZIP transcription factors in plant salt stress. Int. J. Mol. Sci. 2023, 24, 7893. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Zhou, J. Receptor-like cytoplasmic kinases: Central players in plant receptor kinase-mediated signaling. Annu. Rev. Plant Biol. 2018, 69, 267–299. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.W.; Jon, J.H.; Kwak, J.M.; Nam, H.G. Identification of a receptor-like protein kinase gene rapidly induced by abscisic acid, dehydration, high salt, and cold treatments in Arabidopsis thaliana. Plant Physiol. 1997, 113, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Ingram, G.C.; Simon, R.; Carpenter, R.; Coen, E.S. The Antirrhinum ERG gene encodes a protein related to bacterial small GTPases and is required for embryonic viability. Curr. Biol. 1998, 8, 1079–1082. [Google Scholar] [CrossRef]
- Voshol, G.P.; Meyer, V.; Van den Hondel, C.A. GTP-binding protein Era: A novel gene target for biofuel production. BMC Biotechnol. 2015, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Wang, Y.; Sun, Y.; Shan, L.; Chen, P.; Huang, J. Improvement of isolated microspore culture of barley (Hordeum vulgare L.): The effect of floret co-culture. Plant Cell Tissue Organ. Cult. 2008, 93, 21–27. [Google Scholar] [CrossRef]
- Xu, H.; Liu, C.; Lu, R.; Guo, G.; Chen, Z.; He, T.; Gao, R.; Li, Y.; Huang, J. The difference in responses to nitrogen deprivation and re-supply at seedling stage between two barley genotypes differing nitrogen use efficiency. Plant Growth Regul. 2016, 79, 119–126. [Google Scholar] [CrossRef]
- Cao, Q.; Zhang, X.; Gao, X.; Wang, L.; Jia, G. Effects of ploidy level on the cellular, photochemical and photosynthetic characteristics in Lilium FO hybrids. Plant Physiol. Bioch. 2018, 133, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, D.; Tao, Z.; Yang, Y.; Gao, Z.; Zhao, G.; Chang, X. Impacts of nitrogen deficiency on wheat (Triticum aestivum L.) grain during the medium filling stage: Transcriptomic and metabolomic comparisons. Front. Plant Sci. 2021, 12, 674433. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Trapnell, C.; Donaghey, J.; Rinn, J.L.; Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011, 12, 22. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, 1–12. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, 480–484. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, H.; He, T.; Gao, R.; Guo, G.; Lu, R.; Chen, Z.; Liu, C. Comparative analysis of morphology, photosynthetic physiology, and transcriptome between diploid and tetraploid barley derived from microspore culture. Front. Plant Sci. 2021, 12, 626916. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, H.; Halford, N.G.; Guo, G.; Chen, Z.; Li, Y.; Zhou, L.; Liu, C.; Xu, R. Transcriptomic and Metabolomic Analyses Reveal the Importance of Lipid Metabolism and Photosynthesis Regulation in High Salinity Tolerance in Barley (Hordeum vulgare L.) Leaves Derived from Mutagenesis Combined with Microspore Culture. Int. J. Mol. Sci. 2023, 24, 16757. https://doi.org/10.3390/ijms242316757
Xu H, Halford NG, Guo G, Chen Z, Li Y, Zhou L, Liu C, Xu R. Transcriptomic and Metabolomic Analyses Reveal the Importance of Lipid Metabolism and Photosynthesis Regulation in High Salinity Tolerance in Barley (Hordeum vulgare L.) Leaves Derived from Mutagenesis Combined with Microspore Culture. International Journal of Molecular Sciences. 2023; 24(23):16757. https://doi.org/10.3390/ijms242316757
Chicago/Turabian StyleXu, Hongwei, Nigel G. Halford, Guimei Guo, Zhiwei Chen, Yingbo Li, Longhua Zhou, Chenghong Liu, and Rugen Xu. 2023. "Transcriptomic and Metabolomic Analyses Reveal the Importance of Lipid Metabolism and Photosynthesis Regulation in High Salinity Tolerance in Barley (Hordeum vulgare L.) Leaves Derived from Mutagenesis Combined with Microspore Culture" International Journal of Molecular Sciences 24, no. 23: 16757. https://doi.org/10.3390/ijms242316757
APA StyleXu, H., Halford, N. G., Guo, G., Chen, Z., Li, Y., Zhou, L., Liu, C., & Xu, R. (2023). Transcriptomic and Metabolomic Analyses Reveal the Importance of Lipid Metabolism and Photosynthesis Regulation in High Salinity Tolerance in Barley (Hordeum vulgare L.) Leaves Derived from Mutagenesis Combined with Microspore Culture. International Journal of Molecular Sciences, 24(23), 16757. https://doi.org/10.3390/ijms242316757