
TWIST1 Upregulation Is a Potential Target for Reversing Resistance to the CDK4/6 Inhibitor in Metastatic Luminal Breast Cancer Cells
 , , , , , ,
, , , , , ,  , and
, and
Abstract
:1. Introduction
2. Results
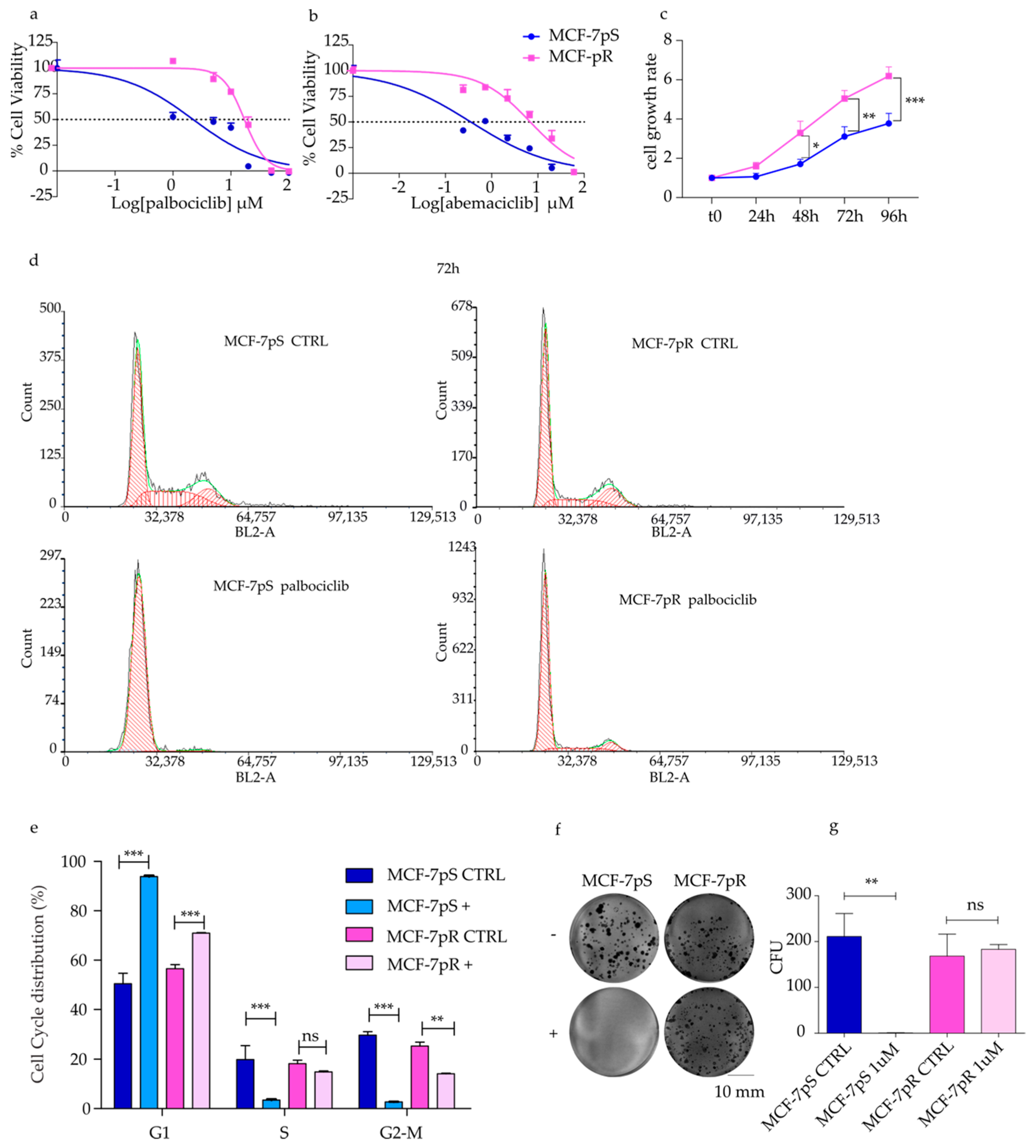
2.1. Generation of Palbociclib-Resistant MCF-7 Cells
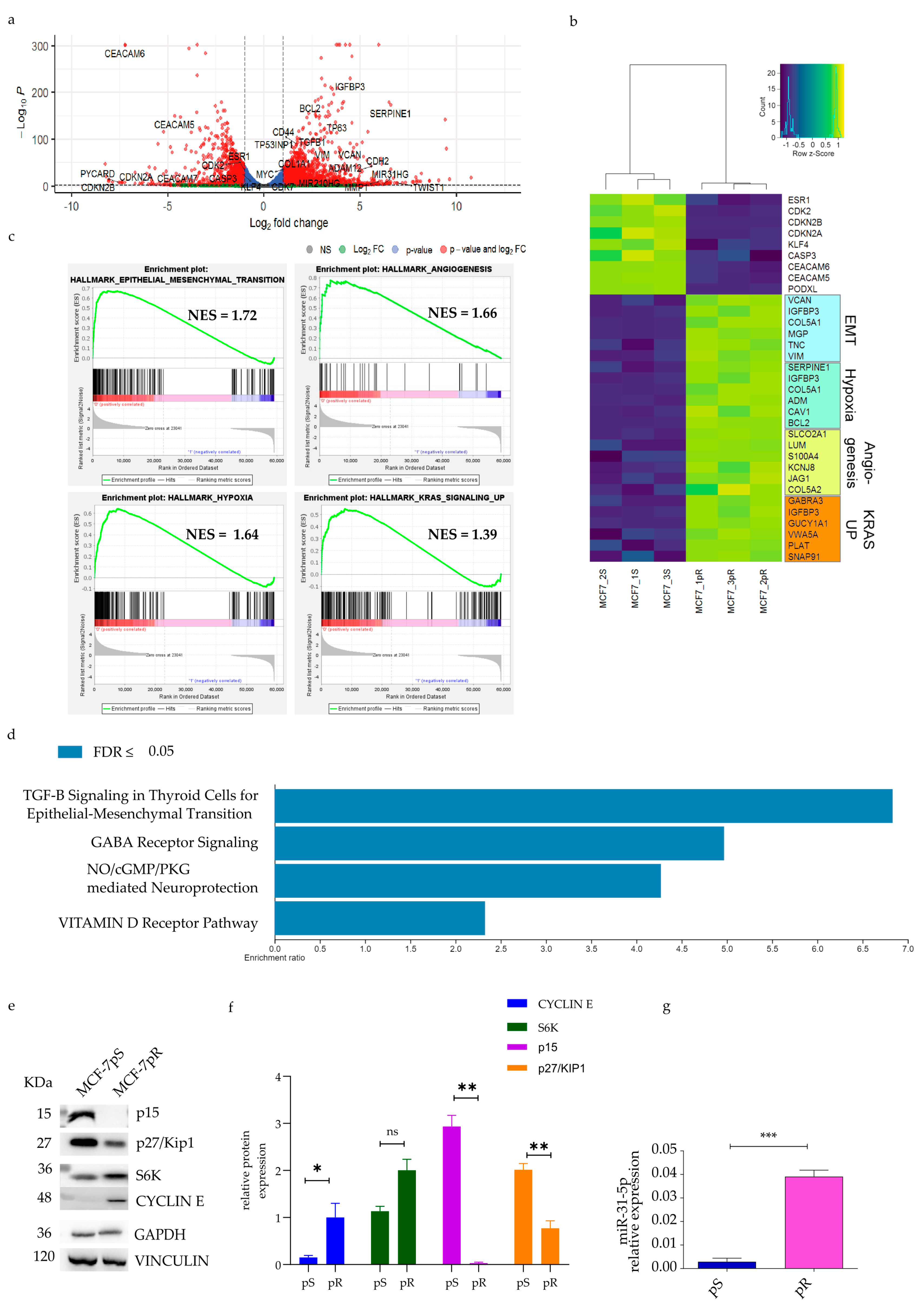
2.2. Pathway Analysis of Palbociclib-Resistant Cells
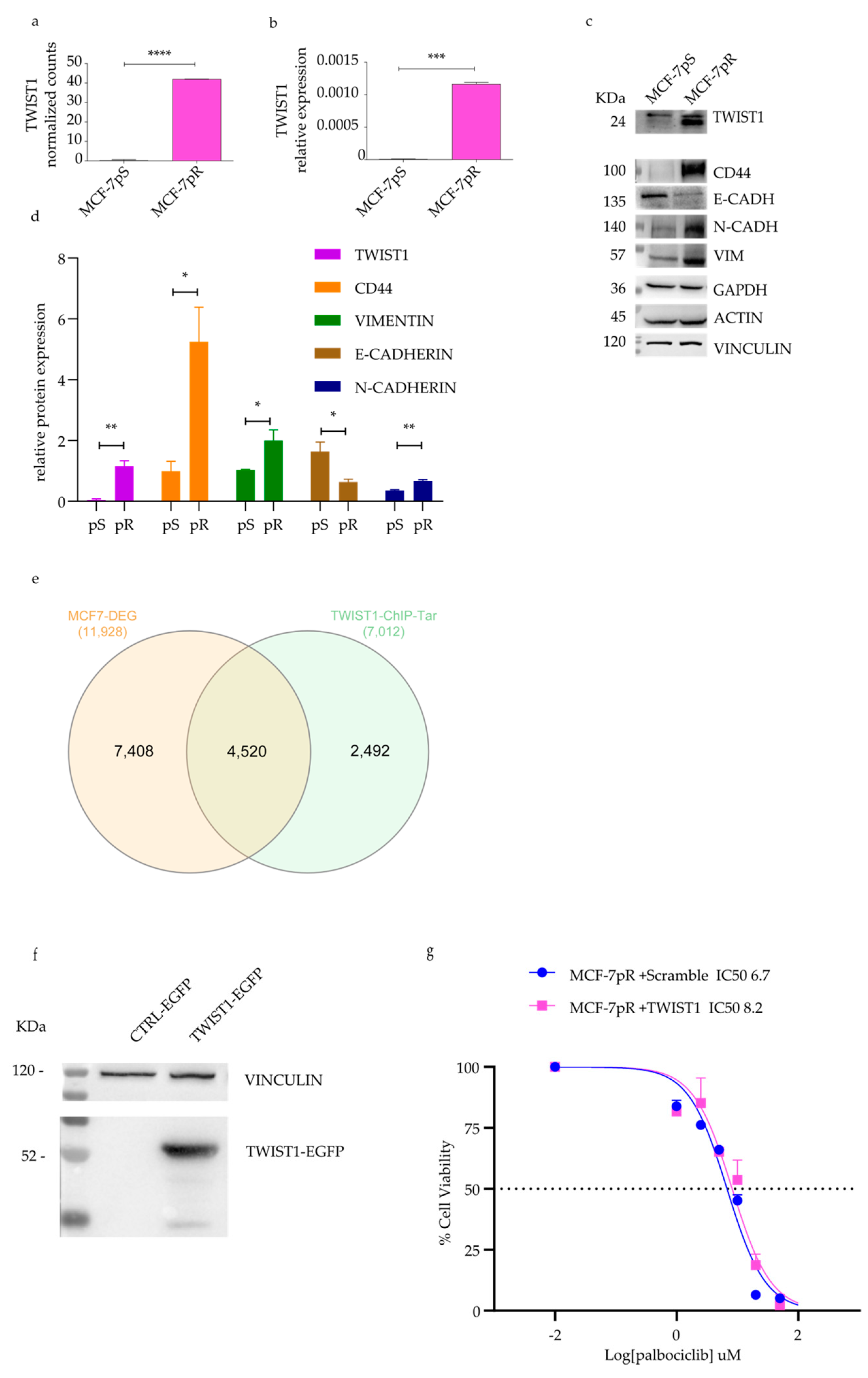
2.3. Detection of TWIST1 Over-Expression in Palbociclib Resistant MCF-7 Cells
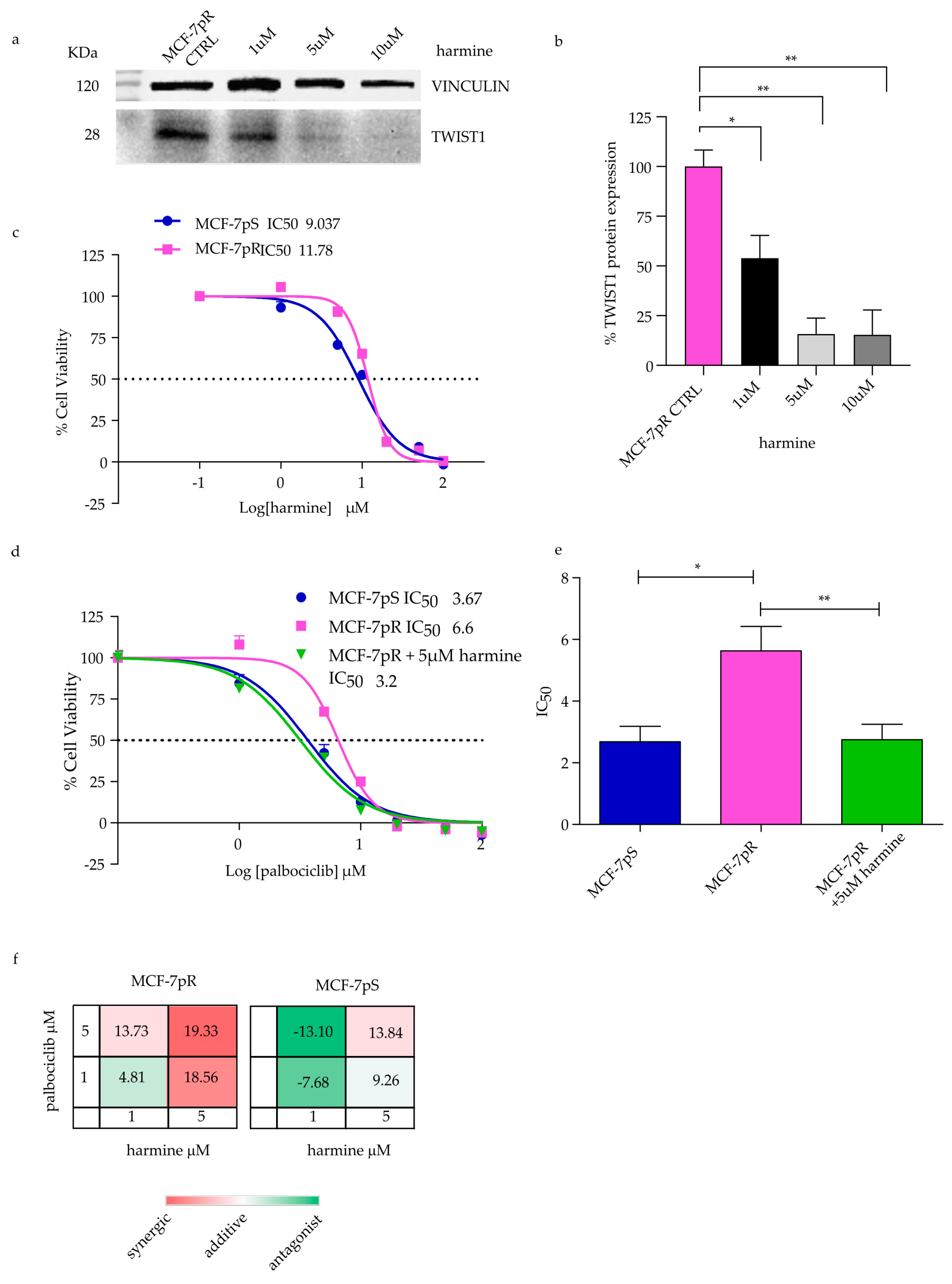
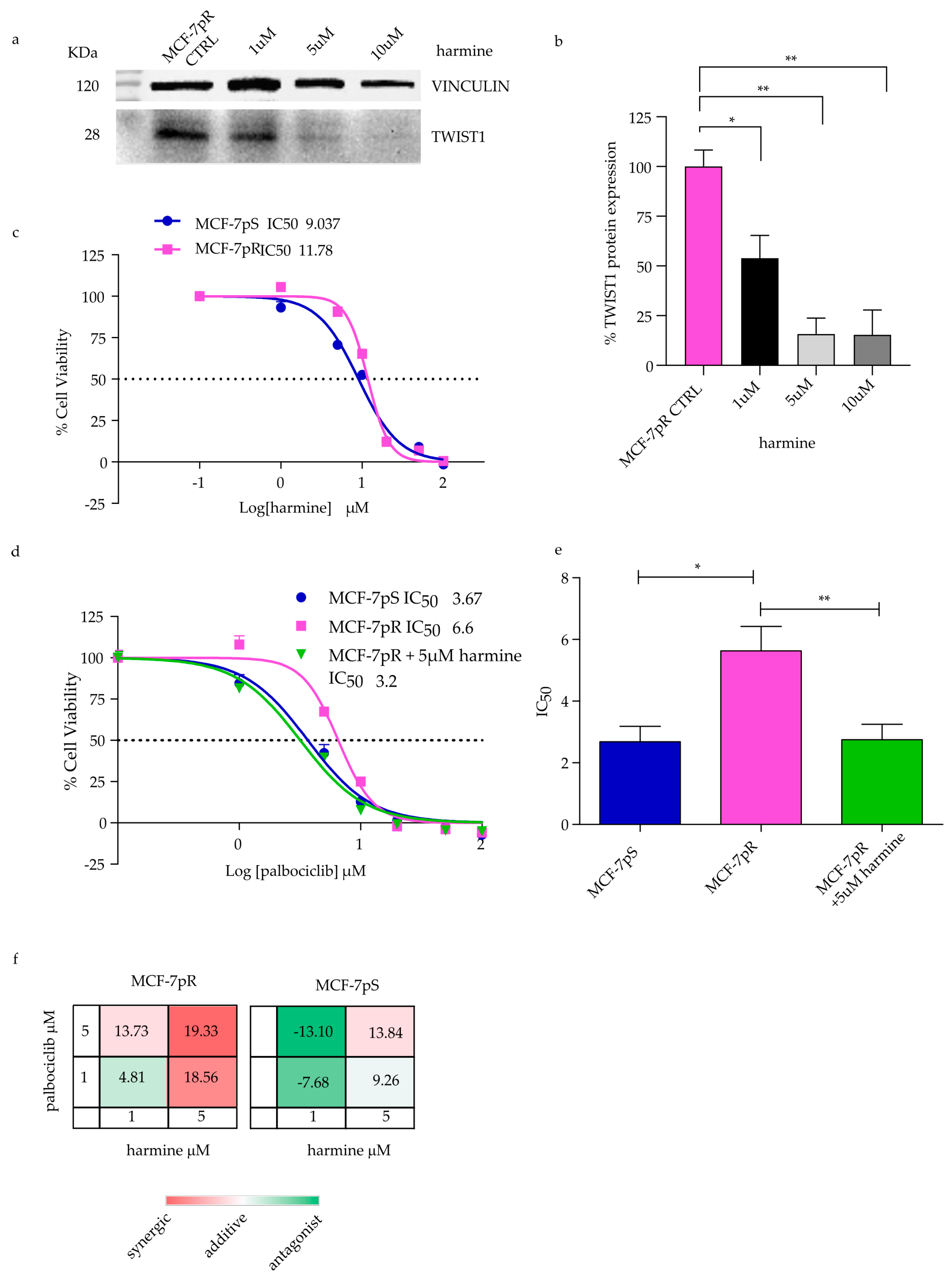
2.4. Overcoming Resistance in MCF-7pR by Targeting TWIST1
3. Discussion
4. Material and Methods
4.1. Cell Lines and Selection of Palbociclib Resistant Cells
4.2. Lentiviral Infection
4.3. Growth Inhibition Assay
4.4. Cell Cycle Analysis
4.5. Clonogenic Survival Assay
4.6. Wound Healing Assay
4.7. RNA Isolation, RNA-seq and Bioinformatic Analysis
4.8. Immunoblotting Analysis
4.9. Quantitative Reverse-Transcription Polymerase Chain Reaction (RT-qPCR)
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Momenimovahed, Z.; Salehiniya, H. Epidemiological characteristics of and risk factors for breast cancer in the world. Breast Cancer Targets Ther. 2019, 11, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 49, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Hart, C.D.; Migliaccio, I.; Malorni, L.; Guarducci, C.; Biganzoli, L.; Di Leo, A. Challenges in the management of advanced, ER-positive, HER2-negative breast cancer. Nat. Rev. Clin. Oncol. 2015, 12, 541–552. [Google Scholar] [CrossRef]
- Yang, H.; Wang, R.; Zeng, F.; Zhao, J.; Peng, S.; Ma, Y.; Chen, S.; Ding, S.; Zhong, L.; Guo, W.; et al. Impact of molecular subtypes on metastatic behavior and overall survival in patients with metastatic breast cancer: A single-center study combined with a large cohort study based on the Surveillance, Epidemiology and End Results database. Oncol. Lett. 2020, 20, 87. [Google Scholar] [CrossRef]
- Piezzo, M.; Chiodini, P.; Riemma, M.; Cocco, S.; Caputo, R.; Cianniello, D.; Di Gioia, G.; Di Lauro, V.; Rella, F.D.; Fusco, G.; et al. Progression-Free Survival and Overall Survival of CDK 4/6 Inhibitors Plus Endocrine Therapy in Metastatic Breast Cancer: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2020, 21, 6400. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Boer, K.; Bondarenko, I.; Patel, R.; Pinter, T.; Schmidt, M.; Shparyk, Y.V.; Thummala, A.; Voitko, N.; Bananis, E.; et al. Overall survival results from the randomized phase 2 study of palbociclib in combination with letrozole versus letrozole alone for first-line treatment of ER+/HER2− advanced breast cancer (PALOMA-1, TRIO-18). Breast Cancer Res. Treat. 2020, 183, 419–428. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Rugo, H.S.; Im, S.-A.; Slamon, D.J.; Harbeck, N.; Bondarenko, I.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; et al. Overall survival (OS) with palbociclib (PAL) + fulvestrant (FUL) in women with hormone receptor–positive (HR+), human epidermal growth factor receptor 2–negative (HER2–) advanced breast cancer (ABC): Updated analyses from PALOMA-3. J. Clin. Oncol. 2021, 39 (Suppl. S15), 1000. [Google Scholar] [CrossRef]
- Cogliati, V.; Capici, S.; Pepe, F.F.; di Mauro, P.; Riva, F.; Cicchiello, F.; Maggioni, C.; Cordani, N.; Cerrito, M.G.; Cazzaniga, M.E. How to Treat HR+/HER2− Metastatic Breast Cancer Patients after CDK4/6 Inhibitors: An Unfinished Story. Life 2022, 12, 378. [Google Scholar] [CrossRef]
- Prat, A.; Chaudhury, A.; Solovieff, N.; Paré, L.; Martinez, D.; Chic, N.; Martínez-Sáez, O.; Brasó-Maristany, F.; Lteif, A.; Taran, T.; et al. Correlative Biomarker Analysis of Intrinsic Subtypes and Efficacy Across the MONALEESA Phase III Studies. J. Clin. Oncol. 2021, 39, 1458–1467. [Google Scholar] [CrossRef]
- Yang, C.; Li, Z.; Bhatt, T.; Dickler, M.; Giri, D.; Scaltriti, M.; Baselga, J.; Rosen, N.; Chandarlapaty, S. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 2017, 36, 2255–2264. [Google Scholar] [CrossRef]
- Brabletz, S.; Schuhwerk, H.; Brabletz, T.; Stemmler, M.P. Dynamic EMT: A multi-tool for tumor progression. EMBO J. 2021, 40, e108647. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.; Trachet, E.; Albassam, M.; Zheng, X.; Leopold, W.R.; Pryer, N.K.; et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 2004, 3, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.; Mahadevan, D. Emerging Role and Targeting of Carcinoembryonic Antigen-related Cell Adhesion Molecule 6 (CEACAM6) in Human Malignancies. Clin. Cancer Drugs 2015, 2, 100–111. [Google Scholar] [CrossRef]
- Li, H.; Yang, B.; Xing, K.; Yuan, N.; Wang, B.; Chen, Z.; He, W.; Zhou, J. A preliminary study of the relationship between breast cancer metastasis and loss of heterozygosity by using exome sequencing. Sci. Rep. 2015, 4, 5460. [Google Scholar] [CrossRef]
- Huskey, A.L.W.; McNeely, I.; Merner, N.D. CEACAM Gene Family Mutations Associated With Inherited Breast Cancer Risk—A Comparative Oncology Approach to Discovery. Front. Genet. 2021, 12, 1368. [Google Scholar] [CrossRef]
- Kaur, B.; Mukhlis, Y.; Natesh, J.; Penta, D.; Musthapa Meeran, S. Identification of hub genes associated with EMT-induced chemoresistance in breast cancer using integrated bioinformatics analysis. Gene 2022, 809, 146016. [Google Scholar] [CrossRef]
- Stevens, L.E.; Peluffo, G.; Qiu, X.; Temko, D.; Fassl, A.; Li, Z.; Trinh, A.; Seehawer, M.; Jovanovic, B.; Aleckovic, M.; et al. JAK-STAT Signaling in Inflammatory Breast Cancer Enables Chemotherapy-Resistant Cell States. Cancer Res. 2023, 83, 264–284. [Google Scholar] [CrossRef]
- Watt, A.C.; Goel, S. Cellular mechanisms underlying response and resistance to CDK4/6 inhibitors in the treatment of hormone receptor-positive breast cancer. Breast Cancer Res. 2022, 24, 17. [Google Scholar] [CrossRef] [PubMed]
- Guiley, K.Z.; Stevenson, J.W.; Lou, K.; Barkovich, K.J.; Kumarasamy, V.; Wijeratne, T.U.; Bunch, K.L.; Tripathi, S.; Knudsen, E.S.; Witkiewicz, A.K.; et al. p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science 2019, 366, eaaw2106. [Google Scholar] [CrossRef]
- Lei, S.L.; Zhao, H.; Yao, H.L.; Chen, Y.; Lei, Z.D.; Liu, K.J.; Yang, Q. Regulatory roles of microRNA-708 and microRNA-31 in proliferation, apoptosis and invasion of colorectal cancer cells. Oncol. Lett. 2014, 8, 1768–1774. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Li, F.; Li, X.; Tian, Y.; Zhang, Y.; Sheng, X.; Song, Y.; Meng, Q.; Yuan, S.; Luan, L.; et al. Author Correction: MiR-31 promotes mammary stem cell expansion and breast tumorigenesis by suppressing Wnt signaling antagonists. Nat. Commun. 2020, 11, 5308. [Google Scholar] [CrossRef] [PubMed]
- Mironchik, Y.; Winnard, P.T., Jr.; Vesuna, F.; Kato, Y.; Wildes, F.; Pathak, A.P.; Kominsky, S.; Artemov, D.; Bhujwalla, Z.; Van Diest, P.; et al. Twist overexpression induces in vivo angiogenesis and correlates with chromosomal instability in breast cancer. Cancer Res. 2005, 65, 10801–10809. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, Y.; Chen, J.X.; Lu, X.Y.; Zhu, W.J.; Qin, L.; Xun, Z.X.; Zheng, Q.Y.; Li, E.M.; Sun, N.; et al. Harmine is an effective therapeutic small molecule for the treatment of cardiac hypertrophy. Acta Pharmacol. Sin. 2022, 43, 50–63. [Google Scholar] [CrossRef]
- Nafie, E.; Lolarga, J.; Lam, B.; Guo, J.; Abdollahzadeh, E.; Rodriguez, S.; Glackin, C.; Liu, J. Harmine inhibits breast cancer cell migration and invasion by inducing the degradation of Twist1. PLoS ONE 2021, 16, e0247652. [Google Scholar] [CrossRef]
- Yochum, Z.A.; Cades, J.; Mazzacurati, L.; Neumann, N.M.; Khetarpal, S.K.; Chatterjee, S.; Wang, H.; Attar, M.A.; Huang, E.H.; Chatley, S.N.; et al. A First-in-Class TWIST1 Inhibitor with Activity in Oncogene-Driven Lung Cancer. Mol. Cancer Res. 2017, 15, 1764–1776. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; Andre, F.; Winer, E.P.; et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Xia, Y.; Liu, Y.; Yang, C.; Simeone, D.M.; Sun, T.T.; DeGraff, D.J.; Tang, M.S.; Zhang, Y.; Wu, X.R. Dominant role of CDKN2B/p15INK4B of 9p21.3 tumor suppressor hub in inhibition of cell-cycle and glycolysis. Nat. Commun. 2021, 12, 2047. [Google Scholar] [CrossRef]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Green, J.L.; Okerberg, E.S.; Sejd, J.; Palafox, M.; Monserrat, L.; Alemayehu, S.; Wu, J.; Sykes, M.; Aban, A.; Serra, V.; et al. Direct CDKN2 Modulation of CDK4 Alters Target Engagement of CDK4 Inhibitor Drugs. Mol. Cancer Ther. 2019, 18, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Ogata, R.; Kishino, E.; Saitoh, W.; Koike, Y.; Kurebayashi, J. Resistance to cyclin-dependent kinase (CDK) 4/6 inhibitors confers cross-resistance to other CDK inhibitors but not to chemotherapeutic agents in breast cancer cells. Breast Cancer 2021, 28, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Iida, M.; Toyosawa, D.; Nakamura, M.; Tsuboi, K.; Tokuda, E.; Niwa, T.; Ishida, T.; Hayashi, S.I. Decreased ER dependency after acquired resistance to CDK4/6 inhibitors. Breast Cancer 2020, 27, 963–972. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, R.; McNair, C.; Schiewer, M.J.; Neupane, N.P.; Brand, L.J.; Augello, M.A.; Li, Z.; Cheng, L.C.; Yoshida, A.; Courtney, S.M.; et al. MAPK Reliance via Acquired CDK4/6 Inhibitor Resistance in Cancer. Clin. Cancer Res. 2018, 24, 4201–4214. [Google Scholar] [CrossRef]
- Meng, D.; Zhao, X.; Yang, Y.C.; Navickas, A.; Helland, C.; Goodarzi, H.; Singh, M.; Bandyopadhyay, S. A bi-steric mTORC1-selective inhibitor overcomes drug resistance in breast cancer. Oncogene 2023, 42, 2207–2217. [Google Scholar] [CrossRef]
- Villa, F.; Crippa, A.; Pelizzoni, D.; Ardizzoia, A.; Scartabellati, G.; Corbetta, C.; Cipriani, E.; Lavitrano, M.; Ardizzoia, A. Progression after First-Line Cyclin-Dependent Kinase 4/6 Inhibitor Treatment: Analysis of Molecular Mechanisms and Clinical Data. Int. J. Mol. Sci. 2023, 24, 14427. [Google Scholar] [CrossRef]
- Westcott, J.M.; Camacho, S.; Nasir, A.; Huysman, M.E.; Rahhal, R.; Dang, T.T.; Riegel, A.T.; Brekken, R.A.; Pearson, G.W. ΔNp63-Regulated Epithelial-to-Mesenchymal Transition State Heterogeneity Confers a Leader–Follower Relationship That Drives Collective Invasion. Cancer Res. 2020, 80, 3933–3944. [Google Scholar] [CrossRef]
- Su, X.; Chakravarti, D.; Cho, M.S.; Liu, L.; Gi, Y.J.; Lin, Y.-L.; Leung, M.L.; El-Naggar, A.; Creighton, C.J.; Suraokar, M.B.; et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 2010, 467, 986–990. [Google Scholar] [CrossRef]
- Park, S.; Lee, S.; Kim, J.; Kim, G.; Park, K.H.; Kim, T.U.; Chung, D.; Lee, H. ΔNp63 to TAp63 expression ratio as a potential molecular marker for cervical cancer prognosis. PLoS ONE 2019, 14, e0214867. [Google Scholar] [CrossRef]
- Whittle, J.R.; Vaillant, F.; Surgenor, E.; Policheni, A.N.; Giner, G.; Capaldo, B.D.; Chen, H.-R.; Liu, H.K.; Dekkers, J.F.; Sachs, N.; et al. Dual Targeting of CDK4/6 and BCL2 Pathways Augments Tumor Response in Estrogen Receptor–Positive Breast Cancer. Clin. Cancer Res. 2020, 26, 4120–4134. [Google Scholar] [CrossRef]
- Liu, M.; Liu, H.; Chen, J. Mechanisms of the CDK4/6 inhibitor palbociclib (PD 0332991) and its future application in cancer treatment. Oncol. Rep. 2018, 39, 901–911. [Google Scholar] [CrossRef]
- Guarducci, C.; Nardone, A.; Feiglin, A.; Migliaccio, I.; Malorni, L.; Bonechi, M.; Benelli, M.; Di Leo, A.; Hodgson, G.; Shapiro, G.; et al. Abstract PD7-12: Inhibition of CDK7 overcomes resistance to CDK4/6 inhibitors in hormone receptor positive breast cancer cells. Cancer Res. 2019, 79 (Suppl. S4), PD7-12. [Google Scholar] [CrossRef]
- Lee, H.-A.; Chu, K.-B.; Moon, E.-K.; Quan, F.-S. Histone Deacetylase Inhibitor-Induced CDKN2B and CDKN2D Contribute to G2/M Cell Cycle Arrest Incurred by Oxidative Stress in Hepatocellular Carcinoma Cells via Forkhead Box M1 Suppression. J. Cancer 2021, 12, 5086–5098. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, X.; Huang, Z.; Xu, L.; Zhu, W.; Liu, P. An ER-associated miRNA signature predicts prognosis in ER-positive breast cancer. J. Exp. Clin. Cancer Res. 2014, 33, 94. [Google Scholar] [CrossRef]
- Stepicheva, N.A.; Song, J.L. Function and regulation of microRNA-31 in development and disease. Mol. Reprod. Dev. 2016, 83, 654–674. [Google Scholar] [CrossRef]
- Schmittgen, T.D. miR-31: A master regulator of metastasis? Future Oncol. 2010, 6, 17–20. [Google Scholar] [CrossRef]
- Eguiarte-Solomon, F.; Blazanin, N.; Rho, O.; Carbajal, S.; Felsher, D.W.; Tran, P.T.; DiGiovanni, J. Twist1 is required for the development of UVB-induced squamous cell carcinoma. Mol. Carcinog. 2021, 60, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, S.; Ceccon, M.; Zappa, M.; Sharma, G.G.; Mastini, C.; Mauri, M.; Nigoghossian, M.; Massimino, L.; Cordani, N.; Farina, F.; et al. Lorlatinib Treatment Elicits Multiple On- and Off-Target Mechanisms of Resistance in ALK-Driven Cancer. Cancer Res. 2018, 78, 6866–6880. [Google Scholar] [CrossRef] [PubMed]
- Lanceta, L.; O’Neill, C.; Lypova, N.; Li, X.; Rouchka, E.; Waigel, S.; Gomez-Gutierrez, J.G.; Chesney, J.; Imbert-Fernandez, Y. Transcriptomic Profiling Identifies Differentially Expressed Genes in Palbociclib-Resistant ER+ MCF7 Breast Cancer Cells. Genes 2020, 11, 467. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yin, X.; Languino, L.R.; Altieri, D.C. Evaluation of drug combination effect using a Bliss independence dose-response surface model. Stat. Biopharm. Res. 2018, 10, 112–122. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antobody | Type | Clone | Code | Origin | Brand | Dilution | Milk/BSA |
|---|---|---|---|---|---|---|---|
| N-CADHERIN | Primary | H-2 | sc-393933 | Mouse | Santa Cruz | 1:200 | BSA 5% |
| E-CADHERIN | Primary | G-10 | sc-8426 | Mouse | Santa Cruz | 1:400 | BSA 5% |
| HCAM (CD44) | Primary | EPR18668 | ab189524 | Rabbit | Abcam | 1:200 | BSA 5% |
| RPS6 | Primary | C-8 | sc-74459 | Mouse | Santa Cruz | 1:400 | MILK 5% |
| TWIST1 | Primary | 2C1a | sc-81417 | Mouse | Santa Cruz | 1:800 | BSA 5% |
| p15 (CDKN2B) | Primary | D-12 | sc-271791 | Mouse | Santa Cruz | 1:500 | MILK 5% |
| CYCLIN-E | Primary | HE12 | sc-247 | Mouse | Santa Cruz | 1:200 | MILK 5% |
| VIMENTIN | Primary | D21H3 | 5741S | Rabbit | Cell Signaling | 1:1000 | BSA 5% |
| p27/KIP1 | Primary | F-8 | sc-1641 | Mouse | Santa Cruz | 1:400 | MILK 5% |
| β-ACTIN | Primary | AC-15 | A1978 | Mouse | SIGMA | 1:2000 | BSA 5% |
| VINCULIN | Primary | V284 | 05-386 | Mouse | Millipore | 1:4000 | BSA 5%/MILK 5% |
| GAPDH | Primary | 0411 | sc-47724 | Mouse | Santa Cruz | 1:3000 | BSA 5%/MILK 5% |
| anti-MOUSE | Secondary | IgG (H/L) | 170-6516 | Goat | BIORAD | 1:2000 | BSA 5% |
| anti-RABBIT | Secondary | IgG (H/L) | 170-6515 | Goat | BIORAD | 1:2000 | BSA 5% |
| Assay ID | Gene Symbol |
|---|---|
| Hs01046816_m1 | Esr1 |
| Hs00958111_m1 | Vim |
| Hs01556702_m1 | Pgr |
| Hs00944025_m1 | Ceacam5 |
| Hs00161904_m1 | Snai2 |
| Hs01047973_m1 | Runx2 |
| Hs00998133_m1 | Tgfb1 |
| Hs00609133_m1 | Col5a1 |
| Hs00153133_m1 | Ptgs2 |
| Hs00181211_m1 | Igfbp3 |
| Hs01053790_m1 | Abcg2 |
| Hs00602051_mH | Fscn1 |
| Hs00187067_m1 | Nr5a2 |
| Hs00899658_m1 | Mmp1 |
| Hs03988977_m1 | Ceacam7 |
| Hs00167155_m1 | Serpine1 |
| Hs00171642_m1 | Vcan |
| Hs00178811_m1 | Ntrk2 |
| Hs01115665_m1 | Tnc |
| Hs01041212_m1 | Bhlhe40 |
| Hs99999018_m1 | Bcl2 |
| Hs00924091_m1 | Cdkn2a |
| Hs00179899_m1 | Mgp |
| Hs00203118_m1 | Pycard |
| Hs99999905_m1 | Gapdh |
| Hs00793225_m1 | Cdkn2b |
| Hs01075861_m1 | CD44 |
| Hs00361486_m1 | Cdk7 |
| Hs00358836_m1 | Klf4 |
| Hs00978339_m1 | ΔNp63 |
| 478012_mir | hsa-miR-31-3p |
| 478015_mir | hsa-miR-31-5p |
| 477970_mir | hsa-miR-210-3p |
| 477860_mir | hsa-miR-16-5p |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cordani, N.; Mologni, L.; Piazza, R.; Tettamanti, P.; Cogliati, V.; Mauri, M.; Villa, M.; Malighetti, F.; Di Bella, C.; Jaconi, M.; et al. TWIST1 Upregulation Is a Potential Target for Reversing Resistance to the CDK4/6 Inhibitor in Metastatic Luminal Breast Cancer Cells. Int. J. Mol. Sci. 2023, 24, 16294. https://doi.org/10.3390/ijms242216294
Cordani N, Mologni L, Piazza R, Tettamanti P, Cogliati V, Mauri M, Villa M, Malighetti F, Di Bella C, Jaconi M, et al. TWIST1 Upregulation Is a Potential Target for Reversing Resistance to the CDK4/6 Inhibitor in Metastatic Luminal Breast Cancer Cells. International Journal of Molecular Sciences. 2023; 24(22):16294. https://doi.org/10.3390/ijms242216294
Chicago/Turabian StyleCordani, Nicoletta, Luca Mologni, Rocco Piazza, Pietro Tettamanti, Viola Cogliati, Mario Mauri, Matteo Villa, Federica Malighetti, Camillo Di Bella, Marta Jaconi, and et al. 2023. "TWIST1 Upregulation Is a Potential Target for Reversing Resistance to the CDK4/6 Inhibitor in Metastatic Luminal Breast Cancer Cells" International Journal of Molecular Sciences 24, no. 22: 16294. https://doi.org/10.3390/ijms242216294
APA StyleCordani, N., Mologni, L., Piazza, R., Tettamanti, P., Cogliati, V., Mauri, M., Villa, M., Malighetti, F., Di Bella, C., Jaconi, M., Cerrito, M. G., Cavaletti, G., Lavitrano, M., & Cazzaniga, M. E. (2023). TWIST1 Upregulation Is a Potential Target for Reversing Resistance to the CDK4/6 Inhibitor in Metastatic Luminal Breast Cancer Cells. International Journal of Molecular Sciences, 24(22), 16294. https://doi.org/10.3390/ijms242216294