
Sedanolide Activates KEAP1–NRF2 Pathway and Ameliorates Hydrogen Peroxide-Induced Apoptotic Cell Death
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
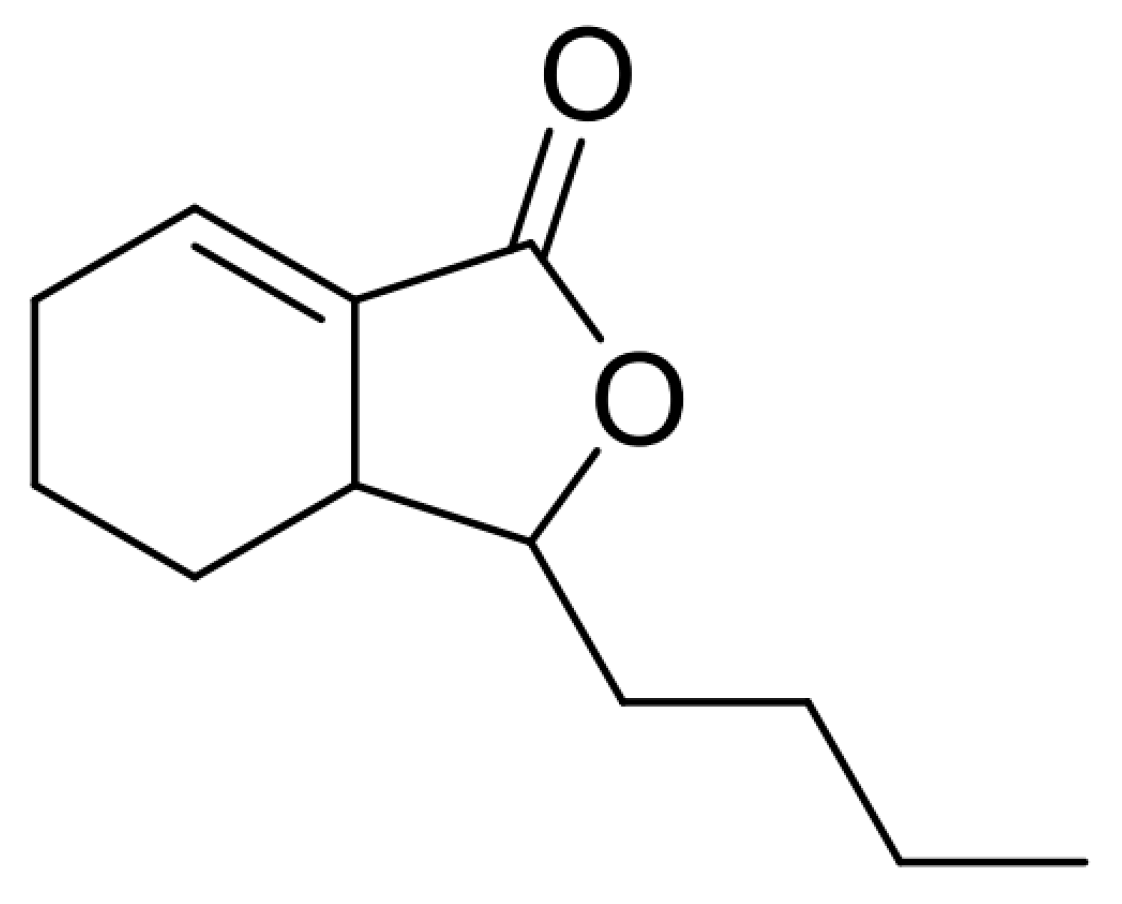
:1. Introduction
2. Results
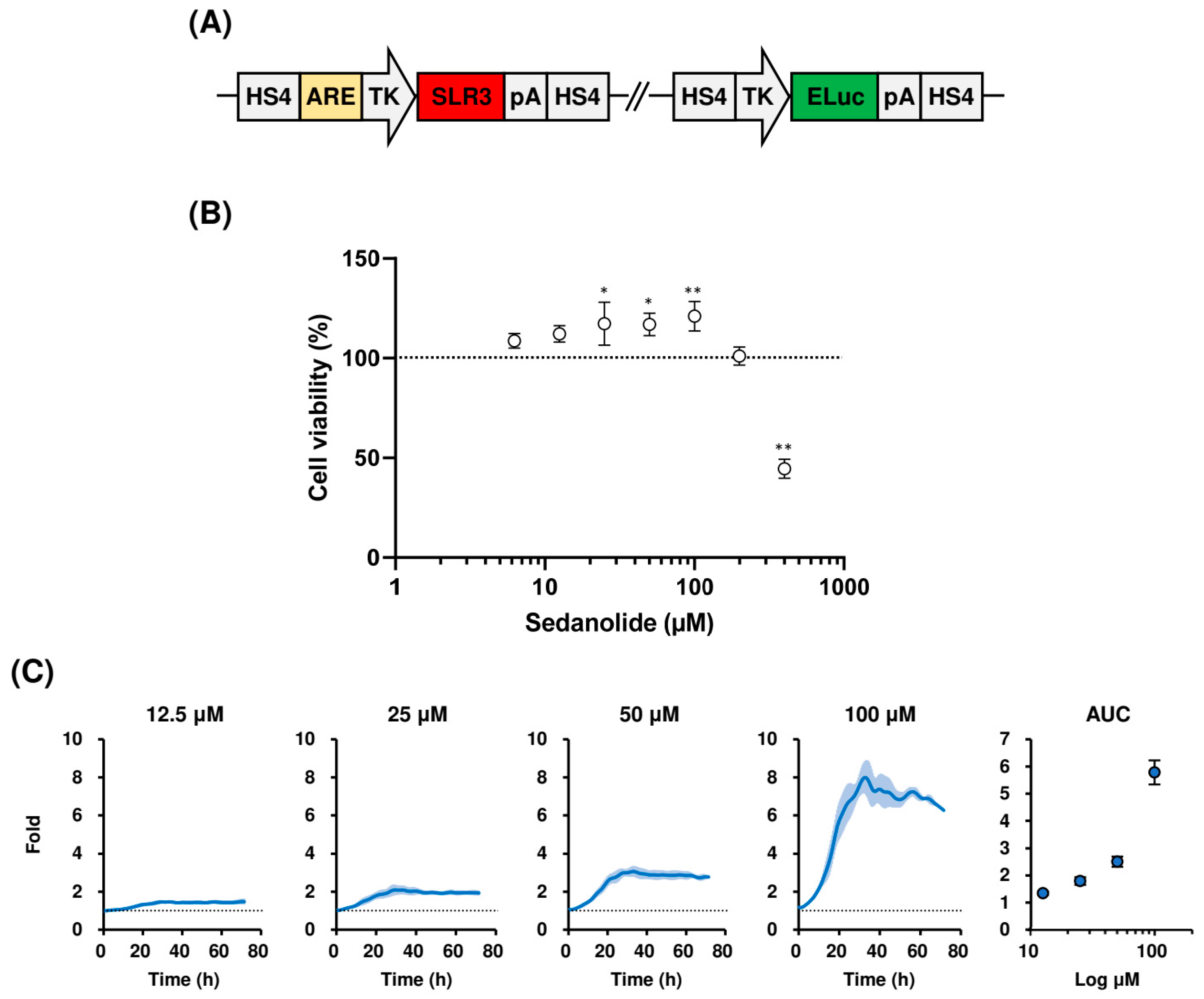
2.1. Sedanolide Activates ARE-Dependent Transcription
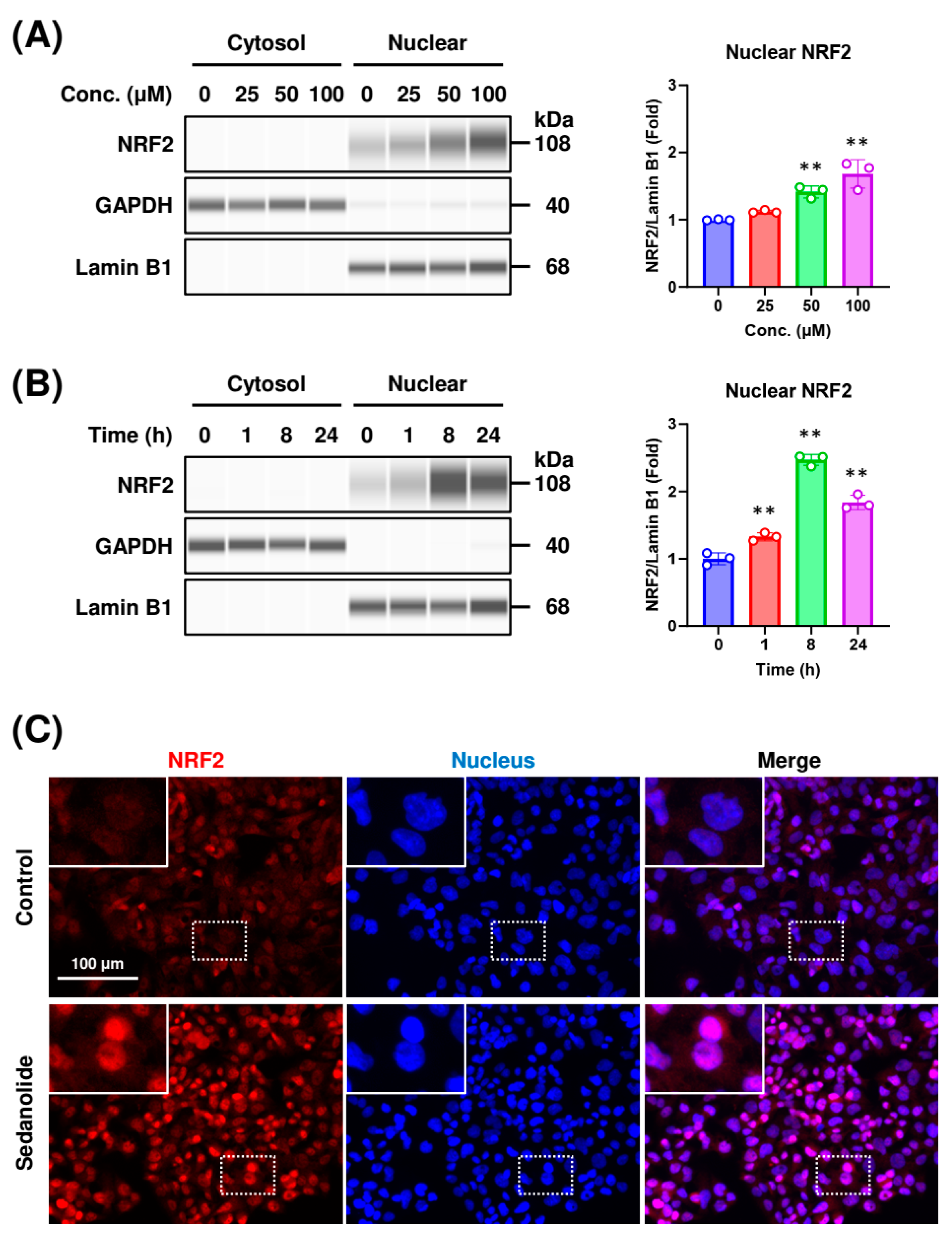
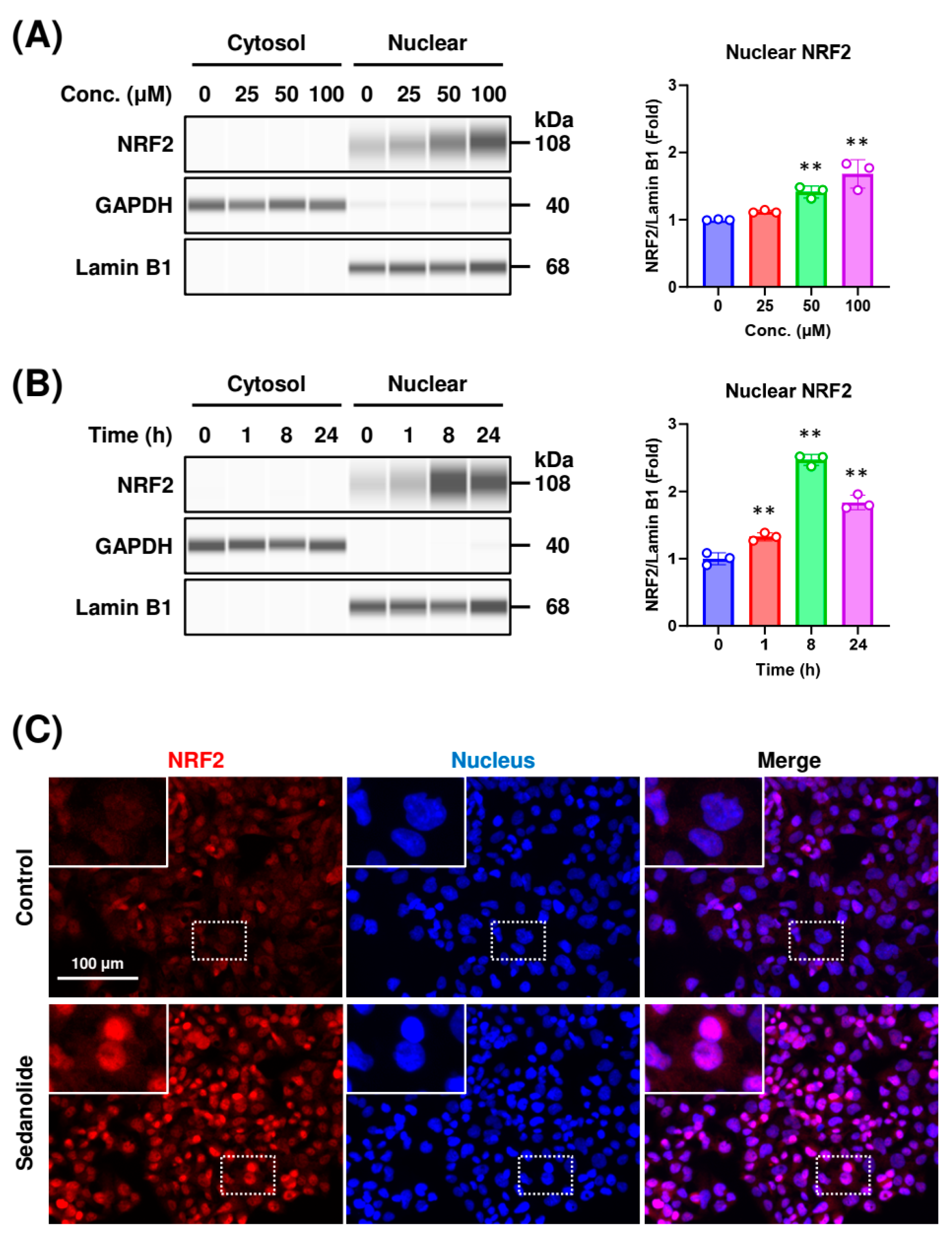
2.2. Sedanolide Induces Nuclear Translocation of NRF2
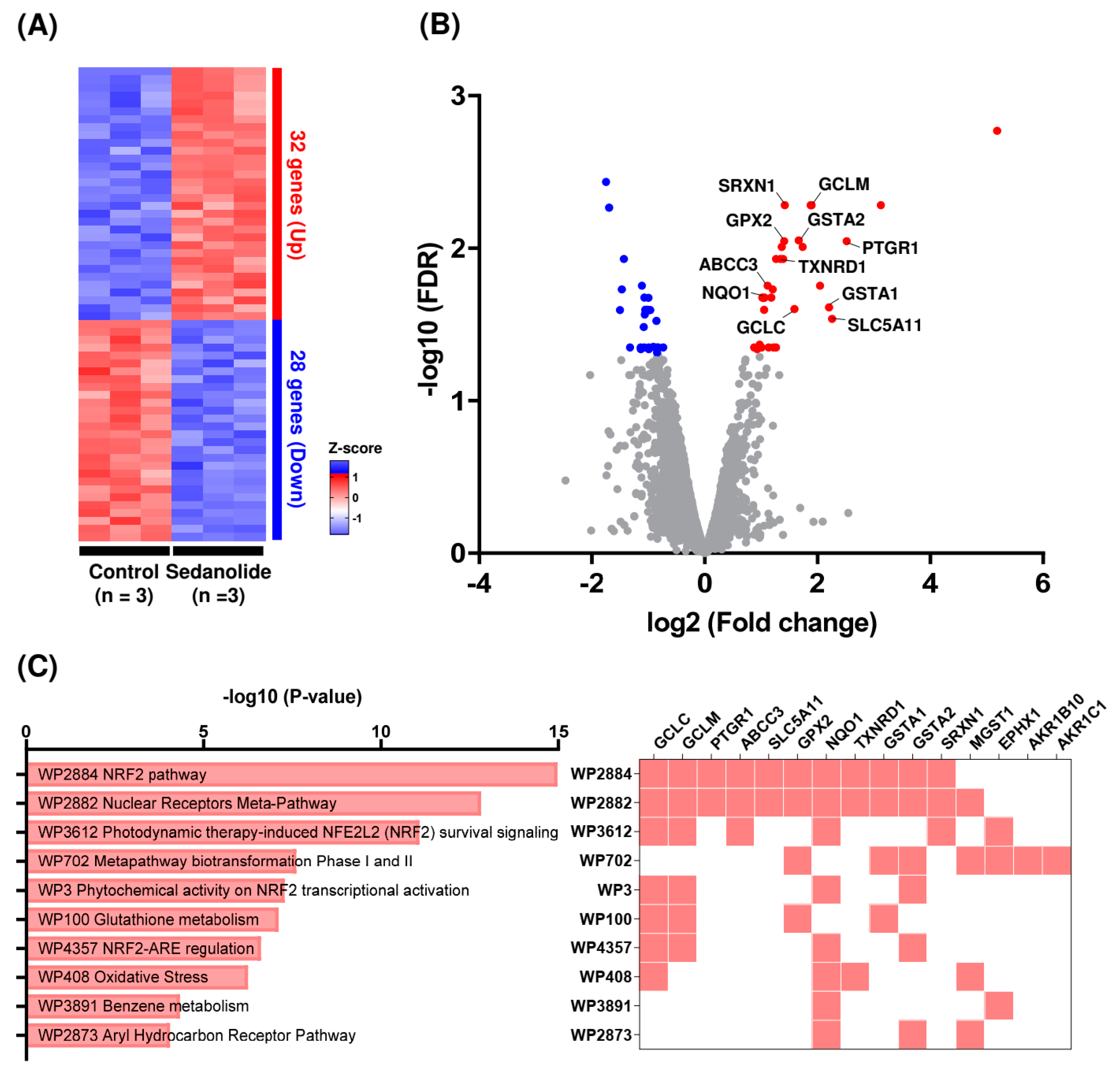
2.3. Sedanolide Activates the NRF2 Pathway
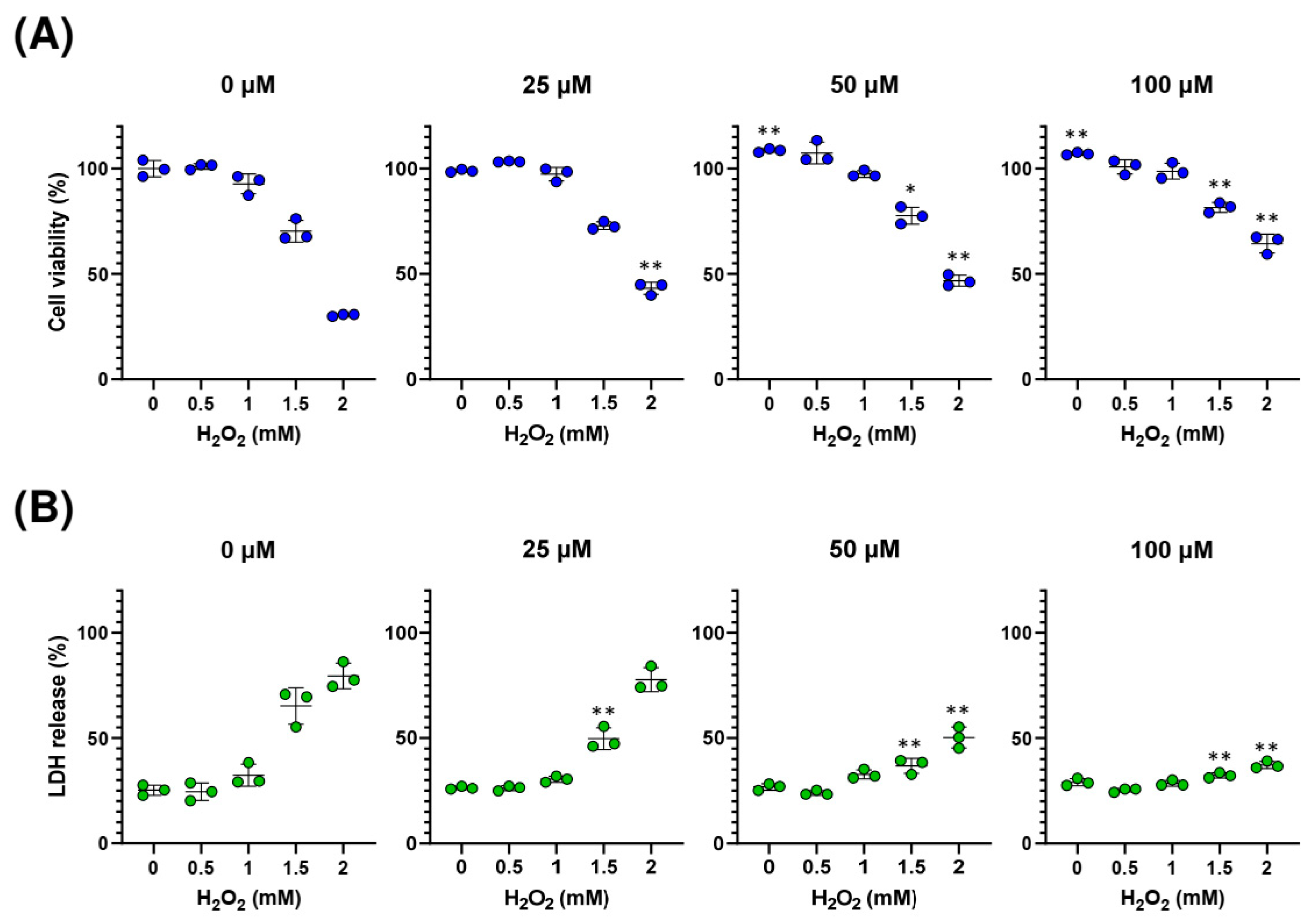
2.4. Sedanolide Protects against H2O2-Induced Cell Death
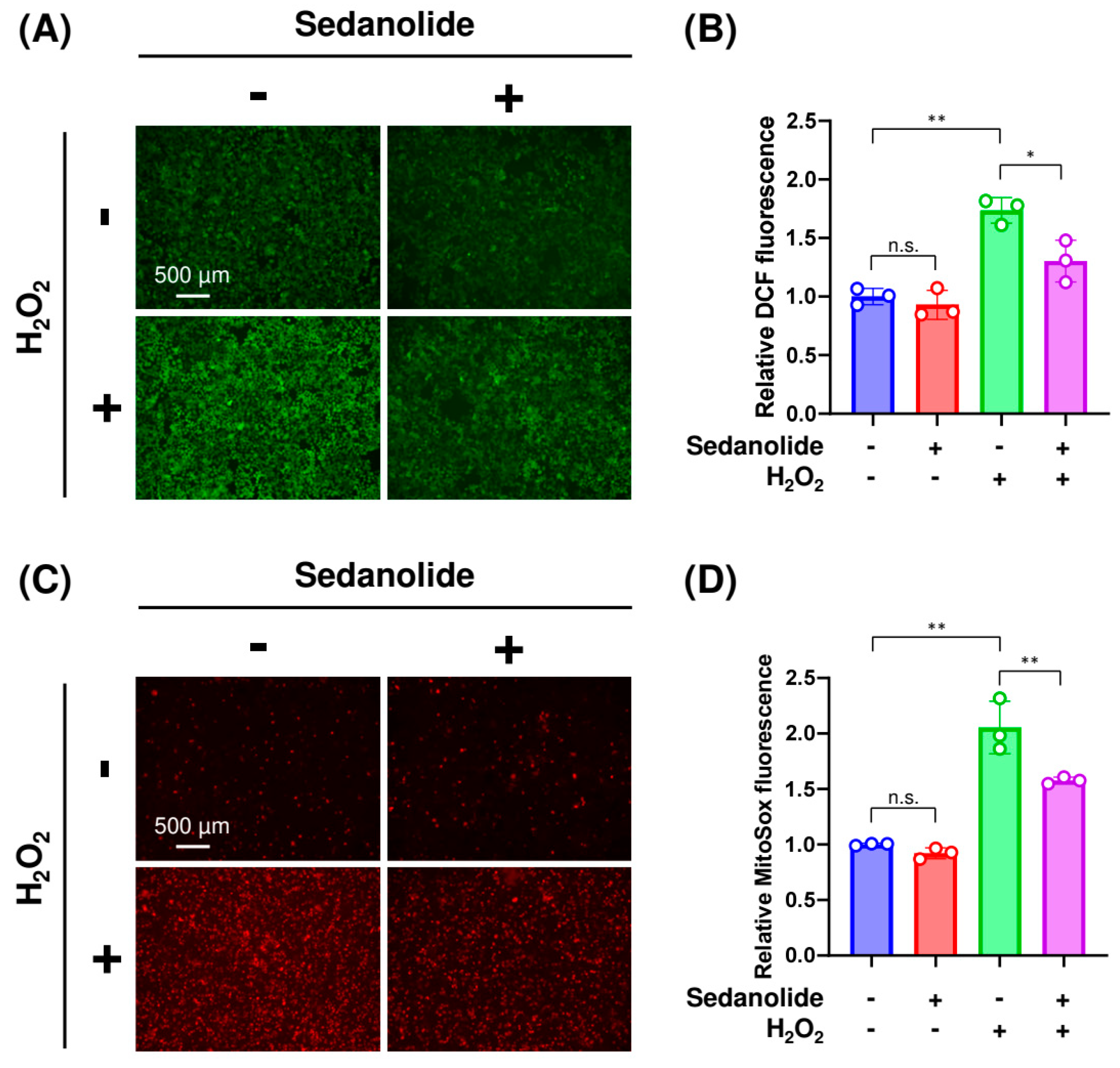
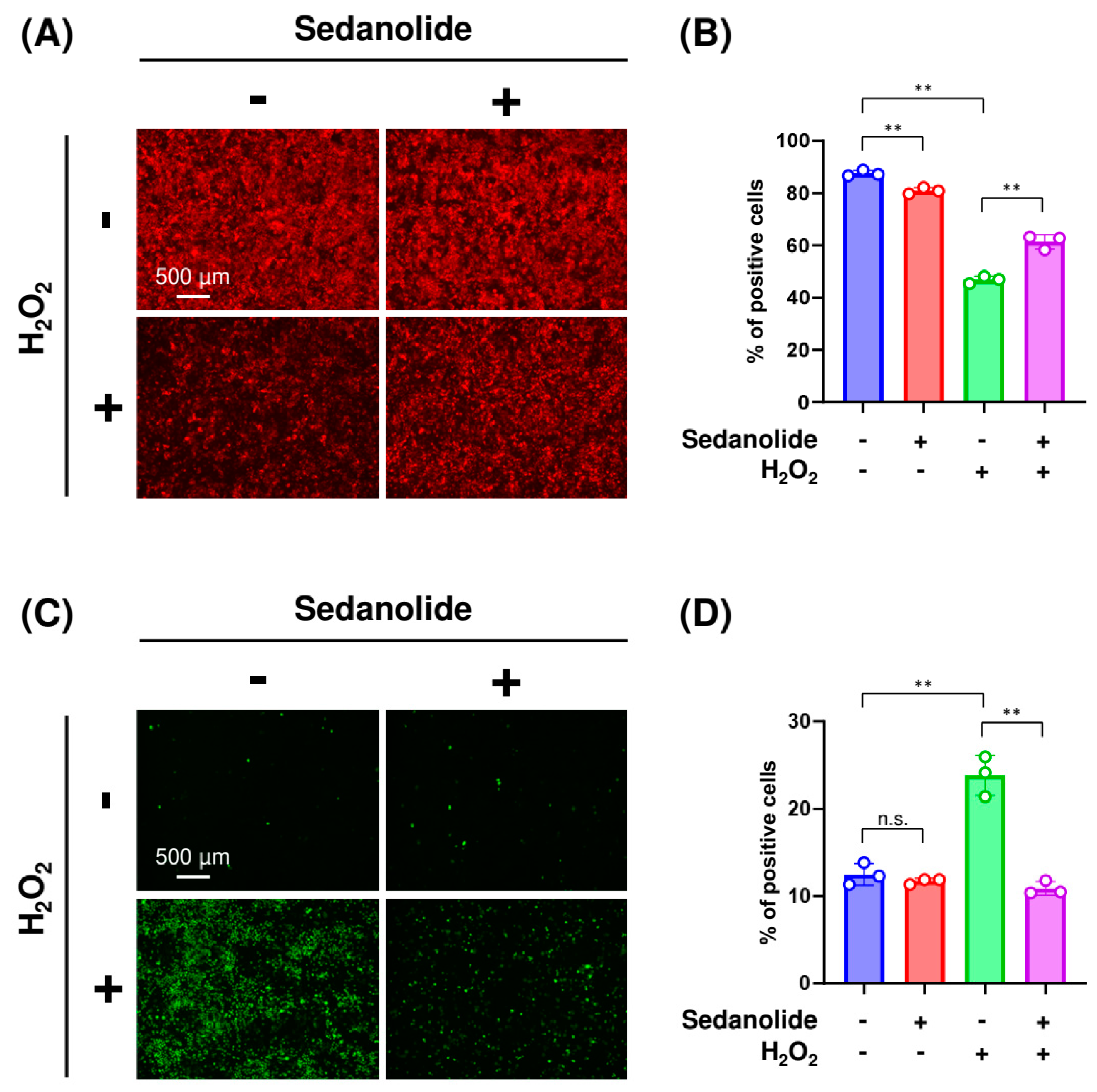
2.5. Sedanolide Reduces Cytosolic and Mitochondrial ROS Levels Increased by H2O2
2.6. Sedanolide Alleviates H2O2-Induced Apoptosis
3. Discussion
4. Materials and Methods
4.1. Generation of Luciferases-Expressing HepG2 Cells
4.2. Cell Culture and Treatment
4.3. Cytotoxicity Assay
4.4. Real-Time Bioluminescence Measurement
4.5. Western Blot Analysis
4.6. Immunofluorescence Analysis
4.7. RNA Sequencing and Data Analysis
4.8. Measurement of Cytosolic and Mitochondrial ROS Level
4.9. Apoptosis Assay
4.10. Measurement of Mitochondrial Membrane Potential Changes
4.11. Quantification of Caspase-3/7 Activity
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Kobayashi, M.; Yamamoto, M. Nrf2–Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv. Enzyme Regul. 2006, 46, 113–140. [Google Scholar] [CrossRef]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kensler, T.W. The role of Keap1 in cellular protective response. Chem. Res. Toxicol. 2005, 18, 1779–1791. [Google Scholar] [CrossRef]
- Zhang, D.D. Mechanistic studies of the Nrf2–Keap1 signaling pathway. Drug Metab. Rev. 2006, 38, 69–89. [Google Scholar] [CrossRef]
- Balogun, E.; Hoque, M.; Gong, P.; Kikkeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-response element. Biochem. J. 2003, 371, 887–895. [Google Scholar] [CrossRef]
- Juge, N.; Mithen, R.F.; Traka, M. Molecular basis for chemoprevention by sulforaphane: A comprehensive review. Cell. Mol. Life Sci. 2007, 64, 1105–1127. [Google Scholar] [CrossRef]
- Ungvari, Z.; Bagi, Z.; Feher, A.; Recchia, F.A.; Sonntag, W.E.; Pearson, K.; de Cabo, R.; Csiszar, A. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H18–H24. [Google Scholar] [CrossRef]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; Leon, R.; Lopez, M.G.; Oliva, B.; et al. Transcription factor NRF2 as a therapeutic target for chronic disease: A systems medicine approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Copple, I.M. Advances and challenges in therapeutic targeting of NRF2. Trends Pharmacol. 2023, 44, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Chan, S.S.; Chung, H.S.; Li, S.L. Chemistry and biological activities of naturally occurring phthalides. Stud. Nat. Prod. Chem. 2005, 32, 611–669. [Google Scholar]
- Wei, X.; Zheng, Y.; Sun, C.; Meng, F.; Wang, Y. Recent advances in natural phthalides: Distribution, chemistry, and biological activities. Fitoterapia 2022, 160, 105223. [Google Scholar] [CrossRef]
- Bjeldanes, L.F.; Kim, I.S. Phthalide components of celery essential oil. J. Org. Chem. 1977, 42, 2333–2335. [Google Scholar] [CrossRef]
- Tang, J.; Zhang, Y.; Hartman, T.G.; Rosen, R.T.; Ho, C.T. Free and glycosidically bound volatile compounds in fresh celery (Apium graveolens L.). J. Agric. Food Chem. 1990, 38, 1937–1940. [Google Scholar] [CrossRef]
- Momin, R.A.; Nair, M.G. Mosquitocidal, nematicidal and antifungal compounds from Apium graveolens L. seeds. J. Agric. Food Chem. 2001, 49, 142–145. [Google Scholar] [CrossRef]
- Momin, R.A.; Nair, M.G. Antioxidant, cyclooxygenase and topoisomerase inhibitory compounds from Apium graveolens Linn. seeds. Phytomedicine 2002, 9, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.Q.; Kenney, P.M.; Zhang, J.; Lam, L.K. Chemoprevention of benzo[a]pyrene-induced forestomach cancer in mice by natural phthalides from celery seed oil. Nutr. Cancer 1993, 19, 77–86. [Google Scholar] [CrossRef]
- Li, S.; Zhuge, A.; Xia, J.; Wang, S.; Lv, L.; Wang, K.; Jiang, H.; Yan, R.; Yang, L.; Bian, X.; et al. Bifidobacterium longum R0175 protects mice against APAP-induced liver injury by modulating the Nrf2 pathway. Free Radic. Biol. Med. 2023, 203, 11–23. [Google Scholar] [CrossRef]
- Takeda, H.; Nakajima, Y.; Yamaguchi, T.; Watanabe, I.; Miyoshi, S.; Nagashio, K.; Sekine, H.; Motohashi, H.; Yano, H.; Tanaka, J. The anti-inflammatory and anti-oxidative effect of a classical hypnotic bromovalerylurea mediated by the activation of NRF2. J. Biochem. 2023, 174, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Yamamoto, M. Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Martens, A.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.; Miller, R.A.; Digles, D.; Lopes, E.N.; Ehrhart, F.; et al. WikiPathways: Connecting communities. Nucleic Acids Res. 2021, 49, D613–D621. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Savitskaya, M.A.; Onishchenko, G.E. Mechanisms of apoptosis. Biochemistry 2015, 80, 1393–1405. [Google Scholar] [CrossRef]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef]
- Tabei, Y.; Murotomi, K.; Umeno, A.; Horie, M.; Tsujino, Y.; Masutani, B.; Yoshida, Y.; Nakajima, Y. Antioxidant properties of 5-hydroxy-4-phenyl-butenolide via activation of Nrf2/ARE signaling pathway. Food Chem. Toxicol. 2017, 107, 129–137. [Google Scholar] [CrossRef]
- Sun, Z.; Zhang, S.; Chan, J.Y.; Zhang, D.D. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol. Cell Biol. 2007, 27, 6334–6349. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Jaiswal, A.K. GSK-3β acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef] [PubMed]
- Papp, L.V.; Lu, J.; Holmgren, A.; Khanna, K.K. From selenium to selenoproteins: Synthesis, identity, and their role in human health. Antioxid. Redox Signal. 2007, 9, 775–806. [Google Scholar] [CrossRef] [PubMed]
- Franklin, C.C.; Backos, D.S.; Mohar, I.; White, C.C.; Forman, H.J.; Kavanagh, T.J. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol. Aspects Med. 2009, 30, 86–98. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, Y.; Miller, M.L.; Shen, D.; Shertzer, H.G.; Stringer, K.F.; Wang, B.; Schneider, S.N.; Nebert, D.W.; Dalton, T.P. Hepatocyte-specific Gclc deletion leads to rapid onset of steatosis with mitochondrial injury and liver failure. Hepatology 2007, 45, 1118–1128. [Google Scholar] [CrossRef]
- Lim, J.; Nakamura, B.N.; Mohar, I.; Kavanagh, T.J.; Luderer, U. Glutamate cycteine ligase modifier subunit (Gclm) null mice have increased ovarian oxidative stress and accelerated age-relate ovarian failure. Endocrinology 2015, 156, 3329–3343. [Google Scholar] [CrossRef]
- Liang, F.Q.; Alssadi, R.; Morehead, P.; Awasthi, Y.C.; Godley, B.F. Enhanced expression of glutathione-S-transferase A1-1 protects against oxidative stress in human retinal pigment epithelial cells. Exp. Eye Res. 2005, 80, 113–119. [Google Scholar] [CrossRef]
- Ramsay, E.E.; Dilda, P.J. Glutathione S-conjugates as prodrugs to target drug-resistant tumors. Front. Pharmacol. 2014, 5, 181. [Google Scholar] [CrossRef]
- Yang, Y.; Dieter, M.Z.; Chen, Y.; Shertzer, H.G.; Nebert, D.W.; Dalton, T.P. Initial characterization of glutamate-cysteine ligase modifier subunit Gclm(-/-) knockout mouse. Novel model system for a severely compromised oxidative stress response. J. Biol. Chem. 2002, 277, 49446–49452. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Talalay, P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch. Biochem. Biophys. 2010, 501, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Arnér, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef]
- Li, X.; He, P.; Wang, X.L.; Zhang, S.; Devejian, N.; Bennett, E.; Cai, C. Sulfiredoxin-1 enhance cardiac progenitor cell survival against oxidative stress via the upregulation of the ERK/NRF2 signaling pathway. Free Radic. Biol. Med. 2018, 123, 8–19. [Google Scholar] [CrossRef]
- Penning, T.M. Aldo-keto reductase regulation by the Nrf2 system: Implications for stress response, chemotherapy drug resistance, and carcinogenesis. Chem. Res. Toxicol. 2017, 30, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Llambi, F. Cell death signaling. Cold Spring Harb. Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef]
- Er, E.; Oliver, L.; Cartron, P.F.; Juin, P.; Manon, S.; Vallette, F.M. Mitochondria as the target of the pro-apoptotic protein Bax. Biochem. Biophys. Acta 2006, 1757, 1301–1311. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, e008656. [Google Scholar] [CrossRef]
- Lakhani, S.A.; Masud, A.; Kuida, K.; Porter Jr, G.A.; Booth, C.J.; Mehal, W.Z.; Inayat, I.; Flavell, R.A. Caspase 3 and 7: Key mediators of mitochondrial events of apoptosis. Science 2006, 311, 847–851. [Google Scholar] [CrossRef]
- McComb, S.; Chan, P.K.; Guinot, A.; Hartmannsdottir, H.; Jenni, S.; Dobay, M.P.; Bourquin, J.P.; Bornhauser, B.C. Efficient apoptosis requires feedback amplification of upstream apoptotic signals by effector caspase-3 or -7. Sci. Adv. 2019, 5, eaau9433. [Google Scholar] [CrossRef]
- Uno, K.; Murotomi, K.; Kazuki, Y.; Oshimura, M.; Nakajima, Y. Bioluminescence-based cytotoxicity assay for simultaneous evaluation of cell viability and membrane damage in human hepatoma HepG2 cells. Luminescence 2018, 33, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Yamazaki, T.; Nishii, S.; Noguchi, T.; Hoshino, H.; Niwa, K.; Viviani, V.R.; Ohmiya, Y. Enhanced beetle luciferase for high-resolution bioluminescence imaging. PLoS ONE 2010, 5, e10011. [Google Scholar] [CrossRef] [PubMed]
- Takiguchi, M.; Kazuki, Y.; Hiramatsu, K.; Abe, S.; Iida, Y.; Takehara, S.; Nishida, T.; Ohbayashi, T.; Wakayama, T.; Oshimura, M. A novel and stable mouse artificial chromosome vector. ACS Synth. Biol. 2014, 3, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Wakuri, S.; Yamakage, K.; Kazuki, Y.; Kazuki, K.; Oshimura, M.; Aburatani, S.; Yasunaga, M.; Nakajima, Y. Correlation between luminescence intensity and cytotoxicity in cell-based cytotoxicity assay using luciferase. Anal. Biochem. 2017, 522, 18–29. [Google Scholar] [CrossRef]
- Noguchi, T.; Michihata, T.; Nakamura, W.; Takumi, T.; Shimizu, R.; Yamamoto, M.; Ikeda, M.; Ohmiya, Y.; Nakajima, Y. Dual-color luciferase mouse directly demonstrates coupled expression of two clock genes. Biochemistry 2010, 49, 8053–8061. [Google Scholar] [CrossRef]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-seq data. BMC Bioinform. 2018, 19, 534. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tabei, Y.; Abe, H.; Suzuki, S.; Takeda, N.; Arai, J.-i.; Nakajima, Y. Sedanolide Activates KEAP1–NRF2 Pathway and Ameliorates Hydrogen Peroxide-Induced Apoptotic Cell Death. Int. J. Mol. Sci. 2023, 24, 16532. https://doi.org/10.3390/ijms242216532
Tabei Y, Abe H, Suzuki S, Takeda N, Arai J-i, Nakajima Y. Sedanolide Activates KEAP1–NRF2 Pathway and Ameliorates Hydrogen Peroxide-Induced Apoptotic Cell Death. International Journal of Molecular Sciences. 2023; 24(22):16532. https://doi.org/10.3390/ijms242216532
Chicago/Turabian StyleTabei, Yosuke, Hiroko Abe, Shingo Suzuki, Nobuaki Takeda, Jun-ichiro Arai, and Yoshihiro Nakajima. 2023. "Sedanolide Activates KEAP1–NRF2 Pathway and Ameliorates Hydrogen Peroxide-Induced Apoptotic Cell Death" International Journal of Molecular Sciences 24, no. 22: 16532. https://doi.org/10.3390/ijms242216532
APA StyleTabei, Y., Abe, H., Suzuki, S., Takeda, N., Arai, J.-i., & Nakajima, Y. (2023). Sedanolide Activates KEAP1–NRF2 Pathway and Ameliorates Hydrogen Peroxide-Induced Apoptotic Cell Death. International Journal of Molecular Sciences, 24(22), 16532. https://doi.org/10.3390/ijms242216532