

Hub Genes in Stable QTLs Orchestrate the Accumulation of Cottonseed Oil in Upland Cotton via Catalyzing Key Steps of Lipid-Related Pathways

 ,
, 
Abstract
:1. Introduction
2. Results
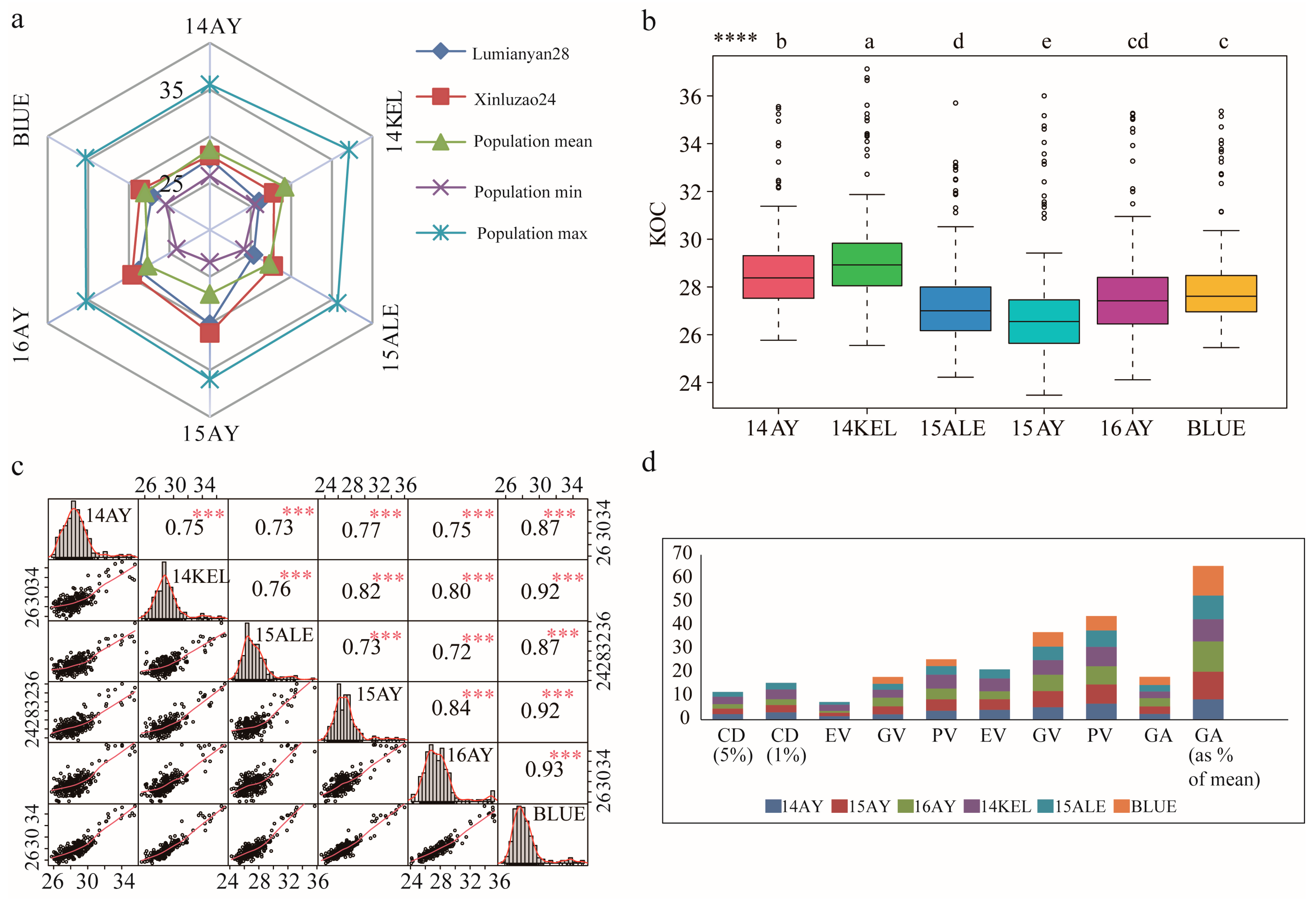
2.1. KOC Phenotypic Characteristics of the Parents and RIL Population
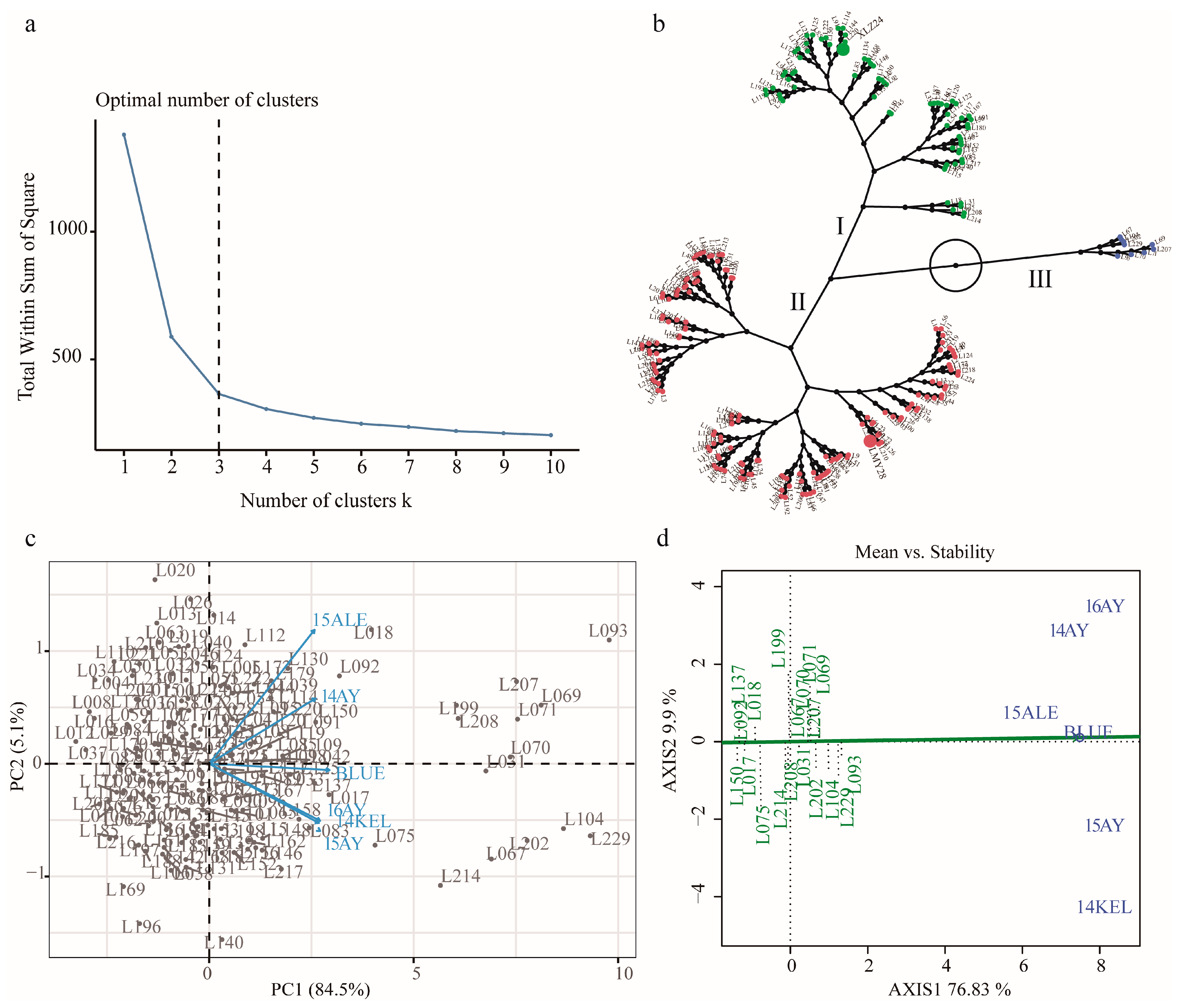
2.2. Hierarchical Cluster Analysis (HCA) and Discrimination of KOC Trait of the RILs
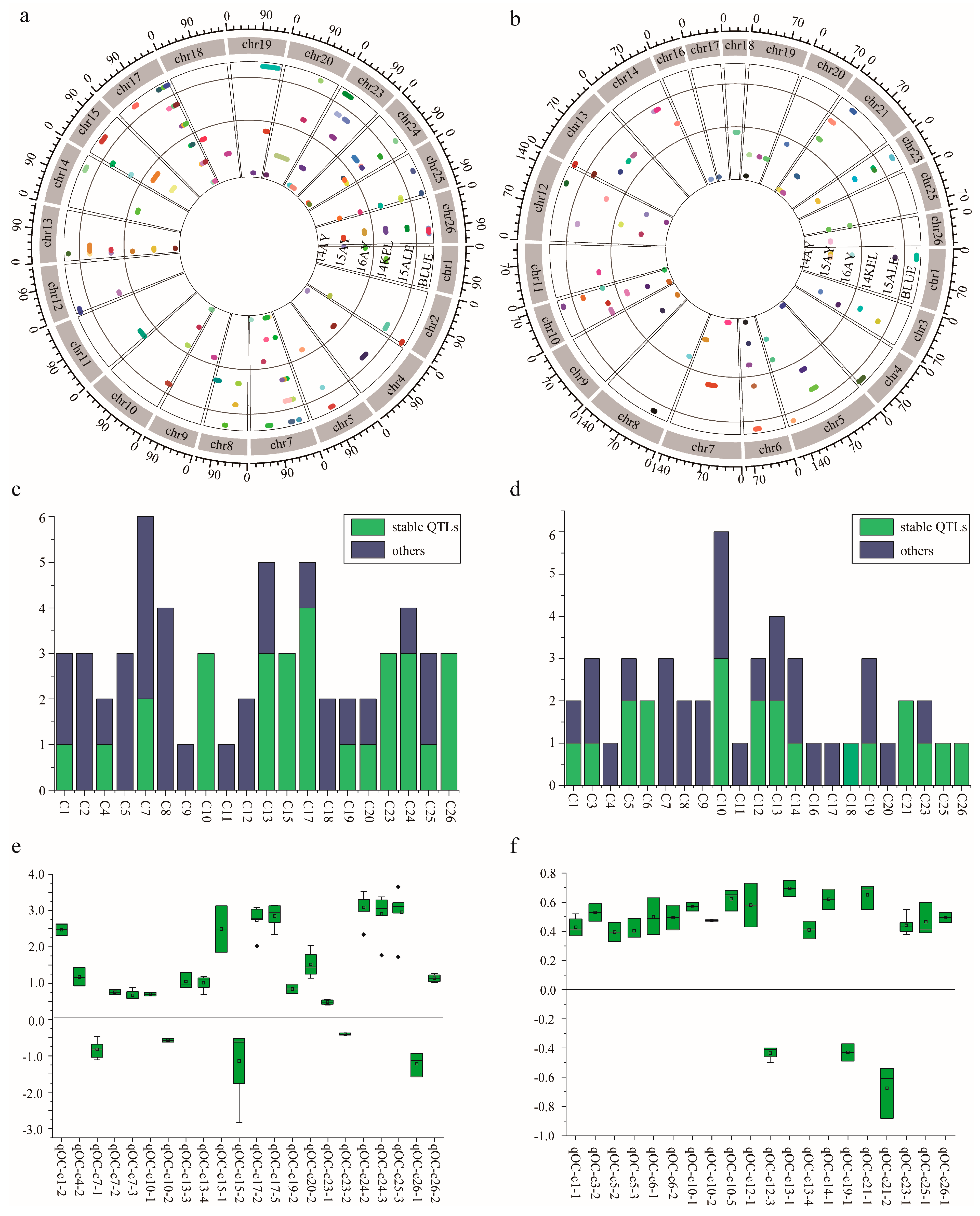
2.3. QTL Mapping of KOC in RIL Population
2.3.1. QTLs Identified via SLAF-Seq Strategy
2.3.2. QTLs Identified via CHIP Strategy
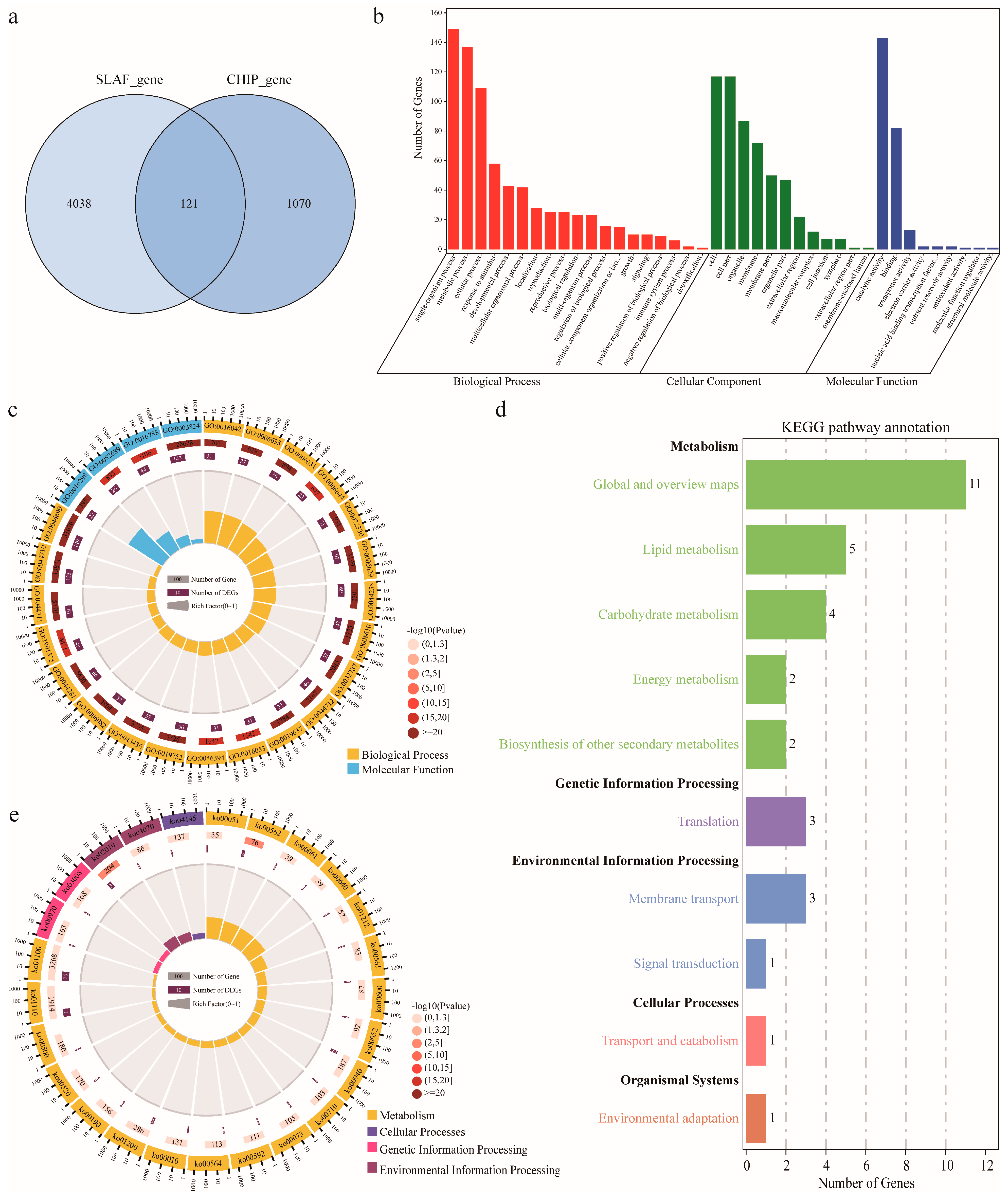
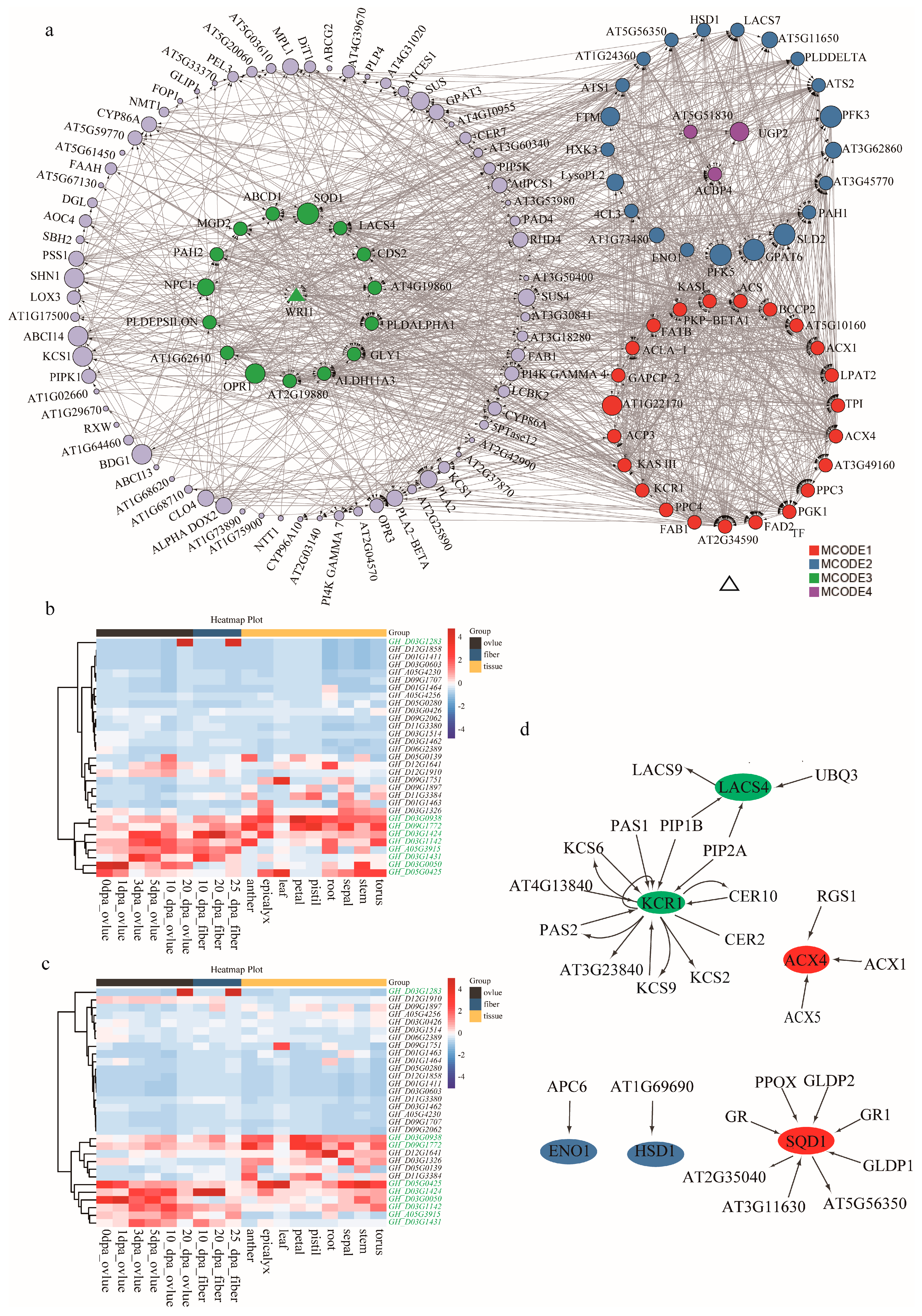
2.4. Candidate Gene Identification and Functional Annotation
2.5. Candidate Gene Expression Verification during Ovule Development
3. Discussion
3.1. Plant Materials and Their Cottonseed KOC Performance across Various Environments
3.2. HDGM and QTL Mapping of Cottonseed KOC
3.3. Candidate Genes Identification and Network Analysis
4. Materials and Methods
4.1. Plant Materials and Field Evaluation
4.2. Phenotypic Trait Evaluation
4.3. Data Analysis
4.4. Linkage Mapping and QTL Identification
4.5. QTL Nomenclature
- Candidate Gene Identification and Functional Annotation
4.6. Differential Expression Profiling and Construction of Co-Expression Network of Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, W.; Sun, Y.; Yang, P.; Cai, X.; Yang, L.; Ma, J.; Ou, Y.; Liu, T.; Ali, I.; Liu, D.; et al. A high density SLAF-seq SNP genetic map and QTL for seed size, oil and protein content in upland cotton. BMC Genom. 2019, 20, 599. [Google Scholar] [CrossRef]
- Wendel, J.F.; Cronn, R.C. Polyploidy and the evolutionary history of cotton. Adv. Agron. 2003, 78, 78004–78008. [Google Scholar]
- Shang, L.; Abduweli, A.; Wang, Y.; Hua, J. Genetic analysis and QTL mapping of oil content and seed index using two recombinant inbred lines and two backcross populations in Upland cotton. Plant Breed. 2016, 135, 224–231. [Google Scholar] [CrossRef]
- Xu, Z.; Li, J.; Guo, X.; Jin, S.; Zhang, X. Metabolic engineering of cottonseed oil biosynthesis pathway via RNA interference. Sci. Rep. 2016, 6, 33342. [Google Scholar] [CrossRef]
- Zhang, Z.; Gong, J.; Zhang, Z.; Gong, W.; Li, J.; Shi, Y.; Liu, A.; Ge, Q.; Pan, J.; Fan, S.; et al. Identification and analysis of oil candidate genes reveals the molecular basis of cottonseed oil accumulation in Gossypium hirsutum L. Theor. Appl. Genet. 2022, 135, 449–460. [Google Scholar] [CrossRef]
- Dhamodaran, G.; Krishnan, R.; Pochareddy, Y.K.; Pyarelal, H.M.; Sivasubramanian, H.; Ganeshram, A.K. A comparative study of combustion, emission, and performance characteristics of rice-bran-, neem-, and cottonseed-oil biodiesels with varying degree of unsaturation. Fuel 2017, 187, 296–305. [Google Scholar] [CrossRef]
- Gong, J.; Peng, Y.; Yu, J.; Pei, W.; Zhang, Z.; Fan, D.; Liu, L.; Xiao, X.; Liu, R.; Lu, Q.; et al. Linkage and association analyses reveal that hub genes in energy-flow and lipid biosynthesis pathways form a cluster in upland cotton. Comput. Struct. Biotechnol. J. 2022, 20, 1841–1859. [Google Scholar] [CrossRef]
- Paterson, A.H.; Wendel, J.F.; Gundlach, H.; Guo, H.; Jenkins, J.; Jin, D.; Llewellyn, D.; Showmaker, K.C.; Shu, S.; Udall, J.; et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 2012, 492, 423–427. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, J.; Fang, L.; Zhang, Z.; Ma, W.; Niu, Y.; Ju, L.; Deng, J.; Zhao, T.; Lian, J.; et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 2019, 51, 739–748. [Google Scholar] [CrossRef]
- Chen, Z.J.; Sreedasyam, A.; Ando, A.; Song, Q.; De Santiago, L.M.; Hulse-Kemp, A.M.; Ding, M.; Ye, W.; Kirkbride, R.C.; Jenkins, J.; et al. Genomic diversifications of five Gossypium allopolyploid species and their impact on cotton improvement. Nat. Genet. 2020, 52, 525–533. [Google Scholar] [CrossRef]
- Ali, I.; Teng, Z.; Bai, Y.; Yang, Q.; Hao, Y.; Hou, J.; Jia, Y.; Tian, L.; Liu, X.; Tan, Z.; et al. A high density SLAF-SNP genetic map and QTL detection for fibre quality traits in Gossypium hirsutum. BMC Genom. 2018, 19, 879. [Google Scholar] [CrossRef]
- Yang, H.; Li, C.; Lam, H.-M.; Clements, J.; Yan, G.; Zhao, S. Sequencing consolidates molecular markers with plant breeding practice. Theor. Appl. Genet. 2015, 128, 779–795. [Google Scholar] [CrossRef]
- Shockey, J.; Dowd, M.; Mack, B.; Gilbert, M.; Scheffler, B.; Ballard, L.; Frelichowski, J.; Mason, C. Naturally occurring high oleic acid cottonseed oil: Identification and functional analysis of a mutant allele of Gossypium barbadense fatty acid desaturase-2. Planta 2017, 245, 611–622. [Google Scholar] [CrossRef]
- Liu, Q.; Singh, S.P.; Brubaker, C.L.; Sharp, P.J.; Green, A.G.; Marshall, D. Molecular cloning and expression of a cDNA encoding a microsomal w-6 fatty acid desaturase from cotton (Gossypium hirsutum). Funct. Plant Biol. 1999, 26, 101–106. [Google Scholar] [CrossRef]
- Sharif, I.; Farooq, J.; Chohan, S.M.; Saleem, S.; Kainth, R.A.; Mahmood, A.; Sarwar, G. Strategies to enhance cottonseed oil contents and reshape fatty acid profile employing different breeding and genetic engineering approaches. J. Integr. Agr. 2019, 18, 2205–2218. [Google Scholar] [CrossRef]
- Chapman, K.D.; Austin-Brown, S.; Sparace, S.A.; Kinney, A.J.; Ripp, K.G.; Pirtle, I.L.; Pirtle, R.M. Transgenic cotton plants with increased seed oleic acid content. J. Am. Oil Chem. Soc. 2001, 78, 941–947. [Google Scholar] [CrossRef]
- Hölzl, G.; Dörmann, P. Chloroplast lipids and their biosynthesis. Annu. Rev. Plant Biol. 2019, 70, 6.1–6.31. [Google Scholar] [CrossRef]
- Shang, X.; Cheng, C.; Ding, J.; Guo, W. Identification of candidate genes from the SAD gene family in cotton for determination of cottonseed oil composition. Mol. Genet. Genom. 2017, 292, 173–186. [Google Scholar] [CrossRef]
- Wang, N.; Ma, J.; Pei, W.; Wu, M.; Li, H.; Li, X.; Yu, S.; Zhang, J.; Yu, J. A genome-wide analysis of the lysophosphatidate acyltransferase (LPAAT) gene family in cotton: Organization, expression, sequence variation, and association with seed oil content and fiber quality. BMC Genom. 2017, 18, 218. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, Z.; Wang, X.; Wang, Y.; Hua, J. Molecular Characterization and Expression Analysis of GhWRI1 in Upland Cotton. J. Plant Biol. 2018, 61, 186–197. [Google Scholar] [CrossRef]
- Du, X.; Liu, S.; Sun, J.; Zhang, G.; Jia, Y.; Pan, Z.; Xiang, H.; He, S.; Xia, Q.; Xiao, S.; et al. Dissection of complicate genetic architecture and breeding perspective of cottonseed traits by genome-wide association study. BMC Genom. 2018, 19, 451. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, Y.; Wang, Y.; Cui, Y.; Liu, Z.; Hua, J. RNA interference of GhPEPC2 enhanced seed oil accumulation and salt tolerance in Upland cotton. Plant Sci. 2018, 271, 52–61. [Google Scholar] [CrossRef]
- Zhu, D.; Le, Y.; Zhang, R.; Li, X.; Lin, Z. A global survey of the gene network and key genes for oil accumulation in cultivated tetraploid cottons. Plant Biotechnol. J. 2020, 19, 1170–1182. [Google Scholar] [CrossRef]
- Liu, R.; Gong, J.; Xiao, X.; Zhang, Z.; Li, J.; Liu, A.; Lu, Q.; Shang, H.; Shi, Y.; Ge, Q.; et al. GWAS Analysis and QTL Identification of Fiber Quality Traits and Yield Components in Upland Cotton Using Enriched High-Density SNP Markers. Front. Plant Sci. 2018, 9, 1067. [Google Scholar] [CrossRef]
- Li-Beisson, Y.; Shorrosh, B.; Beisson, F.; Andersson, M.X.; Arondel, V.; Bates, P.D.; Baud, S.; Bird, D.; Debono, A.; Durrett, T.P.; et al. Acyl-lipid metabolism. Arab. Book 2013, 11, e0161. [Google Scholar] [CrossRef]
- Turquetti-Moraes, D.K.; Moharana, K.C.; Almeida-Silva, F.; Pedrosa-Silva, F.; Venancio, T.M. Integrating omics approaches to discover and prioritize candidate genes involved in oil biosynthesis in soybean. Gene. 2022, 808, 145976. [Google Scholar] [CrossRef]
- Banda, T.; Kumarasamy, M. Application of Multivariate Statistical Analysis in the Development of a Surrogate Water Quality Index (WQI) for South African Watersheds. Water 2020, 12, 1584. [Google Scholar] [CrossRef]
- Rehman, F.; Saeed, A.; Yaseen, M.; Shakeel, A.; Ziaf, K.; Munir, H.; Tariq, S.A.; Raza, M.A.; Riaz, A. Genetic evaluation and characterization using cluster heat map to assess NaCl tolerance in tomato germplasm at the seedling stage. Chil. J. Agr. Res. 2019, 79, 56–65. [Google Scholar] [CrossRef]
- Liu, X.; Teng, Z.; Wang, J.; Wu, T.; Zhang, Z.; Deng, X.; Fang, X.; Tan, Z.; Ali, I.; Liu, D.; et al. Enriching an intraspecific genetic map and identifying QTL for fiber quality and yield component traits across multiple environments in Upland cotton (Gossypium hirsutum L.). Mol. Genet. Genom. 2018, 292, 1281–1306. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Y.; Huang, Y.; Cui, Y.; Hua, J. Gene network of oil accumulation reveals expression profiles in developing embryos and fatty acid composition in Upland cotton. J. Plant Physiol. 2018, 228, 101–112. [Google Scholar] [CrossRef]
- Zhao, W.; Kong, X.; Yang, Y.; Nie, X.; Lin, Z. Association mapping seed kernel oil content in upland cotton using genome-wide SSRs and SNPs. Mol. Breed. 2019, 39, 105. [Google Scholar] [CrossRef]
- Zhu, D.; Li, X.; Wang, Z.; You, C.; Nie, X.; Sun, J.; Zhang, X.; Zhang, D.; Lin, Z. Genetic dissection of an allotetraploid interspecific CSSLs guides interspecific genetics and breeding in cotton. BMC Genom. 2020, 21, 431. [Google Scholar] [CrossRef]
- Huang, A.H.C. Structure of plant seed oil bodies. Curr. Opin. Struct. Biol. 1994, 4, 493–498. [Google Scholar] [CrossRef]
- Kim, H.U.; Hsieh, K.; Ratnayake, C.; Huang, A.H. A novel group of oleosins is present inside the pollen of Arabidopsis. J. Biol. Chem. 2002, 277, 22677–22684. [Google Scholar] [CrossRef]
- Huang, A.H.C. Plant Lipid Droplets and Their Associated Proteins: Potential for Rapid Advances. Plant Physiol. 2018, 176, 1894–1918. [Google Scholar] [CrossRef]
- Walther, T.C.; Chung, J.; Farese, R.V. Lipid Droplet Biogenesis. Ann. Rev. Cell Dev. Biol. 2017, 33, 491–510. [Google Scholar] [CrossRef]
- Baud, S.; Lepiniec, L. Regulation of de novo fatty acid synthesis in maturing oilseeds of Arabidopsis. Plant Physiol. Biochem. 2009, 47, 448–455. [Google Scholar] [CrossRef]
- Cai, G.; Fan, C.; Liu, S.; Yang, Q.; Liu, D.; Wu, J.; Li, J.; Zhou, Y.; Guo, L.; Wang, X. Nonspecific phospholipase C6 increases seed oil production in oilseed Brassicaceae plants. N. Phytol. 2020, 226, 1055–1073. [Google Scholar] [CrossRef]
- Fatland, B.L.; Ke, J.; Anderson, M.D.; Mentzen, W.I.; Cui, L.W.; Allred, C.C.; Johnston, J.L.; Nikolau, B.J.; Wurtele, E.S. Molecular characterization of a heteromeric ATP-citrate lyase that generates cytosolic acetyl-coenzyme A in Arabidopsis. Plant Physiol. 2002, 130, 740–756. [Google Scholar] [CrossRef]
- Fatland, B.L.; Nikolau, B.J.; Wurtele, E.S. Reverse genetic characterization of cytosolic acetyl-CoA generation by ATP-citrate lyase in Arabidopsis. Plant Cell 2005, 17, 182–203. [Google Scholar] [CrossRef]
- Yin, Y.; Guo, Z.; Chen, K.; Tian, T.; Tan, J.; Chen, X.; Chen, J.; Yang, B.; Tang, S.; Peng, K.; et al. Ultra-High α-Linolenic Acid Accumulating Developmental Defective Embryo was Rescued by Lysophosphatidic Acid Acyltransferase 2. Plant J. 2020, 103, 2151–2167. [Google Scholar] [CrossRef]
- Prabhakar, V.; Lottgert, T.; Gigolashvili, T.; Bell, K.; Flugge, U.I.; Hausler, R.E. Molecular and functional characterization of the plastid-localized Phosphoenolpyruvate enolase (ENO1) from Arabidopsis thaliana. FEBS Lett. 2009, 583, 983–991. [Google Scholar] [CrossRef]
- Bao, B.; Chao, H.; Wang, H.; Zhao, W.; Zhang, L.; Raboanatahiry, N.; Wang, X.; Wang, B.; Jia, H.; Li, M. Stable, Environmental Specific and Novel QTL Identification as Well as Genetic Dissection of Fatty Acid Metabolism in Brassica napus. Front. Plant Sci. 2018, 9, 1018. [Google Scholar] [CrossRef]
- Contento, A.L.; Bassham, D.C. Increase in catalase-3 activity as a response to use of alternative catabolic substrates during sucrose starvation. Plant Physiol. Biochem. 2010, 48, 232–238. [Google Scholar] [CrossRef]
- Zhong, Y.; Wang, Y.; Li, P.; Gong, W.; Wang, X.; Yan, H.; Ge, Q.; Liu, A.; Shi, Y.; Shang, H.; et al. Genome-Wide Analysis and Functional Characterization of LACS Gene Family Associated with Lipid Synthesis in Cotton (Gossypium spp.). Int. J. Mol. Sci. 2023, 24, 8530. [Google Scholar] [CrossRef]
- Jessen, D.; Olbrich, A.; Knüfer, J.; Krüger, A.; Hoppert, M.; Polle, A.; Fulda, M. Combined activity of LACS1 and LACS4 is required for proper pollen coat formation in Arabidopsis. Plant J. 2011, 68, 715–726. [Google Scholar] [CrossRef]
- Shimojima, M. Biosynthesis and functions of the plant sulfolipid. Prog. Lipid Res. 2011, 50, 234–239. [Google Scholar] [CrossRef]
- Baud, S.; Dichow, N.R.; Kelemen, Z.; d’Andréa, S.; To, A.; Berger, N.; Canonge, M.; Kronenberger, J.; Viterbo, D.; Dubreucq, B.; et al. Regulation of HSD1 in seeds of Arabidopsis thaliana. Plant Cell Physiol. 2009, 50, 1463–1478. [Google Scholar] [CrossRef]
- Khan, B.R.; Adham, A.R.; Zolman, B.K. Peroxisomal Acyl-CoA oxidase 4 activity differs between Arabidopsis accessions. Plant Mol. Biol. 2012, 78, 45–58. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, Z.; Ge, X.; Lu, L.; Qin, W.; Qanmber, G.; Liu, L.; Wang, Z.; Li, F. Brassinosteroids regulate cotton fiber elongation by modulating very-long-chain fatty acid biosynthesis. Plant Cell 2023, 35, 2114–2131. [Google Scholar] [CrossRef]
- Qu, C.; Jia, L.; Fu, F.; Zhao, H.; Lu, K.; Wei, L.; Xu, X.; Liang, Y.; Li, S.; Wang, R.; et al. Genome-wide association mapping and Identification of candidate genes for fatty acid composition in Brassica napus L. using SNP markers. BMC Genom. 2017, 18, 232. [Google Scholar]
- Kogure, K.; Watanabe, A.; Ito, Y. Interaction of ONION2 ketoacyl CoA synthase with ketoacyl CoA reductase of rice. Mol. Biol. Rep. 2022, 49, 1643–1647. [Google Scholar] [CrossRef]
- Gu, J.; Chao, H.; Wang, H.; Li, Y.; Li, D.; Xiang, J.; Gan, J.; Lu, G.; Zhang, X.; Long, Y.; et al. Identification of the relationship between oil body morphology and oil content by microstructure comparison combining with QTL analysis in Brassica napus. Front. Plant Sci. 2017, 7, 1989. [Google Scholar] [CrossRef]
- Mulichak, A.M.; Theisen, M.J.; Essigmann, B.; Benning, C.; Garavito, R.M. Crystal structure of SQD1, an enzyme involved in the biosynthesis of the plant sulfolipid headgroup donor UDP-sulfoquinovose. Proc. Natl. Acad. Sci. USA 1999, 96, 13097–13102. [Google Scholar] [CrossRef]
- Nakajima, Y.; Umena, Y.; Nagao, R.; Endo, K.; Kobayashi, K.; Akita, F.; Suga, M.; Wada, H.; Noguchi, T.; Shen, J.R. Thylakoid membrane lipid sulfoquinovosyl-diacylglycerol (SQDG) is required for full functioning of photosystem II in Thermosynechococcus elongatus. J. Biol. Chem. 2018, 293, 14786–14797. [Google Scholar] [CrossRef]
- Nguyen, V.C.; Nakamura, Y. Distinctly localized lipid phosphate phosphatases mediate endoplasmic reticulum glycerolipid metabolism in Arabidopsis. Plant Cell 2023, 35, 1548–1571. [Google Scholar] [CrossRef]
- Degenkolbe, T.; Giavalisco, P.; Zuther, E.; Seiwert, B.; Hincha, D.K.; Willmitzer, L. Differential remodeling of the lipidome during cold acclimation in natural accessions of Arabidopsis thaliana. Plant J. 2012, 72, 972–982. [Google Scholar] [CrossRef]
- Hafeez, A.; Ge, Q.; Zhang, Q.; Li, J.; Gong, J.; Liu, R.; Shi, Y.; Shang, H.; Liu, A.; Iqbal, M.S.; et al. Multi-responses of O-methyltransferase genes to salt stress and fiber development of Gossypium species. BMC Plant Biol. 2021, 21, 37. [Google Scholar] [CrossRef]
- Verslues, P.E.; Agarwal, M.; Katiyar-Agarwal, S.; Zhu, J.; Zhu, J.-K. Methods and concepts in quantifying resistance to drought, salt and freezing, abiotic stresses that affect plant water status. Plant J. 2006, 45, 523–539. [Google Scholar] [CrossRef]
- Krawczyk, H.E.; Rotsch, A.H.; Herrfurth, C.; Scholz, P.; Shomroni, O.; Salinas-Riester, G.; Feussner, I.; Ischebeck, T. Heat stress leads to rapid lipid remodeling and transcriptional adaptations in Nicotiana tabacum pollen tubes. Plant Physiol. 2022, 189, 490–515. [Google Scholar] [CrossRef]
- Li, F.; Asami, T.; Wu, X.; Tsang, E.W.T.; Cutler, A.J. A Putative Hydroxysteroid Dehydrogenase Involved in Regulating Plant Growth and Development. Plant Physiol. 2007, 145, 87–97. [Google Scholar] [CrossRef]
- Zhang, Z.; Cheng, Z.-J.; Gan, L.; Zhang, H.; Wu, F.-Q.; Lin, Q.-B.; Wang, J.-L.; Wang, J.; Guo, X.-P.; Zhang, X.; et al. OsHSD1, a hydroxysteroid dehydrogenase, is involved in cuticle formation and lipid homeostasis in rice. Plant Sci. 2016, 249, 35–45. [Google Scholar] [CrossRef]
- Shao, Q.; Liu, X.; Su, T.; Ma, C.; Wang, P. New Insights into the Role of Seed Oil Body Proteins in Metabolism and Plant Development. Front. Plant Sci. 2019, 10, 1568. [Google Scholar] [CrossRef]
- Kunst, L.; Samuels, L. Plant cuticles shine: Advances in wax biosynthesis and export. Curr. Opin. Plant Biol. 2009, 12, 721–727. [Google Scholar] [CrossRef]
- Voelker, T.; Kinney, A.J. Variation in the Biosynthesis of Seed-Storage Lipids. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2001, 52, 335–361. [Google Scholar] [CrossRef]
- Chao, H.; Wang, H.; Wang, X.; Guo, L.; Gu, J.; Zhao, W.; Li, B.; Chen, D.; Raboanatahiry, N.; Li, M. Genetic dissection of seed oil and protein content and identification of networks associated with oil content in Brassica napus. Sci. Rep. 2017, 7, 46295. [Google Scholar] [CrossRef]
- Wang, S.; Basten, C.J.; Zeng, Z. Windows QTL Cartographer; v2.5.; North Carolina State University: Raleigh, NC, USA, 2012; Available online: http://statgen.ncsu.edu/qtlcart/WQTLCart.htm (accessed on 28 August 2022).
- Zeng, Z.-B. Precision mapping of quantitative trait loci. Genetics 1994, 136, 1457–1468. [Google Scholar] [CrossRef]
- Sun, F.-D.; Zhang, J.-H.; Wang, S.-F.; Gong, W.-K.; Shi, Y.-Z.; Liu, A.-Y.; Li, J.-W.; Gong, J.-W.; Shang, H.-H.; Yuan, Y.-L. QTL mapping for fiber quality traits across multiple generations and environments in upland cotton. Mol. Breed. 2012, 30, 569–582. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Environment | Parents | RILs | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LMY 28 | XLZ 24 | Mean ± SD | SE Mean | Range | Variance | CV(%) | Min | Max | Skewness | Kurtosis | H2 | |
| 14AY | 27.59 | 27.95 ** | 28.60 ± 1.71 | 0.11 | 25.77–35.55 | 2.91 | 5.96 | 25.77 | 35.55 | 1.53 | 4.09 | 80.07 |
| 15AY | 30.08 | 31.03 ** | 26.86 ± 2.06 | 0.14 | 23.47–36.00 | 4.22 | 7.65 | 23.47 | 36.00 | 1.91 | 5.46 | 83.81 |
| 16AY | 28.75 | 29.59 ** | 27.71 ± 2.03 | 0.13 | 24.11–35.27 | 4.12 | 7.33 | 24.11 | 35.27 | 1.70 | 4.17 | 94.29 |
| 14KEL | 26.06 | 27.88 ** | 29.20 ± 1.93 | 0.13 | 25.55–37.13 | 3.70 | 6.59 | 25.55 | 37.13 | 1.74 | 4.11 | 87.59 |
| 15ALE | 25.37 | 27.78 ** | 27.32 ± 1.74 | 0.12 | 24.22–35.70 | 3.04 | 6.38 | 24.22 | 35.70 | 1.43 | 3.01 | 83.71 |
| BLUE | 27.17 | 28.59 ** | 28.01 ± 1.73 | 0.11 | 25.46–35.36 | 3.01 | 6.19 | 25.46 | 35.36 | 2.09 | 5.75 | 91.50 |
| Gene ID | Gene Name | Functional Description | Pathways | Localization | MCODE | References |
|---|---|---|---|---|---|---|
| GH_A05G3915 | BCCP2 | Biotin carboxyl carrier protein of heteromeric ACCase | Plastid FA synthesis | Plastid | 1 | [23,26] |
| GH_D03G1283 | HSD1 | Steroleosin | Oil storage | Oil body | 2 | |
| GH_D03G0050 | PLDdelta | Phospholipase D delta | Lipid signaling | 2 | ||
| GH_D03G0938 | LACS4 | lipid biosynthesis and cellular lipid metabolic process | FA elongation | Plastid | 3 | [7] |
| GH_D03G1424/GH_D03G1431 | KCR1 | Ketoacyl CoA reductase | FA elongation | Endomembrane | 1 | GH_D03G1424/GH_D03G1431 |
| GH_D03G1142 | ENO1 | Phosphoenyl pyruvate enolase | Glycolysis | Plastid | 2 | GH_D03G1142 |
| GH_D05G0425 | SQD1 | UDP-Sulfoquinovose synthase | Plastidal glycerolipid, galactolipid and sulfolipid synthesis | 3 | GH_D05G0425 | |
| GH_D09G1772 | ACX4 | Acyl CoA oxidase | beta-oxidation | 1 | GH_D09G1772 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alam, B.; Liu, R.; Gong, J.; Li, J.; Yan, H.; Ge, Q.; Xiao, X.; Pan, J.; Shang, H.; Shi, Y.; et al. Hub Genes in Stable QTLs Orchestrate the Accumulation of Cottonseed Oil in Upland Cotton via Catalyzing Key Steps of Lipid-Related Pathways. Int. J. Mol. Sci. 2023, 24, 16595. https://doi.org/10.3390/ijms242316595
Alam B, Liu R, Gong J, Li J, Yan H, Ge Q, Xiao X, Pan J, Shang H, Shi Y, et al. Hub Genes in Stable QTLs Orchestrate the Accumulation of Cottonseed Oil in Upland Cotton via Catalyzing Key Steps of Lipid-Related Pathways. International Journal of Molecular Sciences. 2023; 24(23):16595. https://doi.org/10.3390/ijms242316595
Chicago/Turabian StyleAlam, Beena, Ruixian Liu, Juwu Gong, Junwen Li, Haoliang Yan, Qun Ge, Xianghui Xiao, Jingtao Pan, Haihong Shang, Yuzhen Shi, and et al. 2023. "Hub Genes in Stable QTLs Orchestrate the Accumulation of Cottonseed Oil in Upland Cotton via Catalyzing Key Steps of Lipid-Related Pathways" International Journal of Molecular Sciences 24, no. 23: 16595. https://doi.org/10.3390/ijms242316595
APA StyleAlam, B., Liu, R., Gong, J., Li, J., Yan, H., Ge, Q., Xiao, X., Pan, J., Shang, H., Shi, Y., Yuan, Y., & Gong, W. (2023). Hub Genes in Stable QTLs Orchestrate the Accumulation of Cottonseed Oil in Upland Cotton via Catalyzing Key Steps of Lipid-Related Pathways. International Journal of Molecular Sciences, 24(23), 16595. https://doi.org/10.3390/ijms242316595