
Computational Prediction of the Interaction of Ivermectin with Fibrinogen
 , ,
, ,  and
and
Abstract
1. Introduction
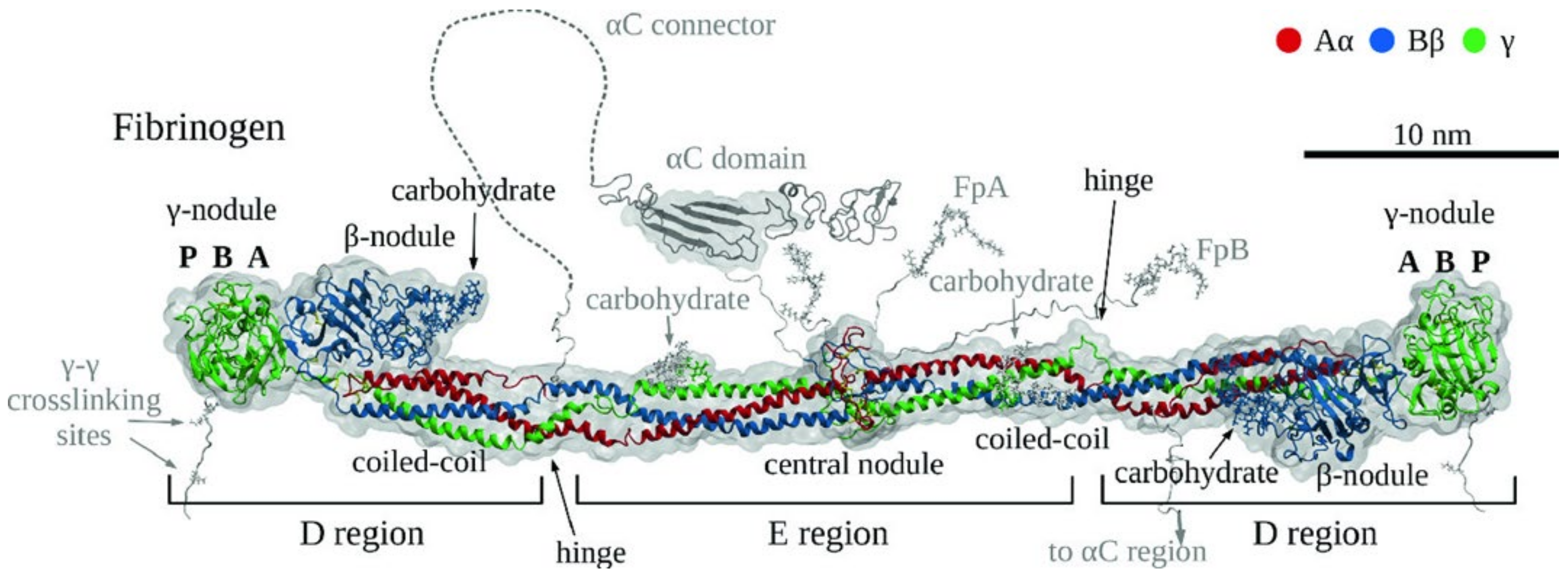
1.1. Structure and Function of Fibrinogen
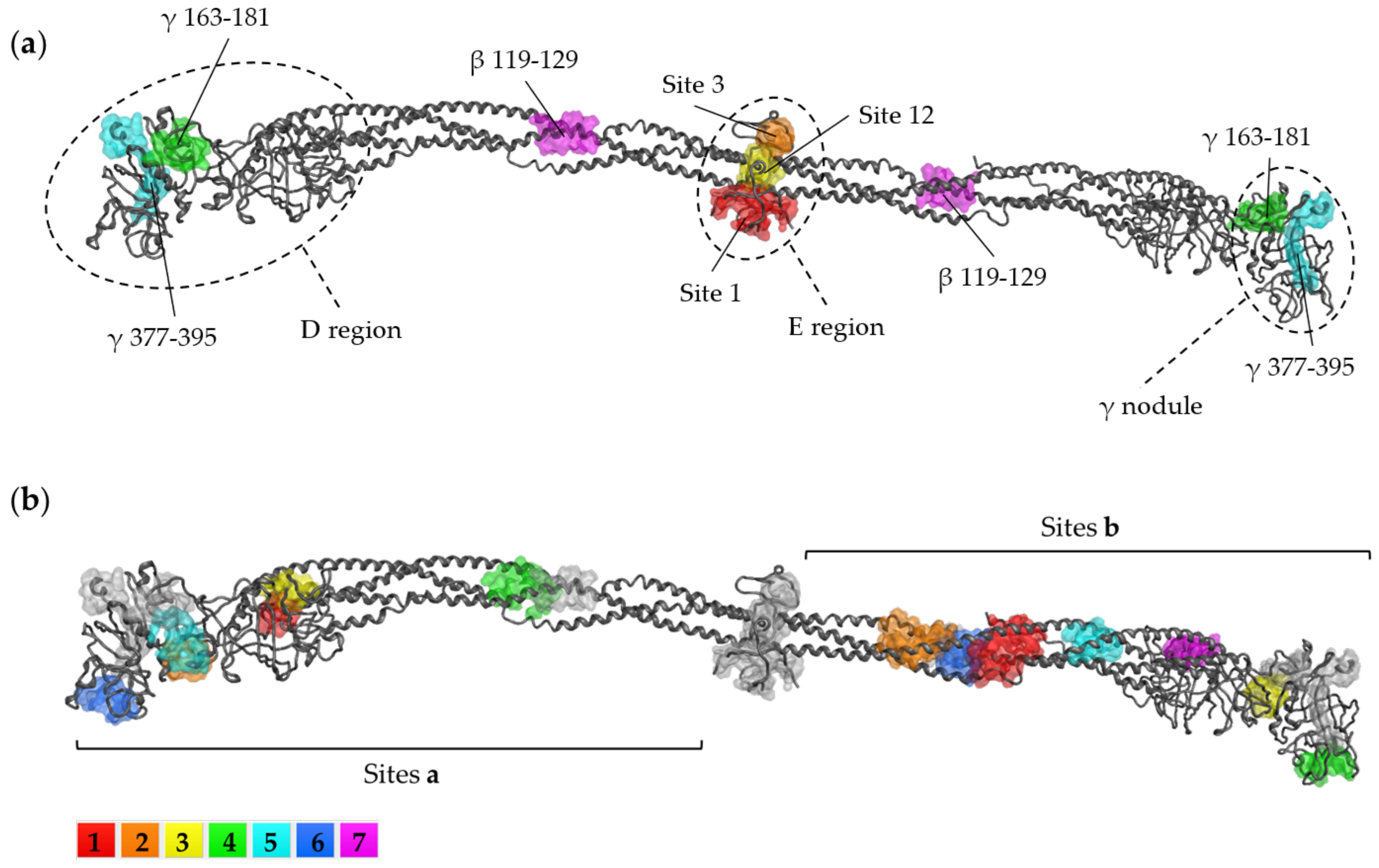
Fibrinogen Domains and Interactions
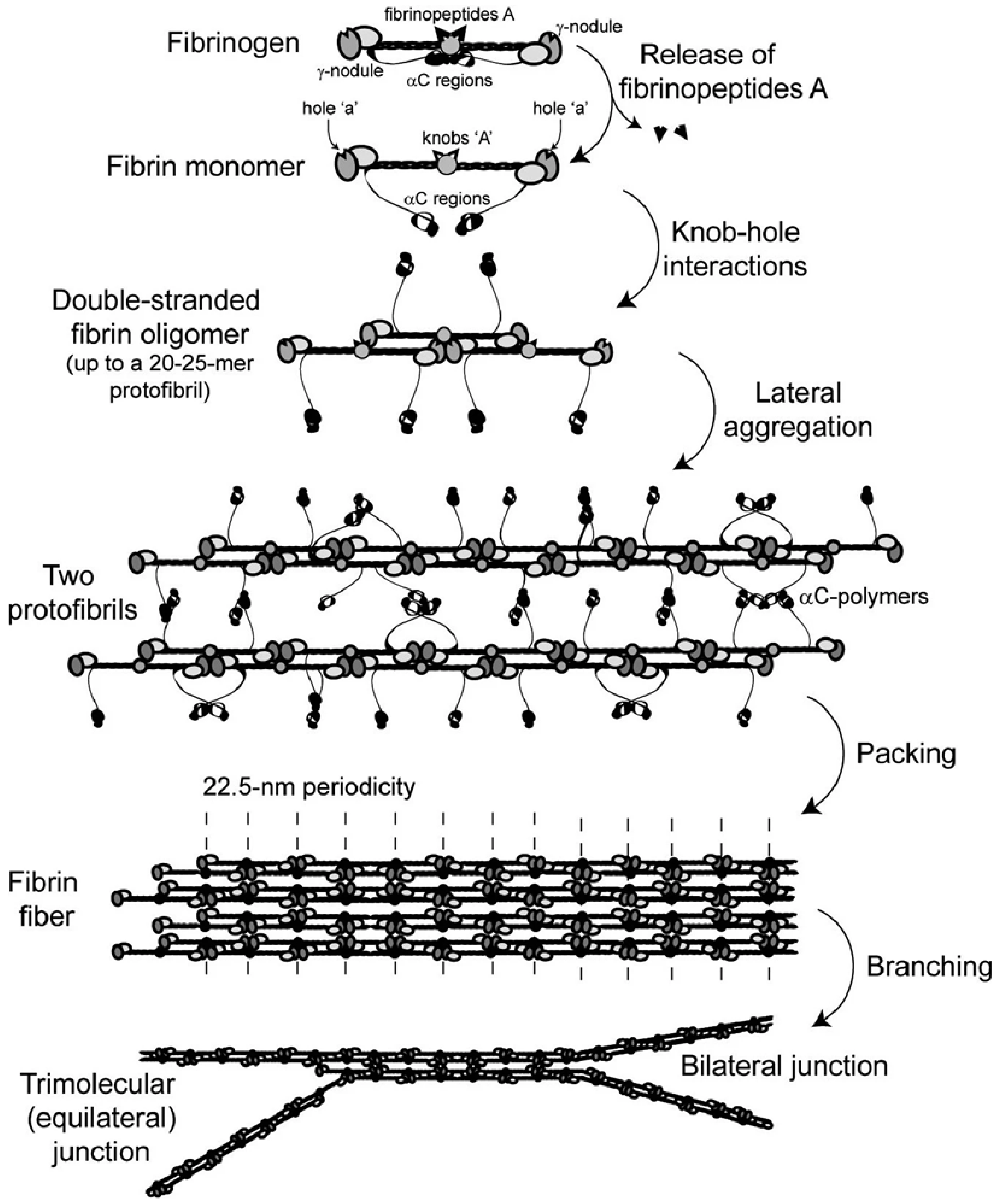
1.2. Fibrin Clot Formation
- Formation of monomeric fibrin: thrombin catalyzes the release of the FpA from the Aα chains, resulting in a fibrin monomer and exposing the ‘knobs A’. The release of FpB is a slower reaction, not essential for polymerization.
- Formation of fibrin protofibrils: the fibrin monomers self-assemble so that the ‘knobs A’ in the E region of one monomer fit into two ‘holes a’ in the D domains of two other adjacent monomers. Longitudinal growth leads to the formation of double-stranded protofibrils.
- Formation of fibrin fibers: protofibrils keep growing linearly and aggregate laterally at the same time to build a fiber, stabilized by ‘knob B–hole b’ bonds and other interactions.
- Formation of a fibrin network: a three-dimensional network architecture is achieved through further longitudinal and lateral growth, as well as branching.
1.3. Clot Properties and Disease
1.3.1. Role of SP in Microclotting
1.3.2. Potential Anti-Thrombo-Inflammatory Action of IVM
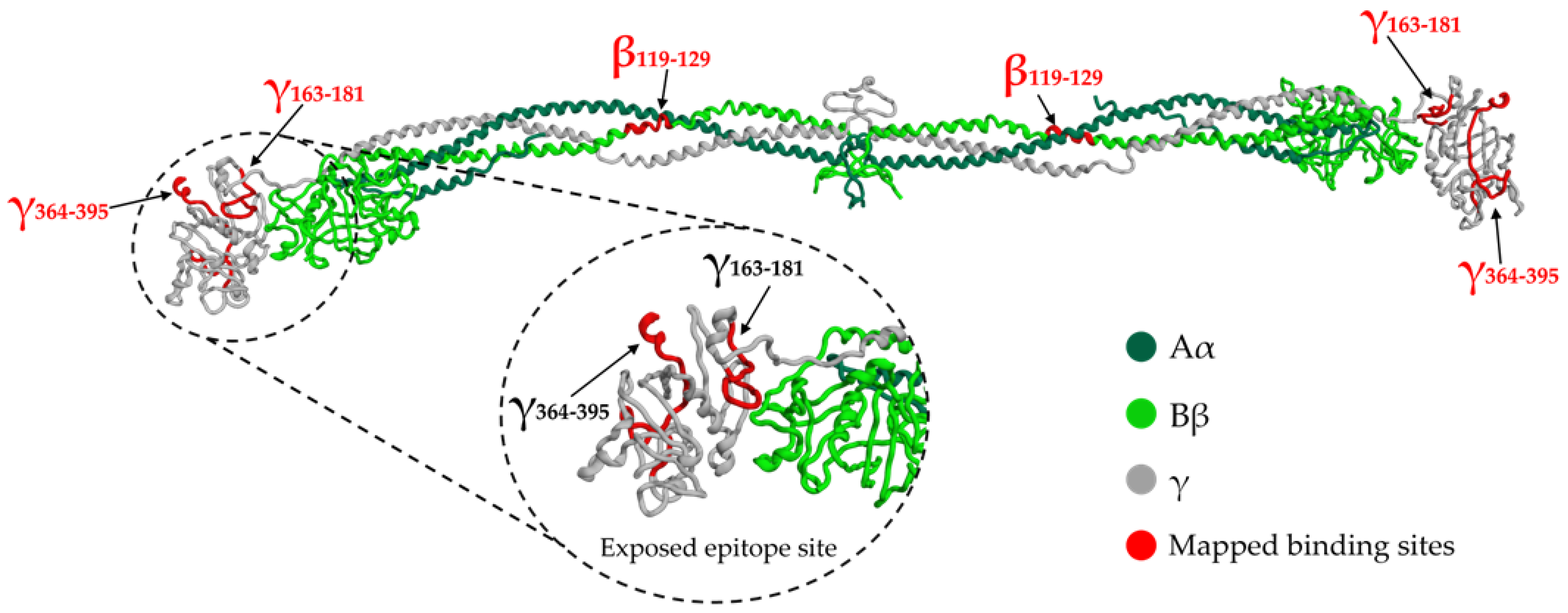
1.4. Overview of Fibrinogen Binding Sites
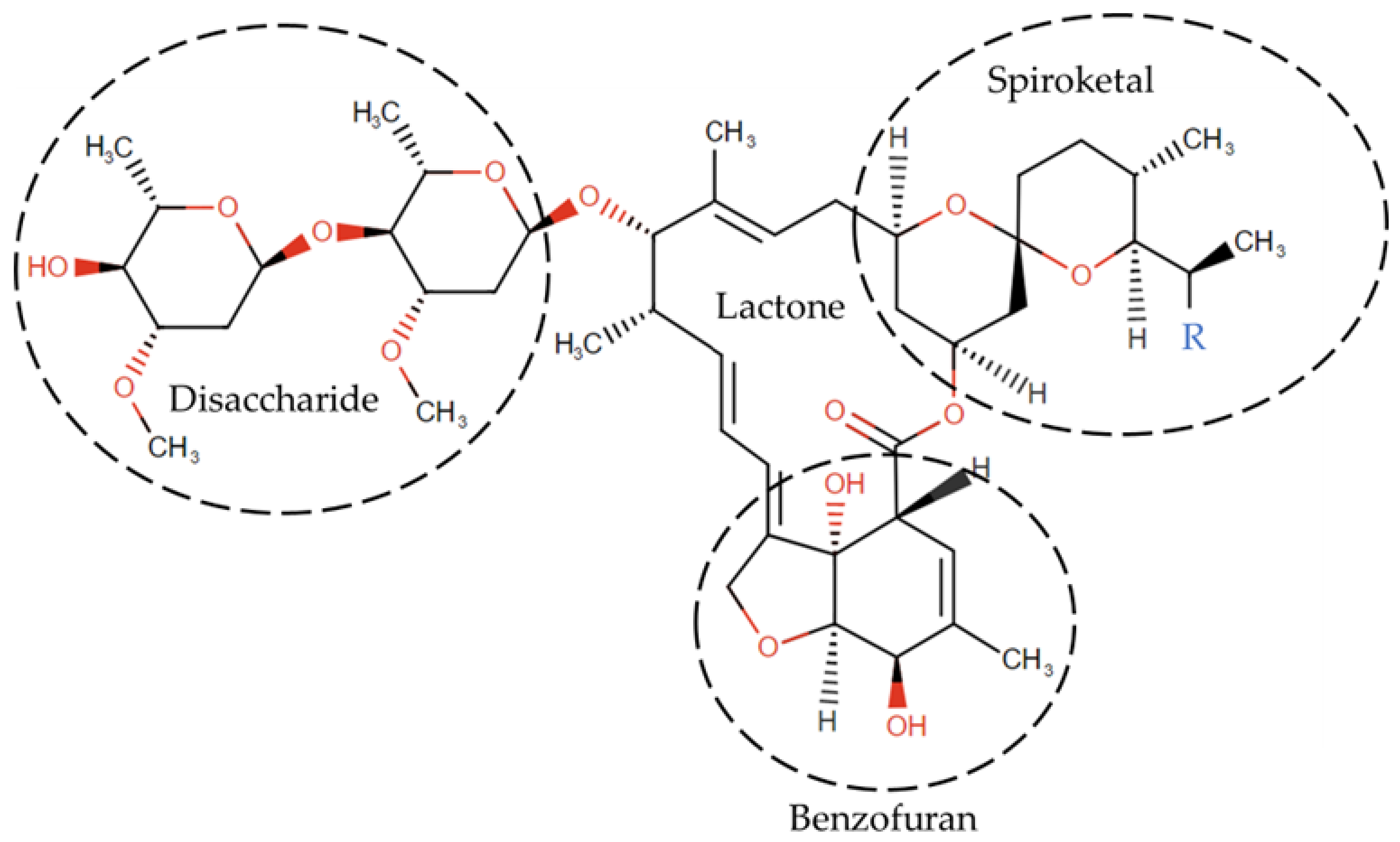
1.5. Structure of IVM
2. Results
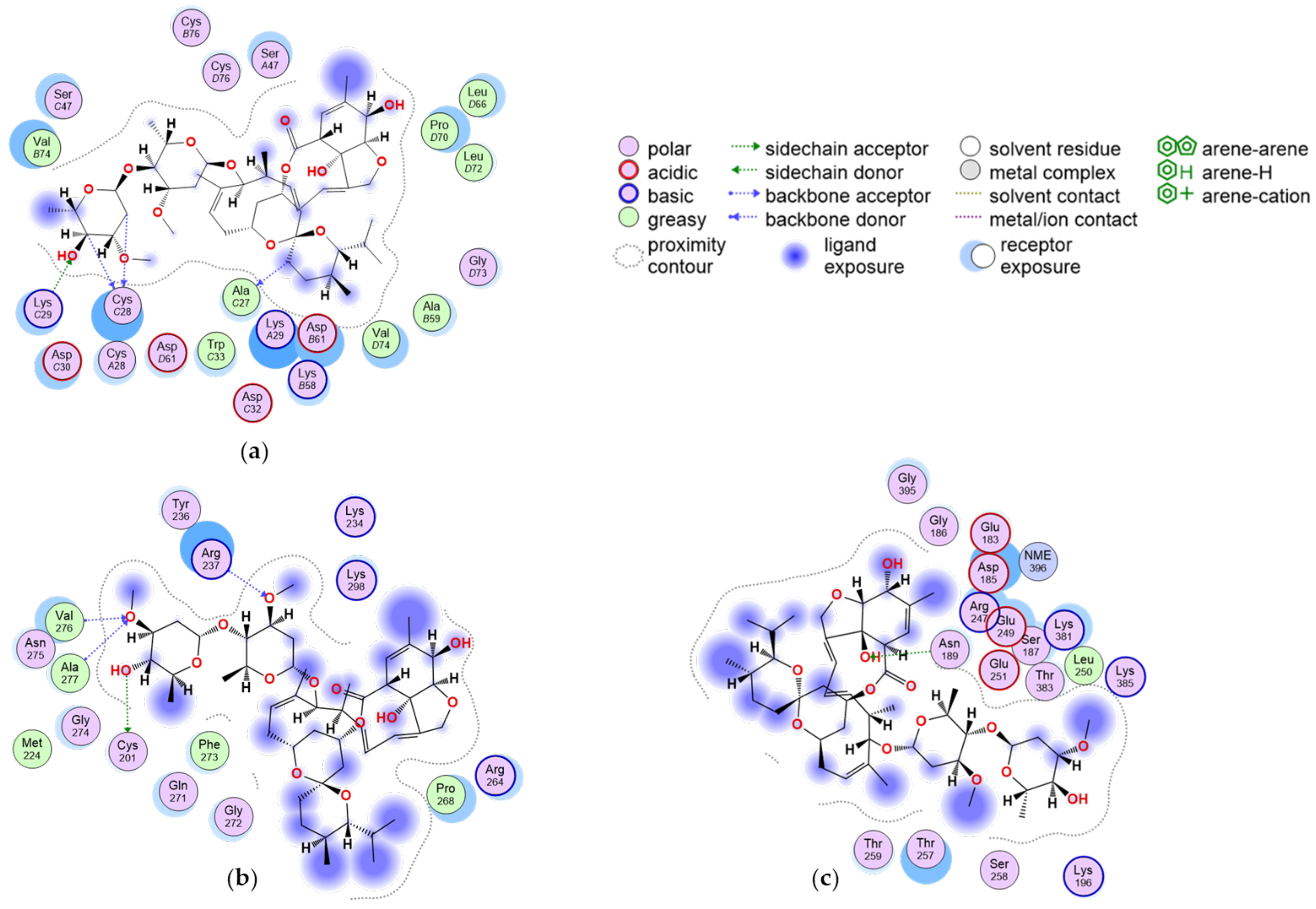
2.1. Molecular Docking
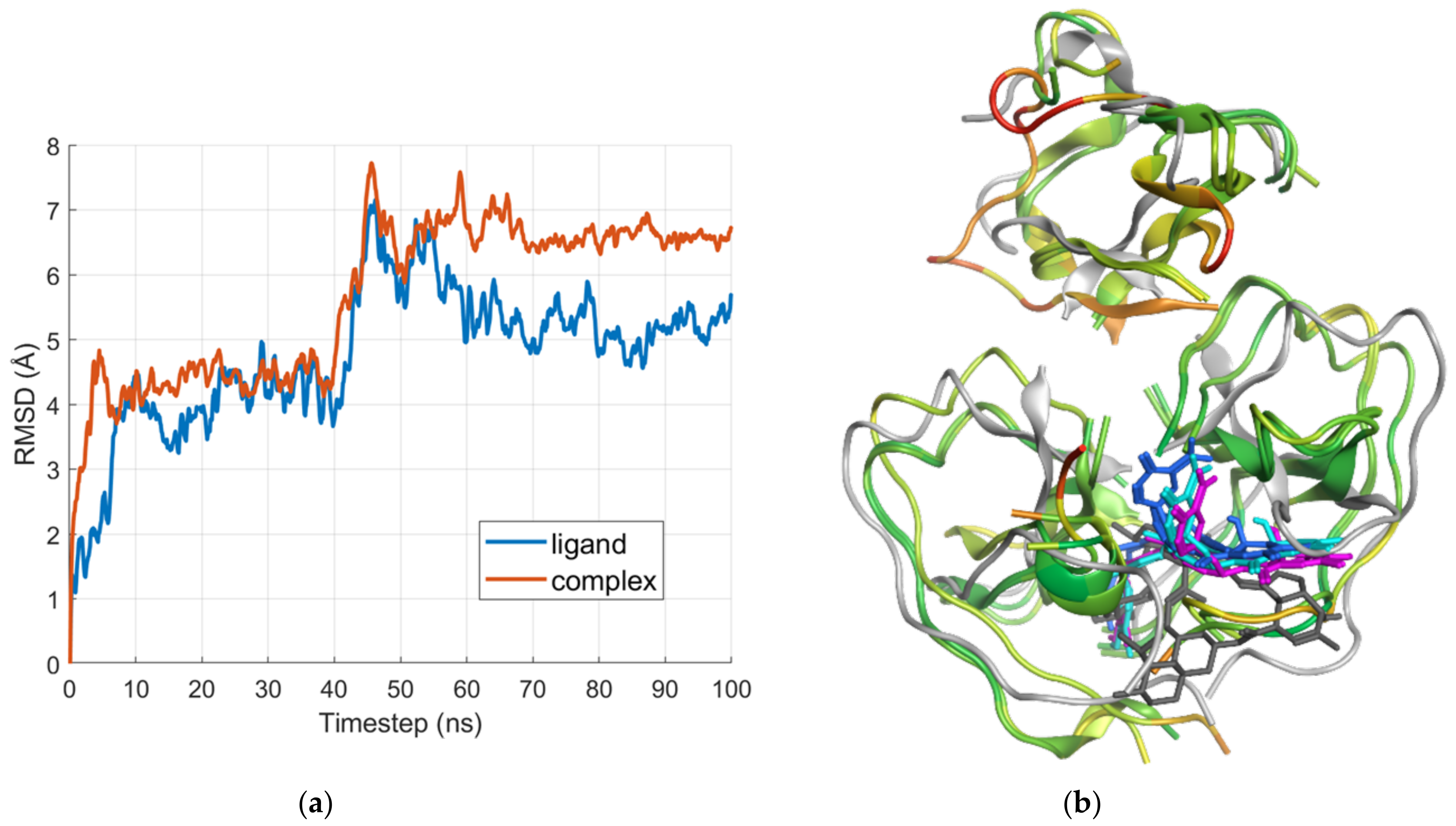
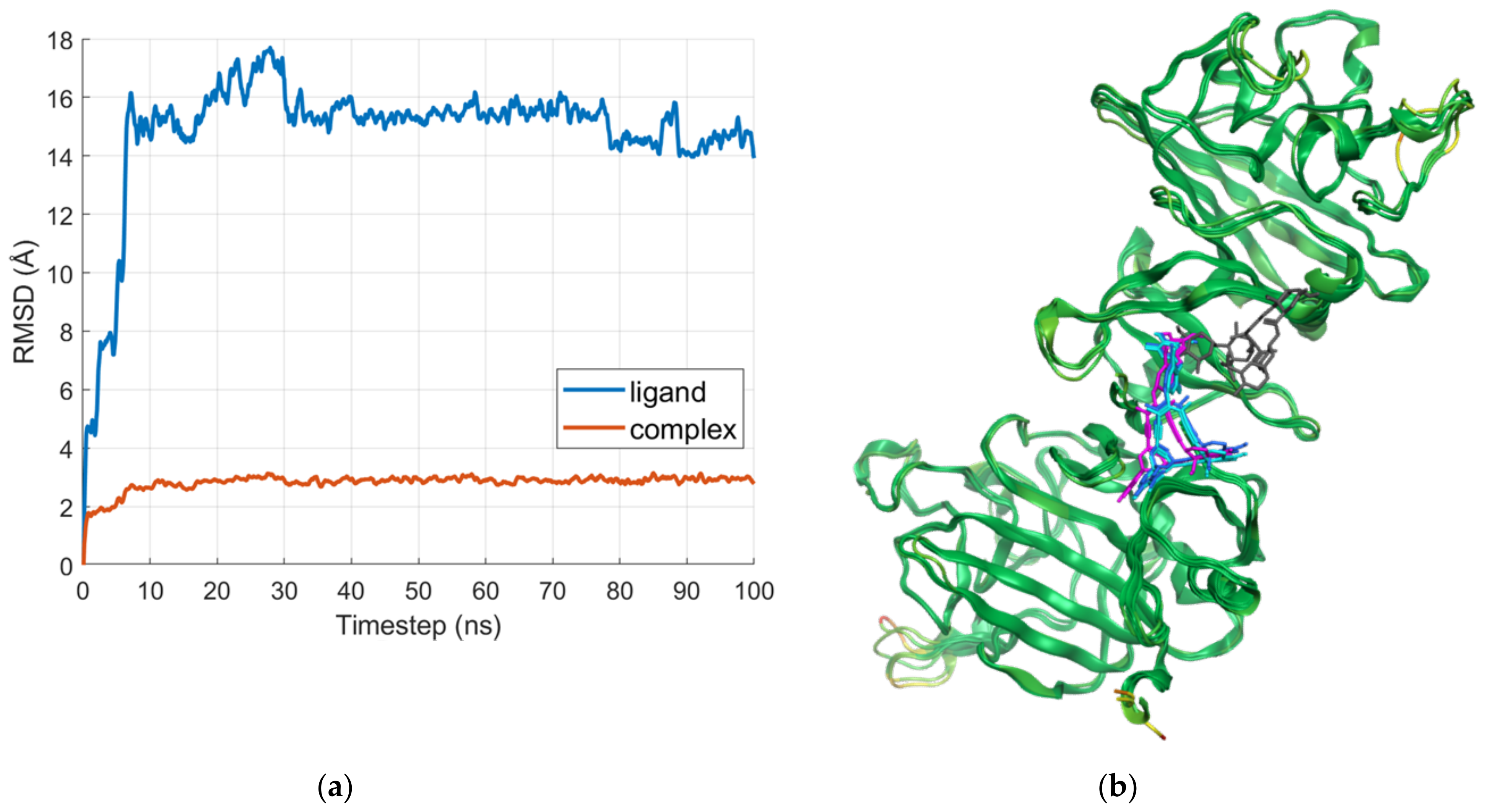
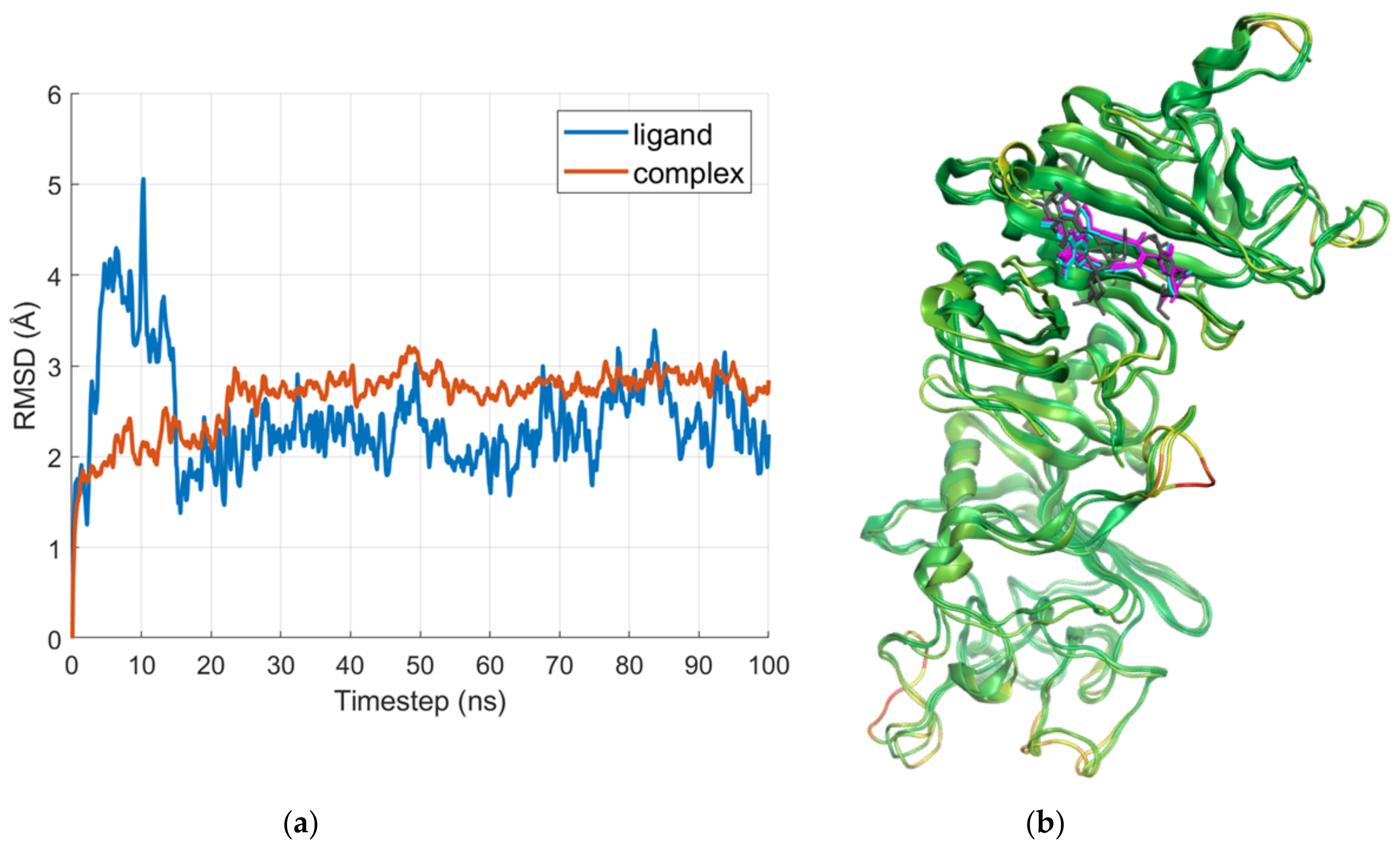
2.2. Molecular Dynamics
2.2.1. E Region
2.2.2. D Region—Gamma1 Site
2.2.3. D Region—Gamma2 Site
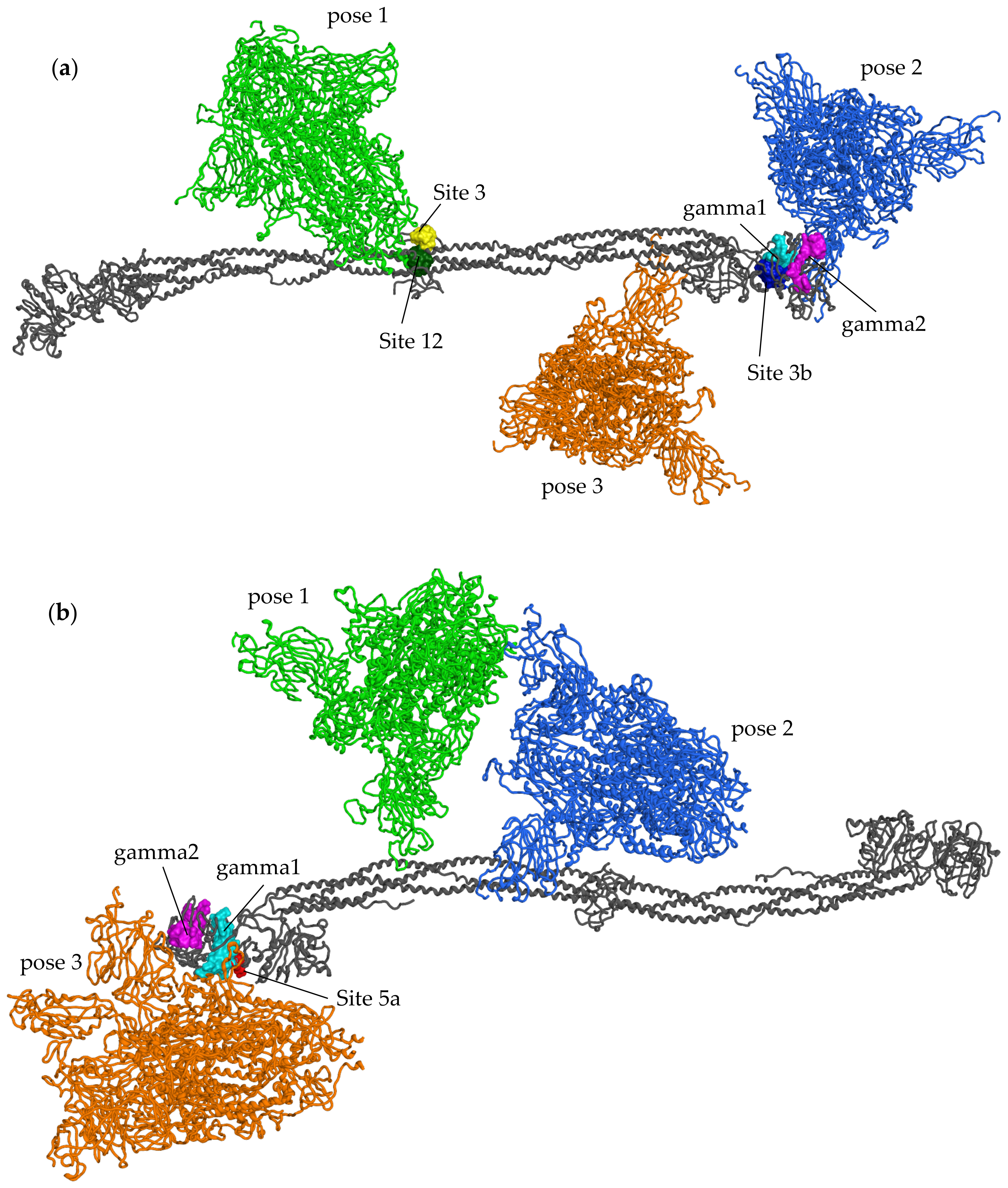
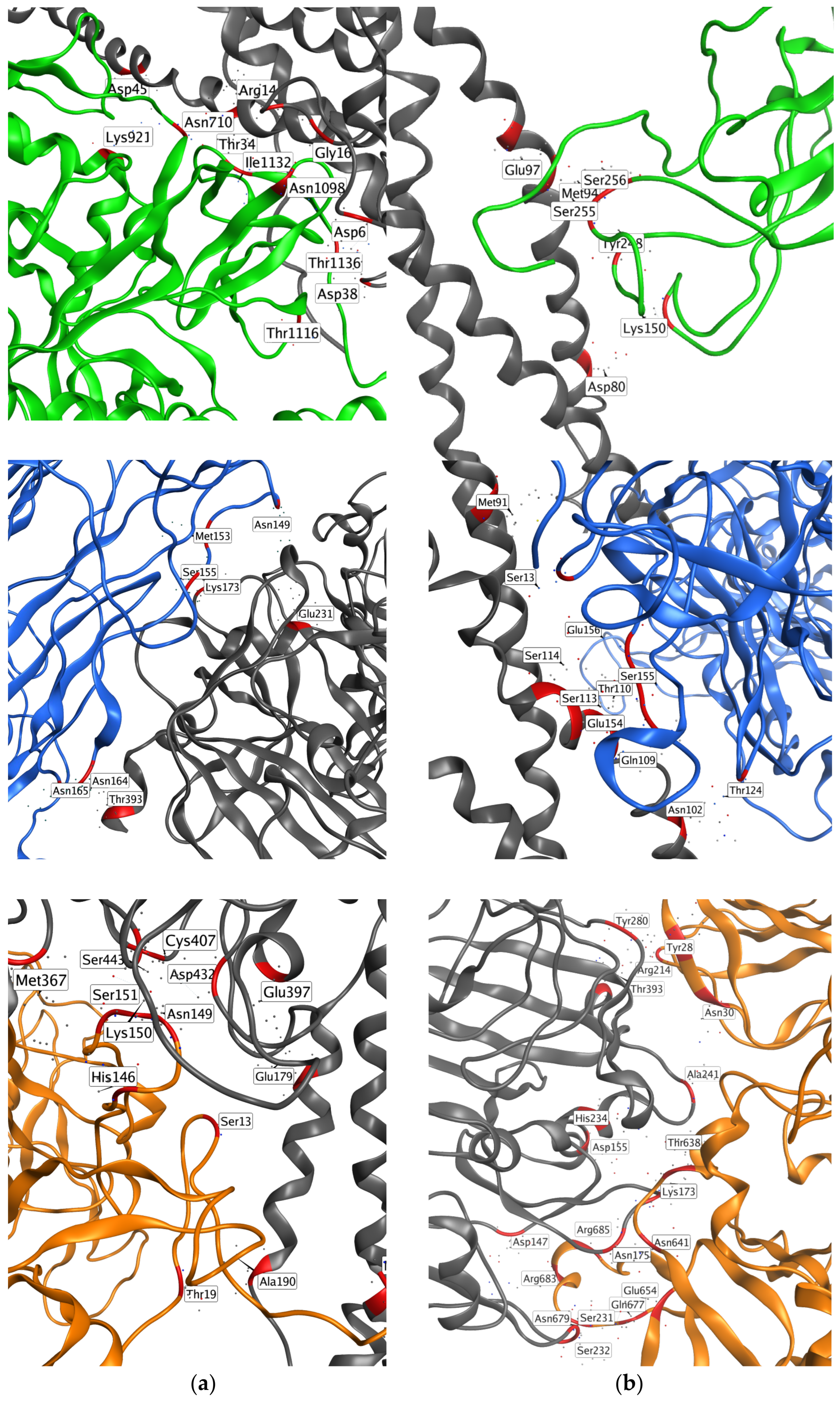
2.3. Protein–Protein Docking
- The S2 subunit of the SP contacts Sites 3 and 12 in the central E region of fibrinogen. In particular, both the SP and the docked pose of IVM_a in Site 12, which had high affinity for fibrinogen in terms of docking score, are predicted to interact with two aspartic acid residues and an arginine residue in the γ chains, forming Site 12 (Asp27 in chain I and Asp6 and Arg14 in chain L);
- The N-terminal domain (NTD) region of the S1 subunit of the SP contacts the γ chain of fibrinogen. Interacting residues belong to both the gamma1 (Leu172 and Lys173) and gamma2 (Phe389, Asn390, Thr393, Ile394) sites, and Site 3b (Lys173 and Glu231) from Site Finder;
- Residues in the S1 subunit of the SP, neighboring the S2 cleavage site, contact chains α in the coiled-coil region and β in the globular D region.
3. Discussion
4. Materials and Methods
4.1. Human Fibrinogen Structure
4.2. Molecular Docking
4.2.1. Identification of Fibrinogen Binding Sites
4.2.2. Ligand Preparation
4.2.3. Molecular Docking Simulations and Analysis of Results
4.3. Molecular Dynamics Simulation
- A minimization with positional restraints of 10 applied to the heavy atoms of the solute;
- A full-structure minimization step;
- A heating phase up to 298 K in the NVT ensemble, using a Berendsen thermostat with a 2.0 picosecond coupling constant, τ;
- An equilibration phase reaching 1.0 bar in the NPT ensemble, with constant pressure and temperature maintained with a Berendsen barostat (employing isotropic position scaling) and a Langevin thermostat, respectively. A pressure relaxation time of 2.0 ps was applied;
- Finally, a 100 ns production run was performed.
4.4. Protein–Protein Docking
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE | atomic contact energy |
| aVdW | attractive van der Waals energy |
| CR3 | complement receptor 3 |
| Fp | fibrinopeptide |
| fs | femtosecond |
| HA | hemagglutination |
| IVM | ivermectin |
| MD | Molecular dynamics |
| ns | nanosecond |
| NTD | N-terminal domain |
| PASC | post-acute sequelae of COVID-19 (long COVID) |
| PDB | Protein Data Bank |
| PLB | propensity for ligand binding |
| PPI | protein–protein interaction |
| ps | picosecond |
| RBC | red blood cell |
| RBD | receptor-binding domain |
| RMSD | root-mean-square deviation |
| rVdW | repulsive van der Waals energy |
| SA | sialic acid |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| SP | spike protein |
Appendix A


References
- Litvinov, R.I.; Pieters, M.; de Lange-Loots, Z.; Weisel, J.W. Fibrinogen and Fibrin. In Macromolecular Protein Complexes III: Structure and Function; Harris, J.R., Marles-Wright, J., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 471–501. ISBN 978-3-030-58971-4. [Google Scholar]
- Ryu, J.K.; Sozmen, E.G.; Dixit, K.; Montano, M.; Matsui, Y.; Liu, Y.; Helmy, E.; Deerinck, T.J.; Yan, Z.; Schuck, R.; et al. SARS-CoV-2 Spike Protein Induces Abnormal Inflammatory Blood Clots Neutralized by Fibrin Immunotherapy. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ogata, A.F.; Maley, A.M.; Wu, C.; Gilboa, T.; Norman, M.; Lazarovits, R.; Mao, C.P.; Newton, G.; Chang, M.; Nguyen, K.; et al. Ultra-Sensitive Serial Profiling of SARS-CoV-2 Antigens and Antibodies in Plasma to Understand Disease Progression in COVID-19 Patients with Severe Disease. Clin. Chem. 2020, 66, 1562–1572. [Google Scholar] [CrossRef]
- Craddock, V.; Mahajan, A.; Spikes, L.; Krishnamachary, B.; Ram, A.K.; Kumar, A.; Chen, L.; Chalise, P.; Dhillon, N.K. Persistent Circulation of Soluble and Extracellular Vesicle-Linked Spike Protein in Individuals with Postacute Sequelae of COVID-19. J. Med. Virol. 2023, 95, e28568. [Google Scholar] [CrossRef] [PubMed]
- Swank, Z.; Senussi, Y.; Manickas-Hill, Z.; Yu, X.G.; Li, J.Z.; Alter, G.; Walt, D.R. Persistent Circulating SARS-CoC-2 Spike Is Associated with Post-Acute COVID-19 Sequelae. Clin. Infect. Dis. 2022, 2022, ciac722. [Google Scholar] [CrossRef]
- Patterson, B.K.; Francisco, E.B.; Yogendra, R.; Long, E.; Pise, A.; Rodrigues, H.; Hall, E.; Herrera, M.; Parikh, P.; Guevara-Coto, J.; et al. Persistence of SARS CoV-2 S1 Protein in CD16+ Monocytes in Post-Acute Sequelae of COVID-19 (PASC) up to 15 Months Post-Infection. Front. Immunol. 2022, 12, 746021. [Google Scholar] [CrossRef] [PubMed]
- Soriano, J.B.; Murthy, S.; Marshall, J.C.; Relan, P.; Diaz, J.V. A Clinical Case Definition of Post-COVID-19 Condition by a Delphi Consensus. Lancet Infect. Dis. 2022, 22, e102–e107. [Google Scholar] [CrossRef]
- Ceban, F.; Ling, S.; Lui, L.M.W.; Lee, Y.; Gill, H.; Teopiz, K.M.; Rodrigues, N.B.; Subramaniapillai, M.; Di Vincenzo, J.D.; Cao, B.; et al. Fatigue and Cognitive Impairment in Post-COVID-19 Syndrome: A Systematic Review and Meta-Analysis. Brain Behav. Immun. 2022, 101, 93–135. [Google Scholar] [CrossRef]
- Aminpour, M.; Cannariato, M.; Preto, J.; Safaeeardebili, M.E.; Moracchiato, A.; Doria, D.; Donato, F.; Zizzi, E.A.; Deriu, M.A.; Scheim, D.E.; et al. In Silico Analysis of the Multi-Targeted Mode of Action of Ivermectin and Related Compounds. Computation 2022, 10, 51. [Google Scholar] [CrossRef]
- Kollman, J.M.; Pandi, L.; Sawaya, M.R.; Riley, M.; Doolittle, R.F. Crystal Structure of Human Fibrinogen. Biochemistry 2009, 48, 3877–3886. [Google Scholar] [CrossRef]
- Klykov, O.; van der Zwaan, C.; Heck, A.J.R.; Meijer, A.B.; Scheltema, R.A. Missing Regions within the Molecular Architecture of Human Fibrin Clots Structurally Resolved by XL-MS and Integrative Structural Modeling. Proc. Natl. Acad. Sci. USA 2020, 117, 1976–1987. [Google Scholar] [CrossRef]
- Zhmurov, A.; Protopopova, A.D.; Litvinov, R.I.; Zhukov, P.; Weisel, J.W.; Barsegov, V. Atomic Structural Models of Fibrin Oligomers. Structure 2018, 26, 857–868.e4. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W. The Mechanical Properties of Fibrin for Basic Scientists and Clinicians. Biophys. Chem. 2004, 112, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Wygrecka, M.; Birnhuber, A.; Seeliger, B.; Michalick, L.; Pak, O.; Schultz, A.-S.; Schramm, F.; Zacharias, M.; Gorkiewicz, G.; David, S.; et al. Altered Fibrin Clot Structure and Dysregulated Fibrinolysis Contribute to Thrombosis Risk in Severe COVID-19. Blood Adv. 2022, 6, 1074–1087. [Google Scholar] [CrossRef] [PubMed]
- Asakura, H.; Ogawa, H. COVID-19-Associated Coagulopathy and Disseminated Intravascular Coagulation. Int. J. Hematol. 2021, 113, 45–57. [Google Scholar] [CrossRef]
- Levi, M.; Thachil, J.; Iba, T.; Levy, J.H. Coagulation Abnormalities and Thrombosis in Patients with COVID-19. Lancet Haematol. 2020, 7, e438–e440. [Google Scholar] [CrossRef]
- Wool, G.D.; Miller, J.L. The Impact of COVID-19 Disease on Platelets and Coagulation. Pathobiology 2021, 88, 15–27. [Google Scholar] [CrossRef]
- Pretorius, E.; Venter, C.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B. Prevalence of Readily Detected Amyloid Blood Clots in ‘Unclotted’ Type 2 Diabetes Mellitus and COVID-19 Plasma: A Preliminary Report. Cardiovasc. Diabetol. 2020, 19, 193. [Google Scholar] [CrossRef]
- Pretorius, E.; Venter, C.; Laubscher, G.J.; Kotze, M.J.; Oladejo, S.O.; Watson, L.R.; Rajaratnam, K.; Watson, B.W.; Kell, D.B. Prevalence of Symptoms, Comorbidities, Fibrin Amyloid Microclots and Platelet Pathology in Individuals with Long COVID/Post-Acute Sequelae of COVID-19 (PASC). Cardiovasc. Diabetol. 2022, 21, 148. [Google Scholar] [CrossRef]
- Picken, M.M. Fibrinogen Amyloidosis: The Clot Thickens! Blood 2010, 115, 2985–2986. [Google Scholar] [CrossRef]
- Kell, D.B.; Laubscher, G.J.; Pretorius, E. A Central Role for Amyloid Fibrin Microclots in Long COVID/PASC: Origins and Therapeutic Implications. Biochem. J. 2022, 479, 537–559. [Google Scholar] [CrossRef]
- Grobbelaar, L.M.; Venter, C.; Vlok, M.; Ngoepe, M.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B.; Pretorius, E. SARS-CoV-2 Spike Protein S1 Induces Fibrin(Ogen) Resistant to Fibrinolysis: Implications for Microclot Formation in COVID-19. Biosci. Rep. 2021, 41, BSR20210611. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Akassoglou, K. Fibrinogen as a Key Regulator of Inflammation in Disease. Semin. Immunopathol. 2012, 34, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Bullard, D.C. Chapter 41—CR3. In The Complement FactsBook, 2nd ed.; Barnum, S., Schein, T., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 435–450. ISBN 978-0-12-810420-0. [Google Scholar]
- Crump, A. Ivermectin: Enigmatic Multifaceted “wonder” Drug Continues to Surprise and Exceed Expectations. J. Antibiot. 2017, 70, 495–505. [Google Scholar] [CrossRef]
- Molyneux, D.H.; Ward, S.A. Reflections on the Nobel Prize for Medicine 2015—The Public Health Legacy and Impact of Avermectin and Artemisinin. Trends Parasitol. 2015, 31, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Santin, A.D.; Scheim, D.E.; McCullough, P.A.; Yagisawa, M.; Borody, T.J. Ivermectin: A Multifaceted Drug of Nobel Prize-Honored Distinction with Indicated Efficacy against a New Global Scourge, COVID-19. New Microbes New Infect. 2021, 43, 100924. [Google Scholar] [CrossRef]
- Guzzo, C.A.; Furtek, C.I.; Porras, A.G.; Chen, C.; Tipping, R.; Clineschmidt, C.M.; Sciberras, D.G.; Hsieh, J.Y.; Lasseter, K.C. Safety, Tolerability, and Pharmacokinetics of Escalating High Doses of Ivermectin in Healthy Adult Subjects. J. Clin. Pharmacol. 2002, 42, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.; Camprubí, D.; Requena-Méndez, A.; Buonfrate, D.; Giorli, G.; Kamgno, J.; Gardon, J.; Boussinesq, M.; Muñoz, J.; Krolewiecki, A. Safety of High-Dose Ivermectin: A Systematic Review and Meta-Analysis. J. Antimicrob. Chemother. 2020, 75, 827–834. [Google Scholar] [CrossRef]
- Nobel Prize Committee. The 2015 Nobel Prize in Physiology or Medicine—Press Release, 5 October 2015; The Nobel Assembly at Karolinska Institutet: Solna, Sweden, 2015. [Google Scholar]
- Molecular Operating Environment (MOE) 2022.02, Chemical Computing Group ULC: 910-1010 Sherbrooke St. W., Montreal, QC H3A 2R7, Canada, 2023.
- Yakovlev, S.; Gorlatov, S.; Ingham, K.; Medved, L. Interaction of Fibrin(Ogen) with Heparin: Further Characterization and Localization of the Heparin-Binding Site. Biochemistry 2003, 42, 7709–7716. [Google Scholar] [CrossRef]
- Li, X.; Duan, H.; Song, Z.; Xu, R. Comparative Study on the Interaction between Fibrinogen and Flavonoids. J. Mol. Struct. 2022, 1262, 132963. [Google Scholar] [CrossRef]
- Shafreen, R.M.; Lakshmi, S.A.; Pandian, S.K.; Park, Y.S.; Kim, Y.M.; Paśko, P.; Deutsch, J.; Katrich, E.; Gorinstein, S. Unraveling the Antioxidant, Binding and Health-Protecting Properties of Phenolic Compounds of Beers with Main Human Serum Proteins: In Vitro and In Silico Approaches. Molecules 2020, 25, 4962. [Google Scholar] [CrossRef]
- Liu, Y.; Tang, X.; Pei, J.; Zhang, L.; Liu, F.; Li, K. Gastrodin Interaction with Human Fibrinogen: Anticoagulant Effects and Binding Studies. Chem. Eur. J. 2006, 12, 7807–7815. [Google Scholar] [CrossRef] [PubMed]
- González-Durruthy, M.; Scanavachi, G.; Rial, R.; Liu, Z.; Cordeiro, M.N.D.S.; Itri, R.; Ruso, J.M. Mapping the Underlying Mechanisms of Fibrinogen Benzothiazole Drug Interactions Using Computational and Experimental Approaches. Int. J. Biol. Macromol. 2020, 163, 730–744. [Google Scholar] [CrossRef]
- González-Durruthy, M.; Rial, R.; Cordeiro, M.N.D.S.; Liu, Z.; Ruso, J.M. Exploring the Conformational Binding Mechanism of Fibrinogen Induced by Interactions with Penicillin β-Lactam Antibiotic Drugs. J. Mol. Liq. 2021, 324, 114667. [Google Scholar] [CrossRef]
- Stubbs, M.T.; Oschkinat, H.; Mayr, I.; Huber, R.; Angliker, H.; Stone, S.R.; Bode, W. The Interaction of Thrombin with Fibrinogen. A structural basis for its specificity. Eur. J. Biochem. 1992, 206, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Syahbanu, F.; Giriwono, P.E.; Tjandrawinata, R.R.; Suhartono, M.T. Molecular Docking of Subtilisin K2, a Fibrin-Degrading Enzyme from Indonesian Moromi, with Its Substrates. Food Sci. Technol. 2022, 42, e61820. [Google Scholar] [CrossRef]
- DrugBank Compound Summary for Ivermectin. Available online: https://go.drugbank.com/drugs/DB00602 (accessed on 23 March 2023).
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for Rigid and Symmetric Docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast Interaction Refinement in Molecular Docking. Proteins 2007, 69, 139–159. [Google Scholar] [CrossRef]
- Scheim, D.E. A Deadly Embrace: Hemagglutination Mediated by SARS-CoV-2 Spike Protein at Its 22 N-Glycosylation Sites, Red Blood Cell Surface Sialoglycoproteins, and Antibody. Int. J. Mol. Sci. 2022, 23, 2558. [Google Scholar] [CrossRef]
- Couzin-Frankel, J. The Mystery of the Pandemic’s ‘Happy Hypoxia’. Science 2020, 368, 455–456. [Google Scholar] [CrossRef]
- Rapkiewicz, A.V.; Mai, X.; Carsons, S.E.; Pittaluga, S.; Kleiner, D.E.; Berger, J.S.; Thomas, S.; Adler, N.M.; Charytan, D.M.; Gasmi, B.; et al. Megakaryocytes and Platelet-Fibrin Thrombi Characterize Multi-Organ Thrombosis at Autopsy in COVID-19: A Case Series. EClinicalMedicine 2020, 24, 100434. [Google Scholar] [CrossRef]
- Price, L.C.; McCabe, C.; Garfield, B.; Wort, S.J. Thrombosis and COVID-19 Pneumonia: The Clot Thickens! Eur. Respir. J. 2020, 56, 2001608. [Google Scholar] [CrossRef] [PubMed]
- Huertas, A.; Montani, D.; Savale, L.; Pichon, J.; Tu, L.; Parent, F.; Guignabert, C.; Humbert, M. Endothelial Cell Dysfunction: A Major Player in SARS-CoV-2 Infection (COVID-19)? Eur. Respir. J. 2020, 56, 2001634. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary Manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Becker, R.C. COVID-19 Update: COVID-19-Associated Coagulopathy. J. Thromb. Thrombolysis 2020, 50, 54–67. [Google Scholar] [CrossRef]
- Mondal, R.; Lahiri, D.; Deb, S.; Bandyopadhyay, D.; Shome, G.; Sarkar, S.; Paria, S.R.; Thakurta, T.G.; Singla, P.; Biswas, S.C. COVID-19: Are We Dealing with a Multisystem Vasculopathy in Disguise of a Viral Infection? J. Thromb. Thrombolysis 2020, 50, 567–579. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement Associated Microvascular Injury and Thrombosis in the Pathogenesis of Severe COVID-19 Infection: A Report of Five Cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Menter, T.; Haslbauer, J.D.; Nienhold, R.; Savic, S.; Hopfer, H.; Deigendesch, N.; Frank, S.; Turek, D.; Willi, N.; Pargger, H.; et al. Postmortem Examination of COVID-19 Patients Reveals Diffuse Alveolar Damage with Severe Capillary Congestion and Variegated Findings in Lungs and Other Organs Suggesting Vascular Dysfunction. Histopathology 2020, 77, 198–209. [Google Scholar] [CrossRef]
- Gattinoni, L.; Coppola, S.; Cressoni, M.; Busana, M.; Rossi, S.; Chiumello, D. COVID-19 Does Not Lead to a “Typical” Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2020, 201, 1299–1300. [Google Scholar] [CrossRef]
- Marini, J.J.; Gattinoni, L. Management of COVID-19 Respiratory Distress. JAMA 2020, 323, 2329–2330. [Google Scholar] [CrossRef]
- Berzuini, A.; Bianco, C.; Migliorini, A.C.; Maggioni, M.; Valenti, L.; Prati, D. Red Blood Cell Morphology in Patients with COVID-19-Related Anaemia. Blood Transfus. 2021, 19, 34–36. [Google Scholar] [CrossRef]
- Lakhdari, N.; Tabet, B.; Boudraham, L.; Laoussati, M.; Aissanou, S.; Beddou, L.; Bensalem, S.; Bellik, Y.; Bournine, L.; Fatmi, S.; et al. Red Blood Cells Injuries and Hypersegmented Neutrophils in COVID-19 Peripheral Blood Film. medRxiv 2020. [Google Scholar] [CrossRef]
- Melkumyants, A.; Buryachkovskaya, L.; Lomakin, N.; Antonova, O.; Serebruany, V. Mild COVID-19 and Impaired Blood Cell–Endothelial Crosstalk: Considering Long-Term Use of Antithrombotics? Thromb. Haemost. 2022, 122, 123–130. [Google Scholar] [CrossRef]
- Kibria, F.; Hysi, E.; Strohm, E.M.; Kolios, M.C. Identification of Red Blood Cell Rouleaux Formation Using Photoacoustic Ultrasound Spectroscopy. In Proceedings of the Photons Plus Ultrasound: Imaging and Sensing 2014, San Francisco, CA, USA, 1–6 February 2014; Volume 8943, p. 894367. [Google Scholar]
- Hysi, E.; Saha, R.K.; Kolios, M.C. Photoacoustic Ultrasound Spectroscopy for Assessing Red Blood Cell Aggregation and Oxygenation. J. Biomed. Opt. 2012, 17, 125006. [Google Scholar] [CrossRef]
- Boschi, C.; Scheim, D.E.; Bancod, A.; Militello, M.; Bideau, M.L.; Colson, P.; Fantini, J.; Scola, B.L. SARS-CoV-2 Spike Protein Induces Hemagglutination: Implications for COVID-19 Morbidities and Therapeutics and for Vaccine Adverse Effects. Int. J. Mol. Sci. 2022, 23, 15480. [Google Scholar] [CrossRef] [PubMed]
- Barshtein, G.; Wajnblum, D.; Yedgar, S. Kinetics of Linear Rouleaux Formation Studied by Visual Monitoring of Red Cell Dynamic Organization. Biophys. J. 2000, 78, 2470–2474. [Google Scholar] [CrossRef] [PubMed]
- Baskurt, O.K.; Meiselman, H.J. Erythrocyte Aggregation: Basic Aspects and Clinical Importance. Clin. Hemorheol. Microcirc. 2013, 53, 23–37. [Google Scholar] [CrossRef]
- Meiselman, H.J. Red Blood Cell Aggregation: 45 Years Being Curious. Biorheology 2009, 46, 1–19. [Google Scholar] [CrossRef]
- Wagner, C.; Steffen, P.; Svetina, S. Aggregation of Red Blood Cells: From Rouleaux to Clot Formation. Comptes Rendus Phys. 2013, 14, 459–469. [Google Scholar] [CrossRef]
- Sabioni, L.; De Lorenzo, A.; Lamas, C.; Muccillo, F.; Castro-Faria-Neto, H.C.; Estato, V.; Tibirica, E. Systemic Microvascular Endothelial Dysfunction and Disease Severity in COVID-19 Patients: Evaluation by Laser Doppler Perfusion Monitoring and Cytokine/Chemokine Analysis. Microvasc. Res. 2021, 134, 104119. [Google Scholar] [CrossRef]
- Biondi, M.A.X.; Gulati, M.; Possick, J.; Joseph, P.; Singh, I.; Lutchmansingh, D. Unexplained dyspnea in a patient with a history of COVID-19. Chest 2021, 160, A2373–A2374. [Google Scholar] [CrossRef]
- Singh, I.; Joseph, P.; Heerdt, P.M.; Cullinan, M.; Lutchmansingh, D.D.; Gulati, M.; Possick, J.D.; Systrom, D.M.; Waxman, A.B. Persistent Exertional Intolerance After COVID-19: Insights from Invasive Cardiopulmonary Exercise Testing. Chest 2022, 161, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Klotz, U.; Ogbuokiri, J.E.; Okonkwo, P.O. Ivermectin Binds Avidly to Plasma Proteins. Eur. J. Clin. Pharmacol. 1990, 39, 607–608. [Google Scholar] [CrossRef] [PubMed]
- Edelsbrunner, H.; Kirkpatrick, D.; Seidel, R. On the Shape of a Set of Points in the Plane. IEEE Trans. Inf. Theory 1983, 29, 551–559. [Google Scholar] [CrossRef]
- Soga, S.; Shirai, H.; Kobori, M.; Hirayama, N. Use of Amino Acid Composition to Predict Ligand-Binding Sites. J. Chem. Inf. Model. 2007, 47, 400–406. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. AMDock: A Versatile Graphical Tool for Assisting Molecular Docking with Autodock Vina and Autodock4. Biol. Direct 2020, 15, 12. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Preto, J.; Gentile, F. Assessing and improving the performance of consensus docking strategies using the DockBox package. J Comput. Aided Mol. Des. 2019, 33, 817–829. [Google Scholar] [CrossRef]
- Darden, T.A.; Duke, R.E.; Giambasu, G.; Gilson, M.K.; Gohlke, H.; Goetz, A.W.; Harris, R.; Izadi, S.; Izmailov, S.A.; Kasavajhala, K.; et al. AMBER 2020. Available online: http://ambermd.org/ (accessed on 15 June 2023).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Site | MOE Site Finder Results |
|---|---|---|
| Heparin | N-terminal regions (residues 15–57) of the two Bβ chains in the E region [32]. | Not modeled |
| Flavonoids (myricetin, rutin, naringin, hesperidin, genistein, puerarin) and phenolic acids (ferulic and caffeic acid) | Hydrophobic cavity of E region near Trp 33 in the N-terminal region of the α chain [33]; residues 19–22 of the γ chain, residues 77–78 of the β chain [34]. | Sites 1 and 12. |
| Gastrodin | Holes’ a’ and ‘b’ in the γ- and β-nodules of the D domain [35]. | Identifiable with the position of the ligand chains Q and R for ‘a’ and S and T for the ‘b’ holes. |
| Benzothiazole and penicillin β-lactam antibiotics (cloxacillin and dicloxacillin) | Hydrophobic cavities of E region (bovine) [36,37]. | Sites 1, 3, 12. |
| Interacting Protein | Site (s) | MOE Site Finder Results |
|---|---|---|
| Thrombin | Fibrinopeptides A and B in the N-termini of chains Aα and Bβ, in the central E region [1,38]. | Not modeled. |
| Plasma transglutaminase | γ–γ crosslinking site, γ-nodule of D domain [1]. | Not modeled. |
| Complement receptor 3 | Residues 377–395 of the γ chain, γ-nodule of D domain [2]. | Not modeled. |
| Subtilisin K2 | Residue PHE117 on the Aα chain, LEU121 and TRP125 on the Bβ chain, and ASP53, PHE54, and THR57 on the γ chain [39]. | Part of Sites 2, 11. |
| Plasmin | Residues 119–129 of the Bβ chain [2]. | Part of Site 11. |
| SARS-CoV-2 SP | Residues 163–181 and 364–395 of the γ chain, in the γ-nodule of D domain; residues 119–129 of the Bβ chain [2]. | γ: part of Sites 7 and 9. Bβ: part of Site 11. |
| Site | Score (kcal/mol) | |
|---|---|---|
| IVM_a | IVM_b | |
| Site 1 | −10.971 | −9.959 |
| Site 3 | −6.567 | −6.426 |
| Site 12 | −10.142 | −5.207 |
| Site | Score (kcal/mol) | |
|---|---|---|
| IVM_a | IVM_b | |
| Site1b | −10.428 | −9.506 |
| Site4a | −9.202 | −8.957 |
| Site6b | −9.155 | −8.223 |
| Site3a | −8.820 | −8.338 |
| Site1a | −8.820 | −6.385 |
| Site5a | −8.800 | −8.453 |
| Site3b | −8.324 | −7.875 |
| Site7b | −8.288 | −8.626 |
| Site2a | −7.673 | −8.056 |
| Site4b | −7.058 | −8.019 |
| Site5b | −6.892 | −6.802 |
| Site6a | −6.844 | −6.675 |
| Site2b | −6.768 | −7.039 |
| Site | Score (kcal/mol) | |
|---|---|---|
| IVM_a | IVM_b | |
| gamma1 (163–181) | −9.281 | −8.915 |
| gamma2a (377–395) | −7.136 | −6.791 |
| gamma2b | −7.448 | −7.834 |
| beta (119–129) | −9.921 | −8.529 |
| Structure | Pose | glob | aVdW | rVdW | ACE |
|---|---|---|---|---|---|
| 6VXX | 1 | −62.17 | −65.02 | 38.94 | 4.11 |
| 2 | −49.83 | −57.34 | 42.31 | −4.01 | |
| 3 | −46.34 | −38.48 | 18.94 | −3.99 | |
| 6VSB | 1 | −54.06 | −36.10 | 43.10 | −9.79 |
| 2 | −55.15 | −40.59 | 23.31 | −3.11 | |
| 3 | −35.48 | −59.01 | 68.62 | −1.01 |
| Chain | N-Terminus Out-Gap | C-Terminus Out-Gap |
|---|---|---|
| A (Aα) | 26 residues | 362 residues |
| B (Bβ) | 57 residues | 3 residues |
| C (γ) | 13 residues | 17 residues |
| D (Aα) | 26 residues | 362 residues |
| E (Bβ) | 57 residues | 3 residues |
| F (γ) | 13 residues | 16 residues |
| G (Aα) | 26 residues | 362 residues |
| H (Bβ) | 57 residues | 3 residues |
| I (γ) | 1 residue | 16 residues |
| J (Aα) | 26 residues | 350 residues |
| K (Bβ) | 57 residues | 3 residues |
| L (γ) | 4 residues | 16 residues |
| Site | PLB | Residues |
|---|---|---|
| 1 | 5.64 | Aα1:(ACE26 ALA27 CYS28 LYS29 ASP32 TRP33 SER47 CYS49 ARG50) Bβ1:(LYS58 ALA59 PRO60 ASP61 CYS65 LEU66 HIS67 ASP69 PRO70 ASP71 GLY73 VAL74 LEU75 CYS76) Aα2:(ALA27 CYS28 LYS29 ASP30 SER31 TRP33 CYS45 PRO46 SER47 CYS49 ARG50 LEU54) Bβ2:(LYS58 ALA59 PRO60 ASP61 PRO70 ASP71 LEU72 GLY73 VAL74 CYS76) |
| 2 | 3.97 | Aα2:(ILE93 ASP97 PHE98 ALA101 ASN102 ARG104 ASP105 ASN106 TYR108 ASN109 SER112 ARG116 HIS201 PRO203 LEU204 ILE205 MET207 LYS208 PRO211) Bβ2:(TRP125 ARG128 GLN129 VAL132 LYS133 ASN135 GLU136 VAL139 TYR142 SER143) γ2:(THR67 TYR68 ASN69 PRO70 ASP71 GLU72 SER73 LYS75 ASN77 MET78 ILE79 ASP80 THR83) |
| 3 | 2.50 | γ1:(CYS8 CYS9 ILE10 TYR18) Bβ2:(CYS80 GLN83) γ2:(ACE4 ARG5 ASP6 CYS8 CYS9 SER17 TYR18 CYS19 PRO20) |
| 4 | 1.91 | Aα1:(ILE156 SER160) Bβ1:(ARG255 ASP257 GLY258 SER259 VAL260 ASP261 PHE262 GLY263 GLU291 GLY399 ASN413 GLY414 ARG415 TYR416) |
| 5 | 1.87 | Bβ1:(PRO204 VAL206 LYS217 GLY218 GLY219 GLU220 THR221 GLU223 TYR225 TYR285) γ1:(VAL202 ASP203 LYS206 ILE209 GLN210 GLU213 GLY214 PHE215 GLY216 HIS217) |
| 6 | 1.72 | Aα2:(LEU73 TYR76 GLN77 ASN79 ASN80 LYS81 SER83 HIS84 THR87 THR88 MET91) Bβ2:(ASN103 VAL104 VAL107 SER108 THR110 SER111 SER114 PHE115 GLN116 TYR117 MET118) γ2:(LEU47 VAL50 GLU51 THR54 VAL57) |
| 7 | 1.63 | Bβ2:(ILE203 PRO204 VAL205 VAL206 SER207 GLY208 LYS209 GLU213 LYS217 LEU226) γ2:(PRO171 LYS173 ALA174 ASN175 GLN176 GLN177 PHE178 LEU179 LYS212 GLU213 LEU218 LEU228 GLU231 LYS232 LEU235) |
| 8 | 1.38 | A α1:(ASP153 ILE156 LYS157 SER160) Bβ1:(ASP261 GLY263 ARG264 TYR378 SER395 LYS396 GLY399 GLY400 GLY401 ARG415 LYS428 HIS429) γ1:(GLU132 GLN136) |
| 9 | 1.32 | γ2:(PHE295 ASP297 ASP298 SER300 ASP301 PHE304 THR305 PHE322 CYS326 GLN329 ASP330 LYS338 CYS339 HIS340 TYR363 ASP364 ILE368 ARG375) |
| 10 | 1.20 | Aα2:(ARG118 ILE119 LEU122 LYS123 LYS125 VAL126 LYS129 VAL130) Bβ2:(GLN150 ILE153 THR156 VAL157 ILE161) γ2:(ILE93 TYR96 GLU97 ILE100 HIS103 ASP104) |
| 11 | 1.08 | Aα2:(THR87 ILE90 MET91 LEU94 ARG95 PHE98) Bβ2:(TYR117 MET118 LEU121 LYS122 TRP125 GLN126 ARG128 GLN129) γ2:(LEU60 ILE61 ILE64 GLN65 THR67 TYR68) |
| 12 | 0.99 | Aα1:(CYS45 PRO46 SER47 GLY48)2:(CYS76 PRO77 THR78 GLY79) Bβ1:(CYS19 PRO20 THR21 THR22) Aα2:(CYS45 PRO46 SER47 GLY48) Bβ2:(CYS76 PRO77 THR78 GLY79 LEU82) γ2:(CYS19 PRO20 THR21 THR22) |
| 13 | 0.99 | Aα2:(ASP153 LYS157) Bβ2:(ASP261 GLY263 ARG264 LYS265 TYR378 SER395 LYS396 GLY399 GLY400 ARG415 LYS428 HIS429) γ2:(LYS125 VAL128 ALA129 GLU132 ALA133 GLN136) |
| Site | Site ID | Box Center Coordinates (Å) | ||
|---|---|---|---|---|
| x | y | z | ||
| E region | 1 | 90.12 | −53.03 | −92.61 |
| 3 | 97.22 | −28.67 | −88.12 | |
| 12 | 91.72 | −38.97 | −92.59 | |
| Chains G, H, I | 1a | −62.20 | −34.62 | −45.53 |
| 2a | −90.42 | −49.52 | −36.75 | |
| 3a | −58.83 | −27.75 | −44.12 | |
| 4a | 10.65 | −26.00 | −83.01 | |
| 5a | −90.47 | −42.66 | −22.91 | |
| 6a | −114.10 | −62.45 | −34.73 | |
| Chains J, K, L | 1b | 173.90 | −45.87 | −107.26 |
| 2b | 142.69 | −43.59 | −96.29 | |
| 3b | 272.20 | −62.61 | −166.68 | |
| 4b | 292.84 | −88.07 | −160.55 | |
| 5b | 203.59 | −44.32 | −114.73 | |
| 6b | 158.42 | −46.21 | −103.52 | |
| 7b | 241.40 | −46.07 | −144.96 | |
| SP-chain I | gamma1 (163–181) | −78.32 | −29.61 | −37.44 |
| gamma2a (377–395) | −99.64 | −29.75 | −41.74 | |
| gamma2b 1 | −107.40 | −31.20 | −44.80 | |
| beta (119–129) | 4.08 | −28.05 | −83.39 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vottero, P.; Tavernini, S.; Santin, A.D.; Scheim, D.E.; Tuszynski, J.A.; Aminpour, M. Computational Prediction of the Interaction of Ivermectin with Fibrinogen. Int. J. Mol. Sci. 2023, 24, 11449. https://doi.org/10.3390/ijms241411449
Vottero P, Tavernini S, Santin AD, Scheim DE, Tuszynski JA, Aminpour M. Computational Prediction of the Interaction of Ivermectin with Fibrinogen. International Journal of Molecular Sciences. 2023; 24(14):11449. https://doi.org/10.3390/ijms241411449
Chicago/Turabian StyleVottero, Paola, Scott Tavernini, Alessandro D. Santin, David E. Scheim, Jack A. Tuszynski, and Maral Aminpour. 2023. "Computational Prediction of the Interaction of Ivermectin with Fibrinogen" International Journal of Molecular Sciences 24, no. 14: 11449. https://doi.org/10.3390/ijms241411449
APA StyleVottero, P., Tavernini, S., Santin, A. D., Scheim, D. E., Tuszynski, J. A., & Aminpour, M. (2023). Computational Prediction of the Interaction of Ivermectin with Fibrinogen. International Journal of Molecular Sciences, 24(14), 11449. https://doi.org/10.3390/ijms241411449