
The Thermodynamic Fingerprints of Ultra-Tight Nanobody–Antigen Binding Probed via Two-Color Single-Molecule Coincidence Detection
 , ,
, ,
Abstract
1. Introduction
2. Results
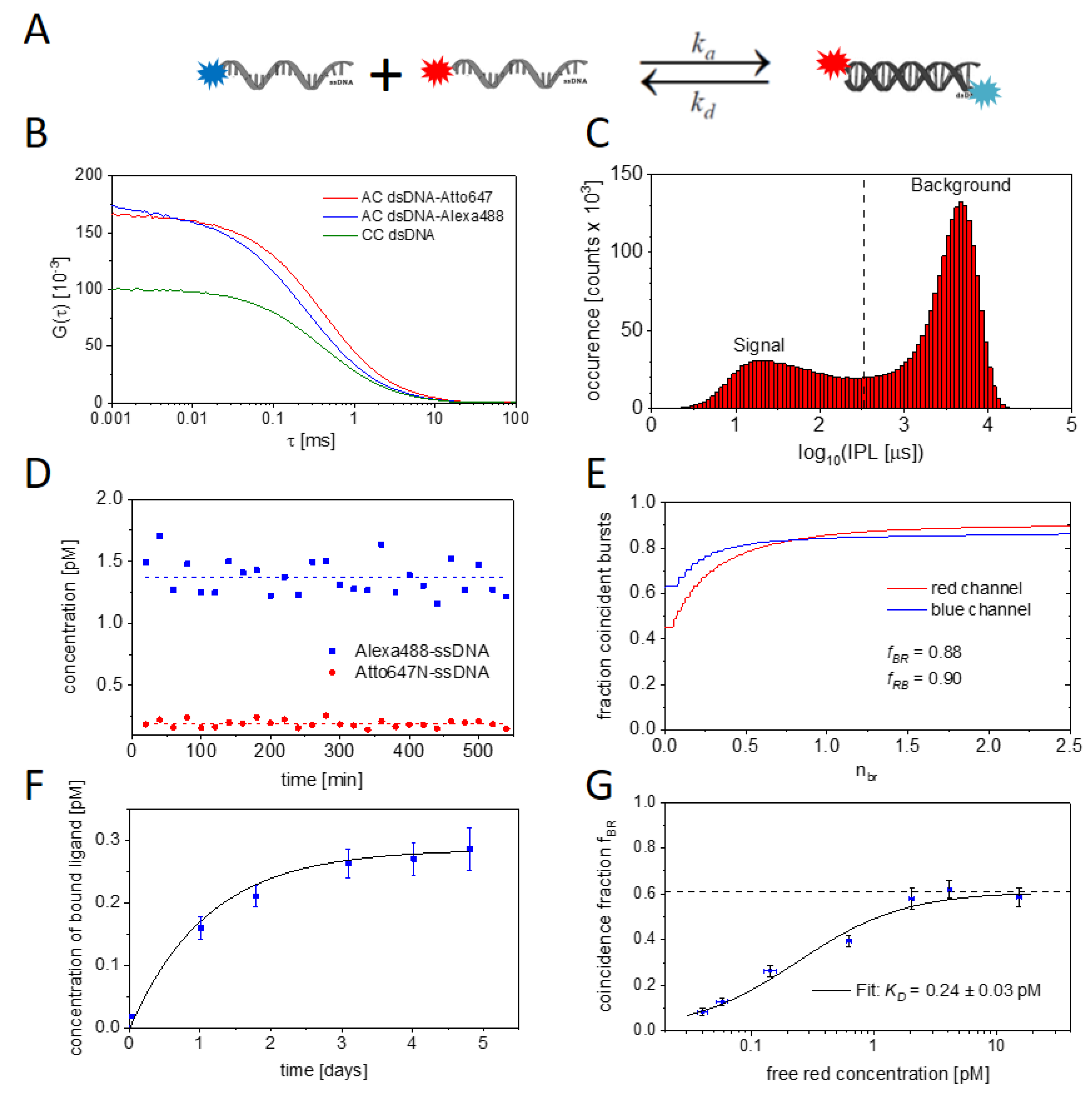
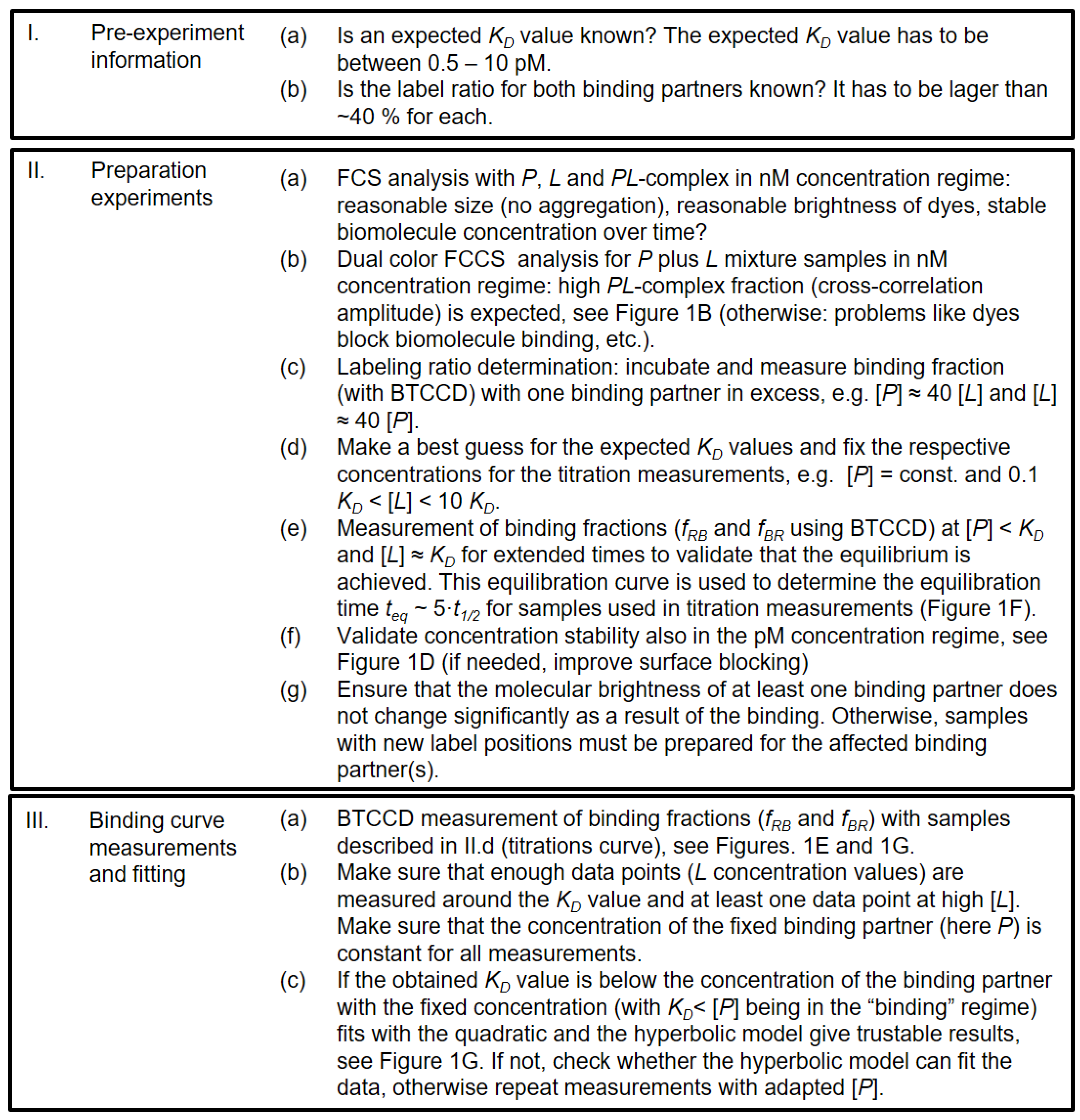
2.1. Experimental and Methodical Design for the Determination of KD Values Using BTCCD
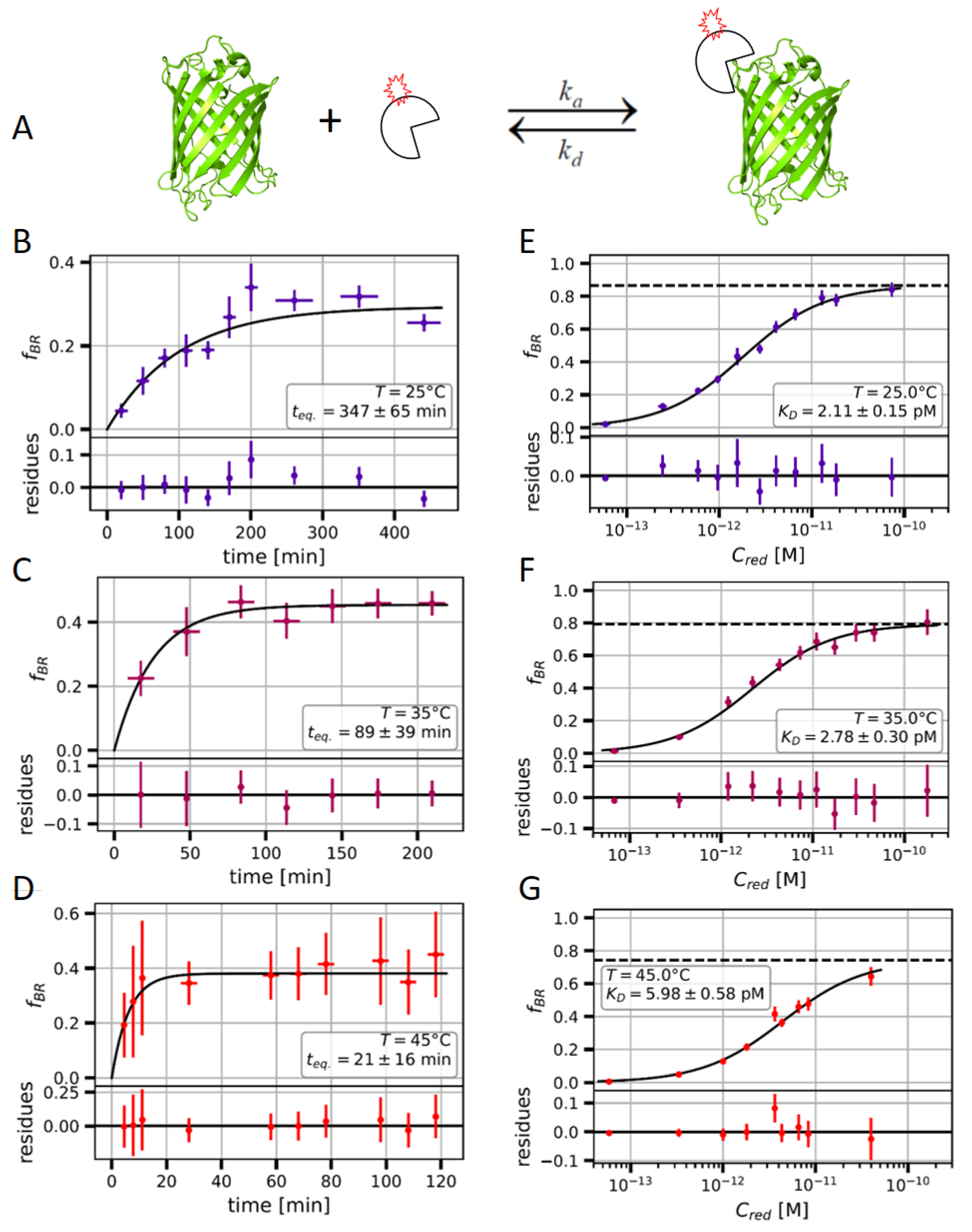
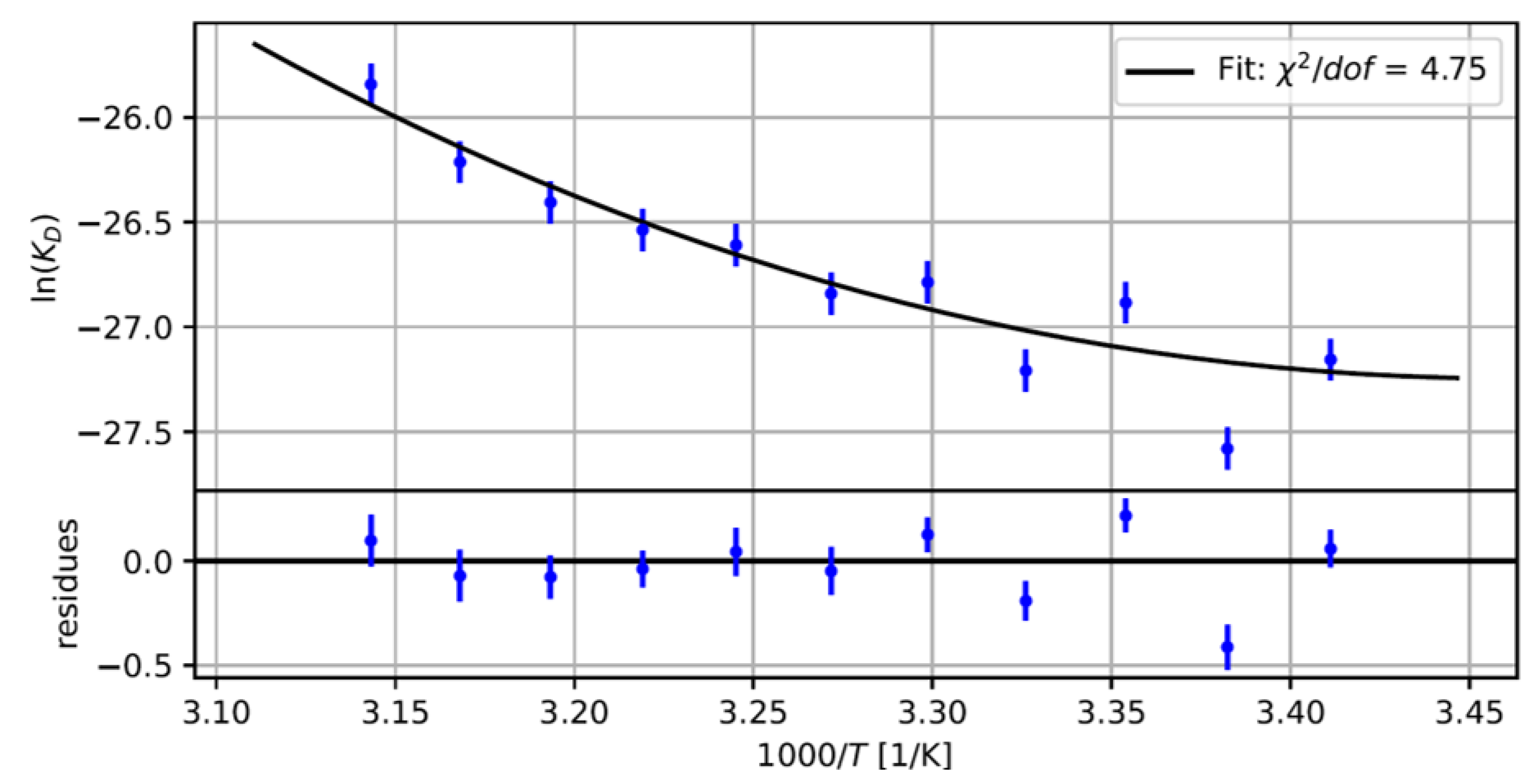
2.2. Measuring the Nanobody-EGFP Binding Affinity and Thermodynamic Analysis
3. Discussion
4. Materials and Methods
4.1. Sample Preparation
4.2. Confocal Microscopy and Data Acquisition
4.3. Burst Analysis and BTCCD
4.4. Corrections for Binding Fractions and Resulting KD Values
4.5. Thermodynamics of the Bi-Molecular Binding
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kuriyan, J.; Konforti, B.; Wemmer, D. The Molecules of Life: Physical and Chemical Principles; Garland Science, Taylor & Francis Group: New York, NY, USA, 2013. [Google Scholar]
- Kastritis, P.L.; Bonvin, A.M. On the Binding Affinity of Macromolecular Interactions: Daring to Ask Why Proteins Interact. J. R. Soc. Interface 2013, 10, 20120835. [Google Scholar] [CrossRef]
- Pollard, T.D. A Guide to Simple and Informative Binding Assays. Mol. Biol. Cell 2010, 21, 4061–4067. [Google Scholar] [CrossRef]
- Sanders, R.C. Biomolecular Ligand-Receptor Binding Studies: Theory, Practice, and Analysis; Vanderbilt University: Nashville, TN, USA, 2010. [Google Scholar]
- Jarmoskaite, I.; AlSadhan, I.; Vaidyanathan, P.P.; Herschlag, D. How to Measure and Evaluate Binding Affinities. eLife 2020, 9, e57264. [Google Scholar] [CrossRef]
- Altschuler, S.E.; Lewis, K.A.; Wuttke, D.S. Practical Strategies for the Evaluation of High-Affinity Protein/Nucleic Acid Interactions. J. Nucleic Acids Investig. 2013, 4, 19–28. [Google Scholar] [CrossRef]
- Chaturvedi, S.K.; Ma, J.; Zhao, H.; Schuck, P. Use of Fluorescence-Detected Sedimentation Velocity to Study High-Affinity Protein Interactions. Nat. Protoc. 2017, 12, 1777–1791. [Google Scholar]
- Pierce, M.M.; Raman, C.S.; Nall, B.T. Isothermal Titration Calorimetry of Protein-Protein Interactions. Methods 1999, 19, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Campoy, A.; Freire, E. Itc in the Post-Genomic Era..? Priceless. Biophys. Chem. 2005, 115, 115–124. [Google Scholar] [CrossRef]
- Wienken, C.J.; Baaske, P.; Rothbauer, U.; Braun, D.; Duhr, S. Protein-Binding Assays in Biological Liquids Using Microscale Thermophoresis. Nat. Commun. 2010, 1, 100. [Google Scholar] [CrossRef]
- Rich, R.L.; Myszka, D.G. Survey of the Year 2007 Commercial Optical Biosensor Literature. J. Mol. Recognit. 2008, 21, 355–400. [Google Scholar] [PubMed]
- Bujalowski, W.M.; Jezewska, M.J. Using Structure-Function Constraints in Fret Studies of Large Macromolecular Complexes. Methods Mol. Biol. 2012, 875, 135–164. [Google Scholar] [PubMed]
- Kriegsmann, J.; Brehs, M.; Klare, J.P.; Engelhard, M.; Fitter, J. Sensory Rhodopsin Ii/Transducer Complex Formation in Detergent and in Lipid Bilayers Studied with Fret. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Oyama, R.; Takashima, H.; Yonezawa, M.; Doi, N.; Miyamoto-Sato, E.; Kinjo, M.; Yanagawa, H. Protein-Protein Interaction Analysis by C-Terminally Specific Fluorescence Labeling and Fluorescence Cross-Correlation Spectroscopy. Nucleic Acids Res. 2006, 34, e102. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ruan, Q.; Tetin, S.Y. Applications of Dual-Color Fluorescence Cross-Correlation Spectroscopy in Antibody Binding Studies. Anal. Biochem. 2008, 374, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Machan, R.; Wohland, T. Recent Applications of Fluorescence Correlation Spectroscopy in Live Systems. FEBS Lett. 2014, 588, 3571–3584. [Google Scholar] [CrossRef]
- Knezevic, J.; Langer, A.; Hampel, P.A.; Kaiser, W.; Strasser, R.; Rant, U. Quantitation of Affinity, Avidity, and Binding Kinetics of Protein Analytes with a Dynamically Switchable Biosurface. J. Am. Chem. Soc. 2012, 134, 15225–15228. [Google Scholar] [CrossRef]
- Andrews, R. DNA Hybridisation Kinetics Using Single-Molecule Fluorescence Imaging. Essays Biochem. 2021, 65, 27–36. [Google Scholar]
- Dupuis, N.F.; Holmstrom, E.D.; Nesbitt, D.J. Single-Molecule Kinetics Reveal Cation-Promoted DNA Duplex Formation through Ordering of Single-Stranded Helices. Biophys. J. 2013, 105, 756–766. [Google Scholar] [CrossRef]
- Zhang, H.D.; Liu, Y.J.; Zhang, K.; Ji, J.; Liu, J.W.; Liu, B.H. Single Molecule Fluorescent Colocalization of Split Aptamers for Ultrasensitive Detection of Biomolecules. Anal. Chem. 2018, 90, 9315–9321. [Google Scholar] [CrossRef]
- Sigmundsson, K.; Masson, G.; Rice, R.H.; Beauchemin, N.; Obrink, B. Determination of Active Concentrations and Association and Dissociation Rate Constants of Interacting Biomolecules: An Analytical Solution to the Theory for Kinetic and Mass Transport Limitations in Biosensor Technology and Its Experimental Verification. Biochemistry 2002, 41, 8263–8276. [Google Scholar] [CrossRef]
- Mohamad, N.R.; Marzuki, N.H.C.; Buang, N.A.; Huyop, F.; Wahab, R.A. An Overview of Technologies for Immobilization of Enzymes and Surface Analysis Techniques for Immobilized Enzymes. Biotechnol. Biotechnol. Equip. 2015, 29, 205–220. [Google Scholar] [CrossRef]
- Edwards, P.R.; Gill, A.; Pollardknight, D.V.; Hoare, M.; Buckle, P.E.; Lowe, P.A.; Leatherbarrow, R.J. Kinetics of Protein-Protein Interactions at the Surface of an Optical Biosensor. Anal. Biochem. 1995, 231, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Ries, J.; Petrasek, Z.; Garcia-Saez, A.J.; Schwille, P. A Comprehensive Framework for Fluorescence Cross-Correlation Spectroscopy. New J. Phys. 2010, 12, 113009. [Google Scholar] [CrossRef]
- Li, H.T.; Ying, L.M.; Green, J.J.; Balasubramanian, S.; Klenerman, D. Ultrasensitive Coincidence Fluorescence Detection of Single DNA Molecules. Anal. Chem. 2003, 75, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- Orte, A.; Clarke, R.; Balasubramanian, S.; Klenerman, D. Determination of the Fraction and Stoichiometry of Femtomolar Levels of Biomolecular Complexes in an Excess of Monomer Using Single-Molecule, Two-Color Coincidence Detection. Anal. Chem. 2006, 78, 7707–7715. [Google Scholar] [CrossRef]
- Goodwin, P.M.; Nolan, R.L.; Cai, H. Quantitative Analysis of Specific Nucleic Acid Sequences by Two-Color Single-Molecule Fluorescence Detection. Manip. Anal. Biomol. Cells Tissues 2003, 4962, 78–88. [Google Scholar]
- Höfig, H.; Yukhnovets, O.; Remes, C.; Kempf, N.; Katranidis, A.; Kempe, D.; Fitter, J. Brightness-Gated Two-Color Coincidence Detection Unravels Two Distinct Mechanisms in Bacterial Protein Translation Initiation. Commun. Biol. 2019, 2, 459. [Google Scholar] [CrossRef] [PubMed]
- Yukhnovets, O.; Höfig, H.; Bustorff, N.; Katranidis, A.; Fitter, J. Impact of Molecule Concentration, Diffusion Rates and Surface Passivation on Single-Molecule Fluorescence Studies in Solution. Biomolecules 2022, 12, 468. [Google Scholar] [CrossRef] [PubMed]
- Bielec, K.; Sozanski, K.; Seynen, M.; Dziekan, Z.; ten Wolde, P.R.; Holyst, R. Kinetics and Equilibrium Constants of Oligonucleotides at Low Concentrations. Hybridization and Melting Study. Phys. Chem. Chem. Phys. 2019, 21, 10798–10807. [Google Scholar] [CrossRef]
- Henry, K.A.; MacKenzie, C.R. Antigen Recognition by Single-Domain Antibodies: Structural Latitudes and Constraints. Mabs 2018, 10, 815–826. [Google Scholar] [CrossRef]
- Prabhu, N.V.; Sharp, K.A. Heat Capacity in Proteins. Annu. Rev. Phys. Chem. 2005, 56, 521–548. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into Protein-Ligand Interactions: Mechanisms, Models; Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Guardiola, S.; Varese, M.; Taules, M.; Diaz-Lobo, M.; Garcia, J.; Giralt, E. Probing the Kinetic and Thermodynamic Fingerprints of Anti-Egf Nanobodies by Surface Plasmon Resonance. Pharmaceuticals 2020, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Stein, J.A.C.; Ianeselli, A.; Braun, D. Kinetic Microscale Thermophoresis for Simultaneous Measurement of Binding Affinity and Kinetics. Angew. Chem.-Int. Ed. 2021, 60, 13988–13995. [Google Scholar] [CrossRef] [PubMed]
- Höfig, H.; Otten, J.; Steffen, V.; Pohl, M.; Arnold, J. Boersma; Jorg Fitter. Genetically Encoded Forster Resonance Energy Transfer-Based Biosensors Studied on the Single-Molecule Level. ACS Sens. 2018, 3, 1462–1470. [Google Scholar] [CrossRef]
- Kubala, M.H.; Kovtun, O.; Alexandrov, K.; Collins, B.M. Structural and Thermodynamic Analysis of the Gfp:Gfp-Nanobody Complex. Protein Sci. A Publ. Protein Soc. 2010, 19, 2389–2401. [Google Scholar] [CrossRef]
- Stites, W.E. Protein-Protein Interactions: Interface Structure, Binding Thermodynamics, and Mutational Analysis. Chem. Rev. 1997, 97, 1233–1250. [Google Scholar] [CrossRef]
- Zavrtanik, U.; Hadzi, S.; Lah, J. Unraveling the Thermodynamics of Ultra-Tight Binding of Intrinsically Disordered Proteins. Front. Mol. Biosci. 2021, 8, 726824. [Google Scholar] [CrossRef]
- Keeble, A.H.; Kirkpatrick, N.; Shimizu, S.; Kleanthous, C. Calorimetric Dissection of Colicin Dnase—Immunity Protein Complex Specificity. Biochemistry 2006, 45, 3243–3254. [Google Scholar] [CrossRef]
- Varese, M.; Guardiola, S.; Garcia, J.; Giralt, E. Enthalpy-Versus Entropy-Driven Molecular Recognition in the Era of Biologics. Chembiochem. A Eur. J. Chem. Biol. 2019, 20, 2981–2986. [Google Scholar] [CrossRef]
- Bee, C.; Abdiche, Y.N.; Pons, J.; Rajpal, A. Determining the Binding Affinity of Therapeutic Monoclonal Antibodies Towards Their Native Unpurified Antigens in Human Serum. PLoS ONE 2013, 8, e80501. [Google Scholar] [CrossRef]
- Lewis, K.A.; Pfaff, D.A.; Earley, J.N.; Altschuler, S.E.; Wuttke, D.S. The Tenacious Recognition of Yeast Telomere Sequence by Cdc13 Is Fully Exerted by a Single Ob-Fold Domain. Nucleic Acids Res. 2014, 42, 475–484. [Google Scholar] [CrossRef]
- Hadzi, S.; Lah, J. The Free Energy Folding Penalty Accompanying Binding of Intrinsically Disordered Alpha-Helical Motifs. Protein Sci. A Publ. Protein Soc. 2022, 31, e4370. [Google Scholar] [CrossRef]
- Drobnak, I.; De Jonge, N.; Haesaerts, S.; Vesnaver, G.; Loris, R.; Lah, J. Energetic Basis of Uncoupling Folding from Binding for an Intrinsically Disordered Protein. J. Am. Chem. Soc. 2013, 135, 1288–1294. [Google Scholar] [CrossRef]
- Borgia, A.; Borgia, M.B.; Bugge, K.; Kissling, V.M.; Heidarsson, P.O.; Fernandes, C.B.; Sottini, A.; Soranno, A.; Buholzer, K.J.; Nettels, D.; et al. Extreme Disorder in an Ultrahigh-Affinity Protein Complex. Nature 2018, 555, 61–66. [Google Scholar] [CrossRef]
- Müller, B.K.; Zaychikov, E.; Bräuchle, C.; Lamb, D.C. Pulsed Interleaved Excitation. Biophys. J. 2005, 89, 3508–3522. [Google Scholar] [CrossRef]
- Watkins, L.P.; Yang, H. Detection of Intensity Change Points in Time-Resolved Single-Molecule Measurements. J. Phys. Chem. B 2005, 109, 617–628. [Google Scholar] [CrossRef]
- Fries, J.R.; Brand, L.; Eggeling, C.; Kollner, M.; Seidel, C.A.M. Quantitative Identification of Different Single Molecules by Selective Time-Resolved Confocal Fluorescence Spectroscopy. J. Phys. Chem. A 1998, 102, 6601–6613. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | KD [pM] | ∆G [kJ/mol] | ∆H [kJ/mol] | T∆S [kJ/mol] | ∆CP [kJ/mol K] |
|---|---|---|---|---|---|
| (1) Nanobody-EGFP 1 | 1.69 | −67.18 | −22.4 | 44.8 | −2.39 |
| (2) Nb1-EGF 2 | 5833 | −47.0 | −44.6 | 2.5 | −2.9 |
| (3) Nb6_EGF 2 | 24,000 | −43.5 | −78.7 | −35.2 | −3.2 |
| (4) HyHEL−5-hen egg lysozyme 3 | 23.49 | −60.67 | −94.56 | −33.93 | −1.42 |
| (5) Ab 5F8–cytochrome C 3 | 64.67 | −58.16 | −90.79 | −32.81 | −0.72 |
| (6) HigA2-HigB2 4 | 3.10 | −65.69 | −144.77 | −79.91 | −1.92 |
| (7) E9-Im9 5 | 0.023 | −77.82 | −43.93 | 33.89 | −1.88 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schedler, B.; Yukhnovets, O.; Lindner, L.; Meyer, A.; Fitter, J. The Thermodynamic Fingerprints of Ultra-Tight Nanobody–Antigen Binding Probed via Two-Color Single-Molecule Coincidence Detection. Int. J. Mol. Sci. 2023, 24, 16379. https://doi.org/10.3390/ijms242216379
Schedler B, Yukhnovets O, Lindner L, Meyer A, Fitter J. The Thermodynamic Fingerprints of Ultra-Tight Nanobody–Antigen Binding Probed via Two-Color Single-Molecule Coincidence Detection. International Journal of Molecular Sciences. 2023; 24(22):16379. https://doi.org/10.3390/ijms242216379
Chicago/Turabian StyleSchedler, Benno, Olessya Yukhnovets, Lennart Lindner, Alida Meyer, and Jörg Fitter. 2023. "The Thermodynamic Fingerprints of Ultra-Tight Nanobody–Antigen Binding Probed via Two-Color Single-Molecule Coincidence Detection" International Journal of Molecular Sciences 24, no. 22: 16379. https://doi.org/10.3390/ijms242216379
APA StyleSchedler, B., Yukhnovets, O., Lindner, L., Meyer, A., & Fitter, J. (2023). The Thermodynamic Fingerprints of Ultra-Tight Nanobody–Antigen Binding Probed via Two-Color Single-Molecule Coincidence Detection. International Journal of Molecular Sciences, 24(22), 16379. https://doi.org/10.3390/ijms242216379