

Progression after First-Line Cyclin-Dependent Kinase 4/6 Inhibitor Treatment: Analysis of Molecular Mechanisms and Clinical Data
 , , , and
, , , and
Abstract
:1. Introduction
2. CDK4/6 Inhibitors’ Resistance Mechanisms
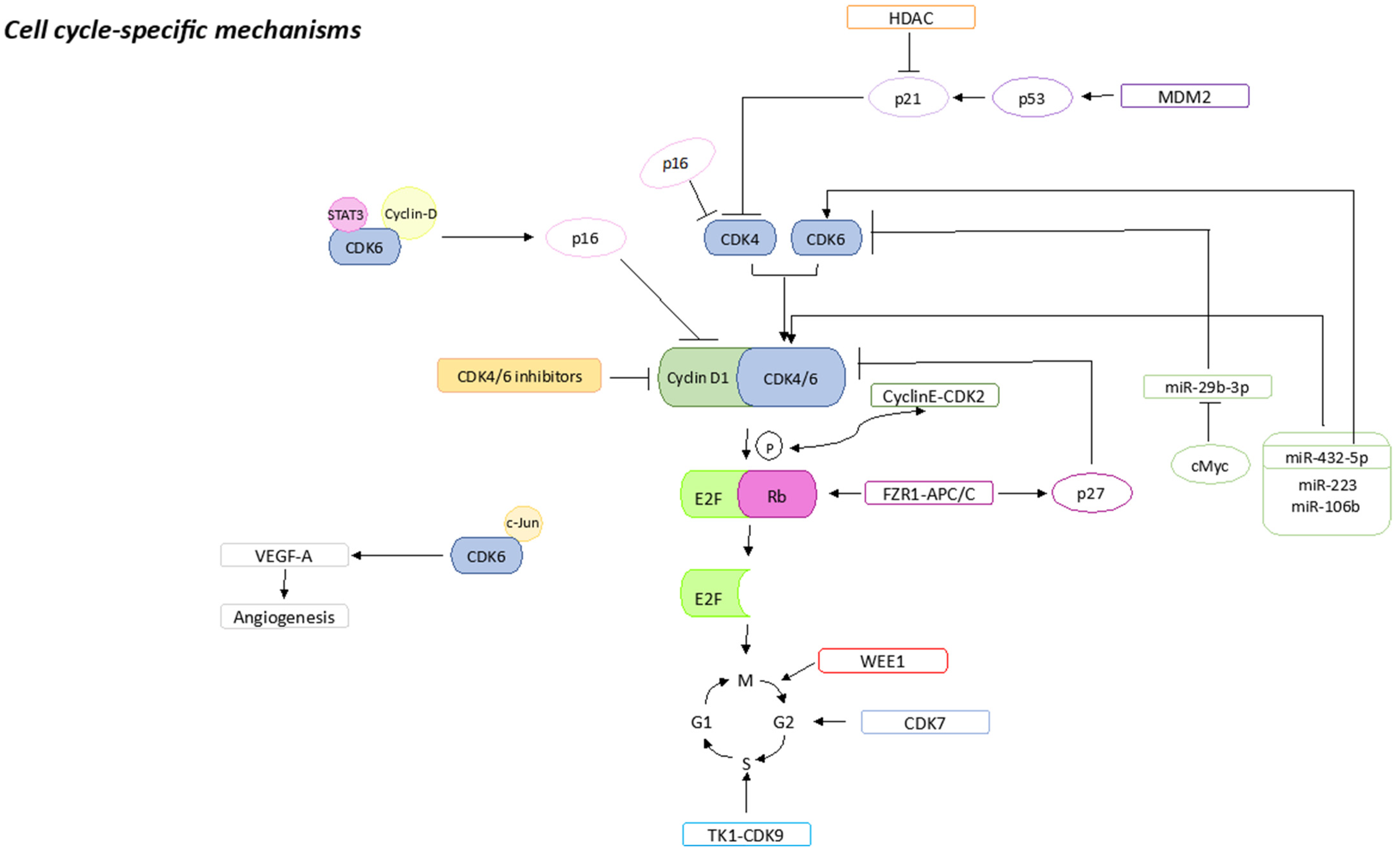
2.1. Cell-Cycle-Specific Mechanisms
2.1.1. pRb Loss or Mutations
2.1.2. p16 Amplification
2.1.3. CDK2 Amplification
2.1.4. E2F Amplification
2.1.5. CDK7 Overexpression
2.1.6. CDK6 Amplification
2.1.7. WEE1 Overexpression
2.1.8. MDM2 Overexpression
2.1.9. HDACs Activation
2.1.10. FZR1 Loss
2.1.11. TK1 Activation
2.1.12. miRNAs Expression
2.1.13. S6K1 Amplification
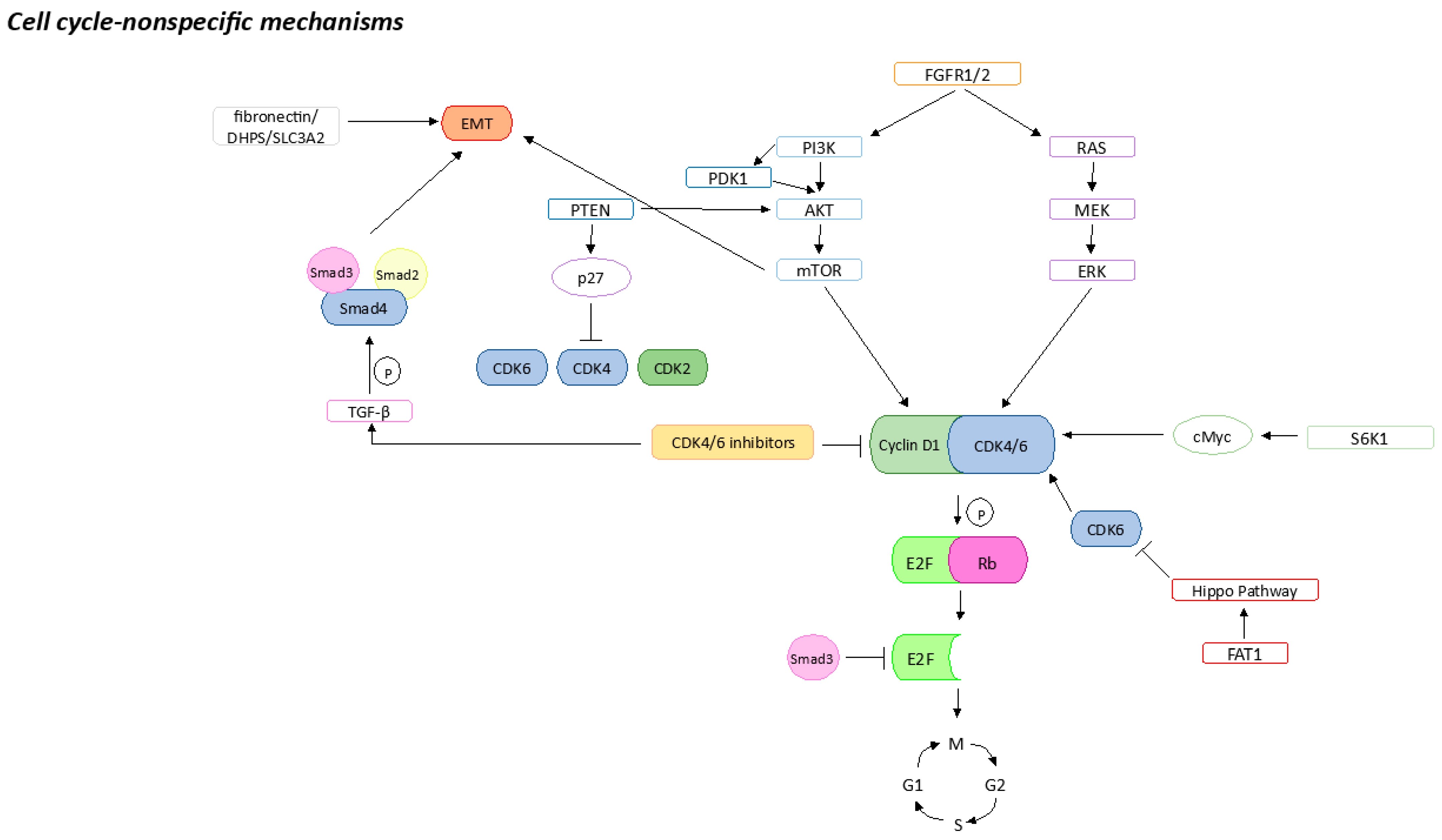
2.2. Cell-Cycle-Non-Specific Mechanisms
2.2.1. FGFR Pathway Activation
2.2.2. PI3K/AKT/mTOR Pathway Activation
2.2.3. MAPK Pathway Activation
2.2.4. Hippo Pathway Inhibition by FAT1
2.2.5. Epithelial–Mesenchymal Transition
2.2.6. Apoptosis Failure
2.2.7. Stemness Properties
3. Short- and Long-Term Adaptation to CDK4/6 Inhibitors
3.1. Short-Term Adaptation to CDK4/6 Inhibitors
3.2. Long-Term Acquisition of CDK4/6 Inhibitors Resistance
4. Types of Subsequent Therapies after Randomized Controlled Trial (RCT) and in the Real World
5. Progression after CDK4/6is
6. Potential Therapeutic Approaches
6.1. Continuation of CDK4/6is

{kind=link}
{kind=link}
| Trial | Number of Patients | Population | Prior CDK4/6is | Subsequent ET | Subsequent CDK4/6is | Efficacy | PFS (Months) |
|---|---|---|---|---|---|---|---|
| MAINTAIN [103] | 119 | Progression on CDK4/6is + ET |
|
| Ribociclib |
|
|
| PACE [104] | 220 | Progression on CDK4/6is + ET after at least 6 months of therapy |
| Fulvestrant | Palbociclib |
|
|
| PALMIRA [105] | 198 | CB during palbociclib | Palbociclib 100% |
| Palbociclib | 6-months CBR 41.9% |
|
| BIOPER [106] | 33 |
| Palbociclib 100% |
| Palbociclib | CBR 34.4% | 2.6 |
6.2. Endocrine Therapy
6.3. Switch to Combined Target Therapies
6.3.1. ET Combined with PI3K Inhibitors
6.3.2. ET Combined with mTOR Inhibitors
6.3.3. ET Combined with Other Targeted Drugs
6.3.4. CDK4/6 Inhibitors plus Other Targeted Drugs
| Target Therapy | Trial | Number of Patients | Population | Prior CDK4/6is | Subsequent TT | Efficacy | PFS (Months) |
|---|---|---|---|---|---|---|---|
| PI3K inhibitor | SOLAR-1 [114] | 572 |
| Any | Fulvestrant + Alpelisib | NA | 7.3 |
| BYLieve [116] | 127 |
| a. CDK4/6i + AI b. CDK4/6i + fulvestrant | a. Fulvestrant + Alpelisib b. Letrozole + Alpelisib |
| a. 8.2 b. 5.6 | |
| MTOR inhibitor | TRINITI-1 [122] | 104 | Progression on a CDK4/6i after ≥4 months of therapy as the last prior treatment regimen | Any | Exemestane + Everolimus + Ribociclib | CBR 41% | 5.7 |
| AKT inhibitor | CAPItello-291 [131] | 708 | Progression during or after treatment with an IA, with or without previous CDK4/6is | Any (69.1% of pts) | a. Capivasertib + fulvestrant b. Placebo + fulvestrant | a. ORR 22.9% b. ORR 12.2% | a. 7.2 b. 3.6 |
| HDAC inhibitor | ACE [132] | 365 | Progression after at least one endocrine therapy | Palbociclib (>1% pts) | a. Tucidinostat + exemestane b. Placebo +exemestane | a. PR 18% SD 56% b. PR 9% SD 54% | a. 7.4 b. 3.8 |
| E2112 phase III [133] | 608 | Progression on AI in the adjuvant or metastatic setting | Any (35% of pts) | a. Etinostat + exemestane b. Placebo + exemestane | a. 5.8% b. 5.6% | a. 3.3 b. 3.1 | |
| BCL-2 inhibitors | VERONICA [136] | 103 | ≤2 prior lines of ET and prior chemotherapy in the locally advanced/mBC setting and progression during/after CDK4/6is | Any | a. Venetoclax + fulvestrant b. Fulvestrant | a. CBR 11.8% b. 13.7% | a. 2.69 b. 1.94 |
| Immune checkpoint inhibitors | PACE [104] | 200 | Prior response to and subsequent progression on CDK4/6is and ET |
| a. Avelumab + Fulvestrant + Palbociclib b. Fulvestrant + Palbociclib c. Fuvestrant | a. 17.9% b. 13.8% c. 10.8% | a. 8.1 b. 4.6 c. 4.8 |
6.3.5. PARP Inhibitors
6.4. Switch to Chemotherapy
| Trial | Number of Patients | Population | Prior CDK4/6is | Subsequent CT | Efficacy | PFS (Months) |
|---|---|---|---|---|---|---|
| 1-US study [91] | 525 | Progression on CDK4/6is | Any | Capecitabine or Taxanes (35.6%) | NA | NA |
| 2-US study [92] | 1210 | Progression on first-line CDK4/6is |
| NA | NA | 3.71 |
| Russian study [144] | 54 | Progression on CDK4/6is |
|
|
| 10.0 |
| DESTINY-Breast04 (HR+ cohort) [145] | 494 | Progression on CT or OT | Any (about 70% of pts) | T-DXd vs. TPC (eribulin, capecitabine, nab-paclitaxel, gemcitabine or paclitaxel) | 52.3% T-DXd vs. 16.3% TPC | 10.1 T-DXd vs. 5.4 TPC |
| TROPiCS-02 [148] | 543 | Progression after CDK4/6is and at least 2 chemotherapeutic agents (including a taxane) | Any (98% of pts) | SG vs. TPC (eribulin, capecitabine, gemcitabine or vinorelbine) | 21% SG vs. 14% TPC | 5.5 SG vs. 4.0 TPC |
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Costa, A.; Norton, L.; Cameron, D.; Cufer, T.; Fallowfield, L.; Francis, P.; Gligorov, J.; Kyriakides, S.; Lin, N.; et al. 1st International Consensus Guidelines for Advanced Breast Cancer (ABC 1). Breast 2012, 21, 242–252. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J. Extending Survival with Chemotherapy in Metastatic Breast Cancer. Oncologist 2005, 10 (Suppl. S3), 20–29. [Google Scholar] [CrossRef]
- Bradley, R.; Burrett, J.; Clarke, M.; Davies, C.; Duane, F.; Evans, V.; Gettins, L.; Godwin, J.; Gray, R.; Liu, H.; et al. Aromatase Inhibitors versus Tamoxifen in Early Breast Cancer: Patient-Level Meta-Analysis of the Randomised Trials. Lancet 2015, 386, 1341–1352. [Google Scholar] [CrossRef]
- Lobbezoo, D.J.A.; Van Kampen, R.J.W.; Voogd, A.C.; Dercksen, M.W.; Van Den Berkmortel, F.; Smilde, T.J.; Van De Wouw, A.J.; Peters, F.P.J.; Van Riel, J.M.G.H.; Peters, N.A.J.B.; et al. Prognosis of Metastatic Breast Cancer Subtypes: The Hormone Receptor/HER2-Positive Subtype Is Associated with the Most Favorable Outcome. Breast Cancer Res. Treat. 2013, 141, 507–514. [Google Scholar] [CrossRef]
- Caswell-Jin, J.L.; Plevritis, S.K.; Tian, L.; Cadham, C.J.; Xu, C.; Stout, N.K.; Sledge, G.W.; Mandelblatt, J.S.; Kurian, A.W. Change in Survival in Metastatic Breast Cancer with Treatment Advances: Meta-Analysis and Systematic Review. JNCI Cancer Spectr. 2018, 2, pky062. [Google Scholar] [CrossRef]
- Carlson, R.W.; Allred, D.C.; Anderson, B.O.; Burstein, H.J.; Edge, S.B.; Farrar, W.B.; Forero, A.; Giordano, S.H.; Goldstein, L.J.; Gradishar, W.J.; et al. Metastatic Breast Cancer, Version 1.2012: Featured Updates to the NCCN Guidelines. J. Natl. Compr. Cancer Netw. 2012, 10, 821–829. [Google Scholar] [CrossRef] [PubMed]
- VanArsdale, T.; Boshoff, C.; Arndt, K.T.; Abraham, R.T. Molecular Pathways: Targeting the Cyclin D-CDK4/6 Axis for Cancer Treatment. Clin. Cancer Res. 2015, 21, 2905–2910. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.-S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; André, F.; Winer, E.P.; et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef]
- Goetz, M.P.; Toi, M.; Campone, M.; Trédan, O.; Bourayou, N.; Sohn, J.; Park, I.H.; Paluch-Shimon, S.; Huober, J.; Chen, S.C.; et al. MONARCH 3: Abemaciclib as Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef]
- Im, S.-A.; Lu, Y.-S.; Bardia, A.; Harbeck, N.; Colleoni, M.; Franke, F.; Chow, L.; Sohn, J.; Lee, K.-S.; Campos-Gomez, S.; et al. Overall Survival with Ribociclib plus Endocrine Therapy in Breast Cancer. N. Engl. J. Med. 2019, 381, 307–316. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.-S.; Sonke, G.S.; Hart, L.; Campone, M.; Petrakova, K.; Winer, E.P.; Janni, W.; et al. Overall Survival with Ribociclib plus Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2022, 386, 942–950. [Google Scholar] [CrossRef]
- Goetz, M.P.; Toi, M.; Huober, J.; Sohn, J.; Tredan, O.; Park, I.H.; Campone, M.; Chen, S.C.; Sanchez, L.M.M.; Paluch-Shimon, S.; et al. LBA15—MONARCH 3: Interim Overall Survival (OS) Results of Abemaciclib plus a Nonsteroidal Aromatase Inhibitor (NSAI) in Patients (Pts) with HR+, HER2-Advanced Breast Cancer (ABC). Ann. Oncol. 2022, 33, S1384. [Google Scholar] [CrossRef]
- Finn, R.S.; Rugo, H.S.; Dieras, V.C.; Harbeck, N.; Im, S.-A.; Gelmon, K.A.; Walshe, J.M.; Martin, M.; Chavez Mac Gregor, M.; Bananis, E.; et al. Overall Survival (OS) with First-Line Palbociclib plus Letrozole (PAL+LET) versus Placebo plus Letrozole (PBO+LET) in Women with Estrogen Receptor–Positive/Human Epidermal Growth Factor Receptor 2–Negative Advanced Breast Cancer (ER+/HER2− ABC): Analyses from PALOMA-2. J. Clin. Oncol. 2022, 40, LBA1003. [Google Scholar] [CrossRef]
- Bockstaele, L.; Bisteau, X.; Paternot, S.; Roger, P.P. Differential Regulation of Cyclin-Dependent Kinase 4 (CDK4) and CDK6, Evidence That CDK4 Might Not Be Activated by CDK7, and Design of a CDK6 Activating Mutation. Mol. Cell. Biol. 2009, 29, 4188–4200. [Google Scholar] [CrossRef] [PubMed]
- Fassl, A.; Geng, Y.; Sicinski, P. CDK4 and CDK6 Kinases: From Basic Science to Cancer Therapy. Science 2022, 375, eabc1495. [Google Scholar] [CrossRef] [PubMed]
- Diehl, J.A. Cycling to Cancer with Cyclin D1. Cancer Biol. Ther. 2002, 1, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.; Infante, J.R. Targeting CDK4/6 in Patients with Cancer. Cancer Treat. Rev. 2016, 45, 129–138. [Google Scholar] [CrossRef]
- Malumbres, M.; Sotillo, R.; Santamaría, D.; Galán, J.; Cerezo, A.; Ortega, S.; Dubus, P.; Barbacid, M. Mammalian Cells Cycle without the D-Type Cyclin-Dependent Kinases Cdk4 and Cdk6. Cell 2004, 118, 493–504. [Google Scholar] [CrossRef]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating Cancer with Selective CDK4/6 Inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The Cyclin-Dependent Kinase 4/6 Inhibitor Palbociclib in Combination with Letrozole versus Letrozole Alone as First-Line Treatment of Oestrogen Receptor-Positive, HER2-Negative, Advanced Breast Cancer (PALOMA-1/TRIO-18): A Randomised Phase 2 Study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zheng, L.; Sun, Z.; Li, J. CDK4/6 Inhibitor Resistance Mechanisms and Treatment Strategies (Review). Int. J. Mol. Med. 2022, 50, 128. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; Edwards, C.D.; Kobzik, L.; Godleski, J.; Richards, W.; Sugarbaker, D.J.; Rollins, B.J. Reciprocal Rb Inactivation and P16INK4 Expression in Primary Lung Cancers and Cell Lines. Cancer Res. 1995, 55, 505–509. [Google Scholar]
- Lukas, J.; Parry, D.; Aagaard, L.; Mann, D.J.; Bartkova, J.; Strauss, M.; Peters, G.; Bartek, J. Retinoblastoma-Protein-Dependent Cell-Cycle Inhibition by the Tumour Suppressor P16. Nature 1995, 375, 503–506. [Google Scholar] [CrossRef]
- Dean, J.L.; McClendon, A.K.; Hickey, T.E.; Butler, L.M.; Tilley, W.D.; Witkiewicz, A.K.; Knudsen, E.S. Therapeutic Response to CDK4/6 Inhibition in Breast Cancer Defined by Ex Vivo Analyses of Human Tumors. Cell Cycle 2012, 11, 2756–2761. [Google Scholar] [CrossRef]
- Palafox, M.; Monserrat, L.; Bellet, M.; Villacampa, G.; Gonzalez-Perez, A.; Oliveira, M.; Brasó-Maristany, F.; Ibrahimi, N.; Kannan, S.; Mina, L.; et al. High P16 Expression and Heterozygous RB1 Loss Are Biomarkers for CDK4/6 Inhibitor Resistance in ER+ Breast Cancer. Nat. Commun. 2022, 13, 5258. [Google Scholar] [CrossRef]
- Gladden, A.B.; Diehl, J.A. Cell Cycle Progression without Cyclin E/CDK2: Breaking down the Walls of Dogma. Cancer Cell 2003, 4, 160–162. [Google Scholar] [CrossRef]
- Etemadmoghadam, D.; Au-Yeung, G.; Wall, M.; Mitchell, C.; Kansara, M.; Loehrer, E.; Batzios, C.; George, J.; Ftouni, S.; Weir, B.A.; et al. Resistance to CDK2 Inhibitors Is Associated with Selection of Polyploid Cells in CCNE1-Amplified Ovarian Cancer. Clin. Cancer Res. 2013, 19, 5960–5971. [Google Scholar] [CrossRef]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef]
- Jin, X.; Ge, L.P.; Li, D.Q.; Shao, Z.M.; Di, G.H.; Xu, X.E.; Jiang, Y.Z. LncRNA TROJAN Promotes Proliferation and Resistance to CDK4/6 Inhibitor via CDK2 Transcriptional Activation in ER+ Breast Cancer. Mol. Cancer 2020, 19, 87. [Google Scholar] [CrossRef] [PubMed]
- Schachter, M.M.; Merrick, K.A.; Larochelle, S.; Hirschi, A.; Zhang, C.; Shokat, K.M.; Rubin, S.M.; Fisher, R.P. A Cdk7-Cdk4 T-Loop Phosphorylation Cascade Promotes G1 Progression. Mol. Cell 2013, 50, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.-A.; Pancholi, S.; Ribas, R.; Gao, Q.; Simigdala, N.; Nikitorowicz-Buniak, J.; Johnston, S.; Dowsett, M. Abstract P3-03-09: Resistance to Palbociclib Depends on Multiple Targetable Mechanisms Highlighting the Potential of Drug Holidays and Drug Switching to Improve Therapeutic Outcome. Cancer Res. 2017, 77 (Suppl. S4). [Google Scholar] [CrossRef]
- Tigan, A.S.; Bellutti, F.; Kollmann, K.; Tebb, G.; Sexl, V. CDK6-a Review of the Past and a Glimpse into the Future: From Cell-Cycle Control to Transcriptional Regulation. Oncogene 2016, 35, 3083–3091. [Google Scholar] [CrossRef] [PubMed]
- Kollmann, K.; Heller, G.; Schneckenleithner, C.; Warsch, W.; Scheicher, R.; Ott, R.G.; Schäfer, M.; Fajmann, S.; Schlederer, M.; Schiefer, A.I.; et al. A Kinase-Independent Function of CDK6 Links the Cell Cycle to Tumor Angiogenesis. Cancer Cell 2016, 30, 359–360. [Google Scholar] [CrossRef]
- Matheson, C.J.; Backos, D.S.; Reigan, P. Targeting WEE1 Kinase in Cancer. Trends Pharmacol. Sci. 2016, 37, 872–881. [Google Scholar] [CrossRef]
- Efeyan, A.; Ortega-Molina, A.; Velasco-Miguel, S.; Herranz, D.; Vassilev, L.T.; Serrano, M. Induction of P53-Dependent Senescence by the MDM2 Antagonist Nutlin-3a in Mouse Cells of Fibroblast Origin. Cancer Res. 2007, 67, 7350–7357. [Google Scholar] [CrossRef]
- Cox, L.S. Multiple Pathways Control Cell Growth and Transformation: Overlapping and Independent Activities of P53 and P21Cip1/Waf1/Sdi1. J. Pathol 1997, 183, 134–140. [Google Scholar] [CrossRef]
- Portman, N.; Chen, J.; Lim, E. MDM2 as a Rational Target for Intervention in CDK4/6 Inhibitor Resistant, Hormone Receptor Positive Breast Cancer. Front. Oncol. 2021, 11, 777867. [Google Scholar] [CrossRef]
- Sabnis, G.J.; Goloubeva, O.; Chumsri, S.; Nguyen, N.; Sukumar, S.; Brodie, A.M.H. Functional Activation of the Estrogen Receptor-α and Aromatase by the HDAC Inhibitor Entinostat Sensitizes ER-Negative Tumors to Letrozole. Cancer Res. 2011, 71, 1893–1903. [Google Scholar] [CrossRef]
- Dash, B.C.; El-Deiry, W.S. Phosphorylation of P21 in G2/M Promotes Cyclin B-Cdc2 Kinase Activity. Mol. Cell. Biol. 2005, 25, 3364–3387. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Jin, X.; Ma, J.; Ding, D.; Huang, Z.; Sheng, H.; Yan, Y.; Pan, Y.; Wei, T.; Wang, L.; et al. HDAC5 Loss Impairs RB Repression of Pro-Oncogenic Genes and Confers CDK4/6 Inhibitor Resistance in Cancer. Cancer Res. 2021, 81, 1486–1499. [Google Scholar] [CrossRef] [PubMed]
- Ramanujan, A.; Tiwari, S. APC/C and Retinoblastoma Interaction: Cross-Talk of Retinoblastoma Protein with the Ubiquitin Proteasome Pathway. Biosci. Rep. 2016, 36, e00377. [Google Scholar] [CrossRef] [PubMed]
- The, I.; Ruijtenberg, S.; Bouchet, B.P.; Cristobal, A.; Prinsen, M.B.W.; Van Mourik, T.; Koreth, J.; Xu, H.; Heck, A.J.R.; Akhmanova, A.; et al. Rb and FZR1/Cdh1 Determine CDK4/6-Cyclin D Requirement in C. Elegans and Human Cancer Cells. Nat. Commun. 2015, 6, 5906. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Liu, W.; Doihara, H.; Wan, Y. Regulation of Skp2-P27 Axis by the Cdh1/Anaphase-Promoting Complex Pathway in Colorectal Tumorigenesis. Am. J. Pathol. 2008, 173, 217–228. [Google Scholar] [CrossRef]
- He, Q.; Zou, L.; Zhang, P.A.; Lui, J.X.; Skog, S.; Fornander, T. The Clinical Significance of Thymidine Kinase 1 Measurement in Serum of Breast Cancer Patients Using Anti-TK1 Antibody. Int. J. Biol. Markers 2000, 15, 139–146. [Google Scholar] [CrossRef]
- Bjöhle, J.; Bergqvist, J.; Gronowitz, J.S.; Johansson, H.; Carlsson, L.; Einbeigi, Z.; Linderholm, B.; Loman, N.; Malmberg, M.; Söderberg, M.; et al. Serum Thymidine Kinase Activity Compared with CA 15-3 in Locally Advanced and Metastatic Breast Cancer within a Randomized Trial. Breast Cancer Res. Treat. 2013, 139, 751–758. [Google Scholar] [CrossRef]
- Del Re, M.; Bertolini, I.; Crucitta, S.; Fontanelli, L.; Rofi, E.; De Angelis, C.; Diodati, L.; Cavallero, D.; Gianfilippo, G.; Salvadori, B.; et al. Overexpression of TK1 and CDK9 in Plasma-Derived Exosomes Is Associated with Clinical Resistance to CDK4/6 Inhibitors in Metastatic Breast Cancer Patients. Breast Cancer Res. Treat. 2019, 178, 57–62. [Google Scholar] [CrossRef]
- Lin, S.L.; Chang, D.C.; Ying, S.Y.; Leu, D.; Wu, D.T.S. MicroRNA MiR-302 Inhibits the Tumorigenecity of Human Pluripotent Stem Cells by Coordinate Suppression of the CDK2 and CDK4/6 Cell Cycle Pathways. Cancer Res. 2010, 70, 9473–9482. [Google Scholar] [CrossRef]
- Pulito, C.; Cristaudo, A.; La Porta, C.; Zapperi, S.; Blandino, G.; Morrone, A.; Strano, S. Oral Mucositis: The Hidden Side of Cancer Therapy. J. Exp. Clin. Cancer Res. 2020, 39, 210. [Google Scholar] [CrossRef]
- Cornell, L.; Wander, S.A.; Visal, T.; Wagle, N.; Shapiro, G.I. MicroRNA-Mediated Suppression of the TGF-β Pathway Confers Transmissible and Reversible CDK4/6 Inhibitor Resistance. Cell Rep. 2019, 26, 2667–2680.e7. [Google Scholar] [CrossRef]
- Ji, W.; Zhang, W.; Wang, X.; Shi, Y.; Yang, F.; Xie, H.; Zhou, W.; Wang, S.; Guan, X. C-Myc Regulates the Sensitivity of Breast Cancer Cells to Palbociclib via c-Myc/MiR-29b-3p/CDK6 Axis. Cell Death Dis. 2020, 11, 760. [Google Scholar] [CrossRef] [PubMed]
- Fingar, D.C.; Blenis, J. Target of Rapamycin (TOR): An Integrator of Nutrient and Growth Factor Signals and Coordinator of Cell Growth and Cell Cycle Progression. Oncogene 2004, 23, 3151–3171. [Google Scholar] [CrossRef] [PubMed]
- Mo, H.; Liu, X.; Xue, Y.; Chen, H.; Guo, S.; Li, Z.; Wang, S.; Li, C.; Han, J.; Fu, M.; et al. S6K1 Amplification Confers Innate Resistance to CDK4/6 Inhibitors through Activating c-Myc Pathway in Patients with Estrogen Receptor-Positive Breast Cancer. Mol. Cancer 2022, 21, 171. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Grose, R. Fibroblast Growth Factor Signalling: From Development to Cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Luqmani, Y.A.; Graham, M.; Coombes, R.C. Expression of Basic Fibroblast Growth Factor, FGFR1 and FGFR2 in Normal and Malignant Human Breast, and Comparison with Other Normal Tissues. Br. J. Cancer 1992, 66, 273–280. [Google Scholar] [CrossRef]
- Turner, N.; Pearson, A.; Sharpe, R.; Lambros, M.; Geyer, F.; Lopez-Garcia, M.A.; Natrajan, R.; Marchio, C.; Iorns, E.; Mackay, A.; et al. FGFR1 Amplification Drives Endocrine Therapy Resistance and Is a Therapeutic Target in Breast Cancer. Cancer Res. 2010, 70, 2085–2094. [Google Scholar] [CrossRef]
- Sobhani, N.; Fassl, A.; Mondani, G.; Generali, D.; Otto, T. Targeting Aberrant FGFR Signaling to Overcome CDK4/6 Inhibitor Resistance in Breast Cancer. Cells 2021, 10, 293. [Google Scholar] [CrossRef]
- Mao, P.; Kusiel, J.; Cohen, O.; Wagle, N. Abstract PD4-01: The Role of FGF/FGFR Axis in Resistance to SERDs and CDK4/6 Inhibitors in ER+ Breast Cancer. Cancer Res. 2018, 78 (Suppl. S4), PD4-01. [Google Scholar] [CrossRef]
- Jansen, V.M.; Bhola, N.E.; Bauer, J.A.; Formisano, L.; Lee, K.M.; Hutchinson, K.E.; Witkiewicz, A.K.; Moore, P.D.; Estrada, M.V.; Sánchez, V.; et al. Kinome-Wide RNA Interference Screen Reveals a Role for PDK1 in Acquired Resistance to CDK4/6 Inhibition in ER-Positive Breast Cancer. Cancer Res. 2017, 77, 2488–2499. [Google Scholar] [CrossRef]
- Michaloglou, C.; Crafter, C.; Siersbaek, R.; Delpuech, O.; Curwen, J.O.; Carnevalli, L.S.; Staniszewska, A.D.; Polanska, U.M.; Cheraghchi-Bashi, A.; Lawson, M.; et al. Combined Inhibition of MTOR and CDK4/6 Is Required for Optimal Blockade of E2F Function and Long-Term Growth Inhibition in Estrogen Receptor-Positive Breast Cancer. Mol. Cancer Ther. 2018, 17, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Ye, W.; Ly, A.; Hosono, Y.; Murchi, E.; Walmsley, C.S.; Huynh, T.; Healy, C.; Peterson, R.; Yanase, S.; et al. PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kα Inhibitors in Breast Cancer. Cancer Discov. 2020, 10, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Jiang, B.; Guo, J.; Shao, H.; Del Priore, I.S.; Chang, Q.; Kudo, R.; Li, Z.; Razavi, P.; Liu, B.; et al. INK4 Tumor Suppressor Proteins Mediate Resistance to CDK4/6 Kinase Inhibitors. Cancer Discov. 2022, 12, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Formisano, L.; Lu, Y.; Servetto, A.; Hanker, A.B.; Jansen, V.M.; Bauer, J.A.; Sudhan, D.R.; Guerrero-Zotano, A.L.; Croessmann, S.; Guo, Y.; et al. Aberrant FGFR Signaling Mediates Resistance to CDK4/6 Inhibitors in ER+ Breast Cancer. Nat. Commun. 2019, 10, 1373. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, L.; Raimondi, F.M.; Pietranera, M.; Di Rocco, A.; Di Benedetto, L.; Miele, E.; Lazzeroni, R.; Cimino, G.; Spinelli, G.P. Assessment of Resistance Mechanisms and Clinical Implications in Patients with KRAS Mutated-Metastatic Breast Cancer and Resistance to CDK4/6 Inhibitors. Cancers 2021, 13, 1928. [Google Scholar] [CrossRef]
- Dey, A.; Varelas, X.; Guan, K.L. Targeting the Hippo Pathway in Cancer, Fibrosis, Wound Healing and Regenerative Medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef]
- Finn, R.S.; Crown, J.P.; Ettl, J.; Schmidt, M.; Bondarenko, I.M.; Lang, I.; Pinter, T.; Boer, K.; Patel, R.; Randolph, S.; et al. Efficacy and Safety of Palbociclib in Combination with Letrozole as First-Line Treatment of ER-Positive, HER2-Negative, Advanced Breast Cancer: Expanded Analyses of Subgroups from the Randomized Pivotal Trial PALOMA-1/TRIO-18. Breast Cancer Res. 2016, 18, 67. [Google Scholar] [CrossRef]
- Li, Z.; Razavi, P.; Li, Q.; Toy, W.; Liu, B.; Ping, C.; Hsieh, W.; Sanchez-Vega, F.; Brown, D.N.; Da Cruz Paula, A.F.; et al. Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell 2018, 34, 893–905.e8. [Google Scholar] [CrossRef]
- Bae, S.N.; Arand, G.; Azzam, H.; Pavasant, P.; Torri, J.; Frandsen, T.L.; Thompson, E.W. Molecular and Cellular Analysis of Basement Membrane Invasion by Human Breast Cancer Cells in Matrigel-Based in Vitro Assays. Breast Cancer Res. Treat. 1993, 24, 241–255. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular Mechanisms of Epithelial-Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Zelivianski, S.; Cooley, A.; Kall, R.; Jeruss, J.S. Cyclin-Dependent Kinase 4-Mediated Phosphorylation Inhibits Smad3 Activity in Cyclin D-Overexpressing Breast Cancer Cells. Mol. Cancer Res. 2010, 8, 1375–1387. [Google Scholar] [CrossRef]
- Yang, J.; Song, K.; Krebs, T.L.; Jackson, M.W.; Danielpour, D. Rb/E2F4 and Smad2/3 Link Survivin to TGF-Beta-Induced Apoptosis and Tumor Progression. Oncogene 2008, 27, 5326–5338. [Google Scholar] [CrossRef]
- Du, B.; Shim, J.S. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef] [PubMed]
- Geller, C.; Maddela, J.; Tuplano, R.; Runa, F.; Adamian, Y.; Güth, R.; Soto, G.O.; Tomaneng, L.; Cantor, J.; Kelber, J.A. Fibronectin, DHPS and SLC3A2 Signaling Cooperate to Control Tumor Spheroid Growth, Subcellular EIF5A1/2 Distribution and CDK4/6 Inhibitor Resistance. bioRxiv 2023. [Google Scholar] [CrossRef]
- Johnston, S.; Puhalla, S.; Wheatley, D.; Ring, A.; Barry, P.; Holcombe, C.; Boileau, J.F.; Provencher, L.; Robidoux, A.; Rimawi, M.; et al. Randomized Phase II Study Evaluating Palbociclib in Addition to Letrozole as Neoadjuvant Therapy in Estrogen Receptor-Positive Early Breast Cancer: PALLET Trial. J. Clin. Oncol. 2019, 37, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Whittle, J.R.; Vaillant, F.; Surgenor, E.; Policheni, A.N.; Giner, G.; Capaldo, B.D.; Chen, H.R.; Liu, H.K.; Dekkers, J.F.; Sachs, N.; et al. Dual Targeting of CDK4/6 and BCL2 Pathways Augments Tumor Response in Estrogen Receptor-Positive Breast Cancer. Clin. Cancer Res. 2020, 26, 4120–4134. [Google Scholar] [CrossRef]
- Zeng, X.; Liu, C.; Yao, J.; Wan, H.; Wan, G.; Li, Y.; Chen, N. Breast Cancer Stem Cells, Heterogeneity, Targeting Therapies and Therapeutic Implications. Pharmacol. Res. 2021, 163, 105320. [Google Scholar] [CrossRef]
- Wang, S.; Bei, Y.; Tian, Q.; He, J.; Wang, R.; Wang, Q.; Sun, L.; Ke, J.; Xie, C.; Shen, P. PFKFB4 Facilitates Palbociclib Resistance in Oestrogen Receptor-Positive Breast Cancer by Enhancing Stemness. Cell Prolif. 2023, 56, e13337. [Google Scholar] [CrossRef]
- Cetin, B.; Wabl, C.A.; Gumusay, O. CDK4/6 Inhibitors: Mechanisms of Resistance and Potential Biomarkers of Responsiveness in Breast Cancer. Future Oncol. 2022, 18, 1143–1157. [Google Scholar] [CrossRef]
- Guan, X.; LaPak, K.M.; Hennessey, R.C.; Yu, C.Y.; Shakya, R.; Zhang, J.; Burd, C.E. Stromal Senescence By Prolonged CDK4/6 Inhibition Potentiates Tumor Growth. Mol. Cancer Res. 2017, 15, 237–249. [Google Scholar] [CrossRef]
- Xu, B.; Krie, A.; De, P.; Williams, C.; Elsey, R.; Klein, J.; Leyland-Jones, B. Utilizing Tumor and Plasma Liquid Biopsy in Treatment Decision Making for an Estrogen Receptor-Positive Advanced Breast Cancer Patient. Cureus 2017, 9, e1408. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, R.; Spring, L.; O’Shaughnessy, J.; Lacroix, L.; Bailleux, C.; Scott, V.; Dubois, J.; Nagy, R.J.; Lanman, R.B.; Iafrate, A.J.; et al. Polyclonal RB1 Mutations and Acquired Resistance to CDK 4/6 Inhibitors in Patients with Metastatic Breast Cancer. Ann. Oncol. 2018, 29, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, Z.; Bhatt, T.; Dickler, M.; Giri, D.; Scaltriti, M.; Baselga, J.; Rosen, N.; Chandarlapaty, S. Acquired CDK6 Amplification Promotes Breast Cancer Resistance to CDK4/6 Inhibitors and Loss of ER Signaling and Dependence. Oncogene 2017, 36, 2255–2264. [Google Scholar] [CrossRef] [PubMed]
- Gacche, R.N.; Assaraf, Y.G. Redundant Angiogenic Signaling and Tumor Drug Resistance. Drug Resist. Updat. 2018, 36, 47–76. [Google Scholar] [CrossRef]
- Caldon, C.E.; Sergio, C.M.; Schütte, J.; Boersma, M.N.; Sutherland, R.L.; Carroll, J.S.; Musgrove, E.A. Estrogen Regulation of Cyclin E2 Requires Cyclin D1 but Not C-Myc. Mol. Cell. Biol. 2009, 29, 4623–4639. [Google Scholar] [CrossRef]
- Papadimitriou, M.C.; Pazaiti, A.; Iliakopoulos, K.; Markouli, M.; Michalaki, V.; Papadimitriou, C.A. Resistance to CDK4/6 Inhibition: Mechanisms and Strategies to Overcome a Therapeutic Problem in the Treatment of Hormone Receptor-Positive Metastatic Breast Cancer. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119346. [Google Scholar] [CrossRef]
- Tripathy, D.; Im, S.A.; Colleoni, M.; Franke, F.; Bardia, A.; Harbeck, N.; Hurvitz, S.A.; Chow, L.; Sohn, J.; Lee, K.S.; et al. Ribociclib plus Endocrine Therapy for Premenopausal Women with Hormone-Receptor-Positive, Advanced Breast Cancer (MONALEESA-7): A Randomised Phase 3 Trial. Lancet Oncol. 2018, 19, 904–915. [Google Scholar] [CrossRef]
- Sledge, G.W.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination with Fulvestrant in Women with HR+/HER2− Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus Palbociclib versus Fulvestrant plus Placebo for Treatment of Hormone-Receptor-Positive, HER2-Negative Metastatic Breast Cancer That Progressed on Previous Endocrine Therapy (PALOMA-3): Final Analysis of the Multicentre, Double-Blind, Phase 3 Randomised Controlled Trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar] [CrossRef]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Val Bianchi, G.; Esteva, F.J.; Martín, M.; et al. Phase III Randomized Study of Ribociclib and Fulvestrant in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: MONALEESA-3. J. Clin. Oncol. 2018, 36, 2465–2472. [Google Scholar] [CrossRef]
- Princic, N.; Aizer, A.; Tang, D.H.; Smith, D.M.; Johnson, W.; Bardia, A. Predictors of Systemic Therapy Sequences Following a CDK 4/6 Inhibitor-Based Regimen in Post-Menopausal Women with Hormone Receptor Positive, HEGFR-2 Negative Metastatic Breast Cancer. Curr. Med. Res. Opin. 2019, 35, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.M.; Handorf, E.A.; Montero, A.J.; Goldstein, L.J. Systemic Therapies Following Progression on First-Line CDK4/6-Inhibitor Treatment: Analysis of Real-World Data. Oncologist 2022, 27, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, W.; Gong, C.; Zheng, Y.; Ouyang, Q.; Xie, N.; Qu, Q.; Ge, R.; Wang, B. A Multicenter Analysis of Treatment Patterns and Clinical Outcomes of Subsequent Therapies after Progression on Palbociclib in HR+/HER2− Metastatic Breast Cancer. Ther. Adv. Med Oncol. 2021, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bashour, S.I.; Doostan, I.; Keyomarsi, K.; Valero, V.; Ueno, N.T.; Brown, P.H.; Litton, J.K.; Koenig, K.B.; Karuturi, M.; Abouharb, S.; et al. Rapid Breast Cancer Disease Progression Following Cyclin Dependent Kinase 4 and 6 Inhibitor Discontinuation. J. Cancer 2017, 8, 2004–2009. [Google Scholar] [CrossRef] [PubMed]
- West, M.; Kaempf, A.; Goodyear, S.; Kartika, T.; Ribkoff, J.; Mitri, Z.I. Real-World Analysis of Disease Progression after CDK 4/6 Inhibitor (CDKi) Therapy in Patients with Hormone Receptor Positive (HR+)/HER2− Metastatic Breast Cancer (MBC). J. Clin. Oncol. 2021, 39, e13030. [Google Scholar] [CrossRef]
- Evaluation of Anticancer Medicinal Products in Man—Scientific Guideline | European Medicines Agency. Available online: https://www.ema.europa.eu/en/evaluation-anticancer-medicinal-products-man-scientific-guideline (accessed on 11 July 2023).
- Munzone, E.; Pagan, E.; Bagnardi, V.; Montagna, E.; Cancello, G.; Dellapasqua, S.; Iorfida, M.; Mazza, M.; Colleoni, M. Systematic Review and Meta-Analysis of Post-Progression Outcomes in ER+/HER2− Metastatic Breast Cancer after CDK4/6 Inhibitors within Randomized Clinical Trials. ESMO Open 2021, 6, 100332. [Google Scholar] [CrossRef]
- Mariotti, V.; Khong, H.T.; Soliman, H.H.; Costa, R.L.; Fisher, S.; Boulware, D.; Han, H.S. Efficacy of Abemaciclib (Abema) after Palbociclib (Palbo) in Patients (Pts) with Metastatic Breast Cancer (MBC). J. Clin. Oncol. 2019, 37, e12521. [Google Scholar] [CrossRef]
- Eziokwu, A.S.; Varella, L.; Kruse, M.L.; Jia, X.; Moore, H.C.F.; Budd, G.T.; Abraham, J.; Montero, A.J. Real-World Evidence Evaluating Continuation of CDK4/6 Inhibitors beyond First Progression in Hormone Receptor-Positive (HR+) Metastatic Breast Cancer. J. Clin. Oncol. 2019, 37, e12538. [Google Scholar] [CrossRef]
- Tamragouri, K.; Cobleigh, M.A.; Rao, R.D. Abemaciclib with or without Fulvestrant for the Treatment of Hormone Receptor-Positive and HER2-Negative Metastatic Breast Cancer with Disease Progression Following Prior Treatment with Palbociclib. J. Clin. Oncol. 2019, 37, e12533. [Google Scholar] [CrossRef]
- dos Anjos, C.H.; Razavi, P.; Herbert, J.; Colon, J.; Gill, K.; Modi, S.; Bromberg, J.; Dang, C.T.; Liu, D.; Norton, L.; et al. A Large Retrospective Analysis of CDK 4/6 Inhibitor Retreatment in ER+ Metastatic Breast Cancer (MBC). J. Clin. Oncol. 2019, 37, 1053. [Google Scholar] [CrossRef]
- Wander, S.A.; Zangardi, M.; Niemierko, A.; Kambadakone, A.; Kim, L.S.; Xi, J.; Pandey, A.K.; Spring, L.; Stein, C.; Juric, D.; et al. A Multicenter Analysis of Abemaciclib after Progression on Palbociclib in Patients (Pts) with Hormone Receptor-Positive (HR+)/HER2− Metastatic Breast Cancer (MBC). J. Clin. Oncol. 2019, 37, 1057. [Google Scholar] [CrossRef]
- Kalinsky, K.; Accordino, M.K.; Chiuzan, C.; Mundi, P.S.; Sakach, E.; Sathe, C.; Ahn, H.; Trivedi, M.S.; Novik, Y.; Tiersten, A.; et al. Randomized Phase II Trial of Endocrine Therapy with or without Ribociclib After Progression on Cyclin-Dependent Kinase 4/6 Inhibition in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer: MAINTAIN Trial. J. Clin. Oncol. 2023, 41, JCO2202392. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.L.; Wander, S.A.; Regan, M.M.; DeMichele, A.; Forero-Torres, A.; Rimawi, M.F.; Ma, C.X.; Cristofanilli, M.; Anders, C.K.; Bartlett, C.H.; et al. Palbociclib after CDK and Endocrine Therapy (PACE): A Randomized Phase II Study of Fulvestrant, Palbociclib, and Avelumab for Endocrine Pre-Treated ER+/HER2− Metastatic Breast Cancer. J. Clin. Oncol. 2018, 36, TPS1104. [Google Scholar] [CrossRef]
- Llombart-Cussac, A.; Harper-Wynne, C.; Perello, A.; Hennequin, A.; Fernandez, A.; Colleoni, M.; Carañana, V.; Quiroga, V.; Medioni, J.; Iranzo, V.; et al. Second-Line Endocrine Therapy (ET) with or without Palbociclib (P) Maintenance in Patients (Pts) with Hormone Receptor-Positive (HR[+])/Human Epidermal Growth Factor Receptor 2-Negative (HER2[-]) Advanced Breast Cancer (ABC): PALMIRA Trial. J. Clin. Oncol. 2023, 41, 1001. [Google Scholar] [CrossRef]
- Albanell, J.; Pérez-García, J.M.; Gil-Gil, M.; Curigliano, G.; Ruíz-Borrego, M.; Comerma, L.; Gibert, J.; Bellet, M.; Bermejo, B.; Calvo, L.; et al. Palbociclib Rechallenge for Hormone Receptor-Positive/HER-Negative Advanced Breast Cancer: Findings from the Phase II BioPER Trial. Clin. Cancer Res. 2023, 29, 67–80. [Google Scholar] [CrossRef]
- Karacin, C.; Oksuzoglu, B.; Demirci, A.; Keskinkılıç, M.; Baytemür, N.K.; Yılmaz, F.; Selvi, O.; Erdem, D.; Avşar, E.; Paksoy, N.; et al. Efficacy of Subsequent Treatments in Patients with Hormone-Positive Advanced Breast Cancer Who Had Disease Progression under CDK 4/6 Inhibitor Therapy. BMC Cancer 2023, 23, 136. [Google Scholar] [CrossRef]
- Robertson, J.F.R.; Bondarenko, I.M.; Trishkina, E.; Dvorkin, M.; Panasci, L.; Manikhas, A.; Shparyk, Y.; Cardona-Huerta, S.; Cheung, K.L.; Philco-Salas, M.J.; et al. Fulvestrant 500 Mg versus Anastrozole 1 Mg for Hormone Receptor-Positive Advanced Breast Cancer (FALCON): An International, Randomised, Double-Blind, Phase 3 Trial. Lancet 2016, 388, 2997–3005. [Google Scholar] [CrossRef]
- Chia, S.; Gradishar, W.; Mauriac, L.; Bines, J.; Amant, F.; Federico, M.; Fein, L.; Romieu, G.; Buzdar, A.; Robertson, J.F.R.; et al. Double-Blind, Randomized Placebo Controlled Trial of Fulvestrant Compared with Exemestane after Prior Nonsteroidal Aromatase Inhibitor Therapy in Postmenopausal Women with Hormone Receptor-Positive, Advanced Breast Cancer: Results from EFECT. J. Clin. Oncol. 2008, 26, 1664–1670. [Google Scholar] [CrossRef]
- Bidard, F.C.; Kaklamani, V.G.; Neven, P.; Streich, G.; Montero, A.J.; Forget, F.; Mouret-Reynier, M.A.; Sohn, J.H.; Taylor, D.; Harnden, K.K.; et al. Elacestrant (Oral Selective Estrogen Receptor Degrader) Versus Standard Endocrine Therapy for Estrogen Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: Results from the Randomized Phase III EMERALD Trial. J. Clin. Oncol. 2022, 40, 3246–3256. [Google Scholar] [CrossRef]
- Bardia, A.; Bidard, F.-C.; Neven, P.; Streich, G.; Montero, A.J.; Forget, F.; Mouret-Reynier, M.-A.; Sohn, J.H.; Taylor, D.; Harnden, K.K.; et al. Abstract GS3-01: GS3-01 EMERALD Phase 3 Trial of Elacestrant versus Standard of Care Endocrine Therapy in Patients with ER+/HER2− Metastatic Breast Cancer: Updated Results by Duration of Prior CDK4/6is in Metastatic Setting. Cancer Res. 2023, 83, GS3-01. [Google Scholar] [CrossRef]
- Oliveira, M.; Hamilton, E.P.; Incorvati, J.; de la Heras, B.B.; Calvo, E.; García-Corbacho, J.; Ruiz-Borrego, M.; Vaklavas, C.; Turner, N.C.; Ciruelos, E.M.; et al. Serena-1: Updated Analyses from a Phase 1 Study (Parts C/D) of the next-Generation Oral SERD Camizestrant (AZD9833) in Combination with Palbociclib, in Women with ER-Positive, HER2-Negative Advanced Breast Cancer. J. Clin. Oncol. 2022, 40, 1032. [Google Scholar] [CrossRef]
- Mosele, F.; Stefanovska, B.; Lusque, A.; Tran Dien, A.; Garberis, I.; Droin, N.; Le Tourneau, C.; Sablin, M.P.; Lacroix, L.; Enrico, D.; et al. Outcome and Molecular Landscape of Patients with PIK3CA-Mutated Metastatic Breast Cancer. Ann. Oncol. 2020, 31, 377–386. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Ciruelos, E.M.; Juric, D.; Loibl, S.; Campone, M.; Mayer, I.A.; Rubovszky, G.; Yamashita, T.; Kaufman, B.; Lu, Y.S.; et al. Alpelisib plus Fulvestrant for PIK3CA-Mutated, Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor-2-Negative Advanced Breast Cancer: Final Overall Survival Results from SOLAR-1. Ann. Oncol. 2021, 32, 208–217. [Google Scholar] [CrossRef] [PubMed]
- CHMP Piqray—Annex I Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/piqray-epar-product-information_en.pdf (accessed on 11 July 2023).
- Rugo, H.S.; Lerebours, F.; Ciruelos, E.; Drullinsky, P.; Borrego, M.R.; Neven, P.; Park, Y.H.; Prat, A.; Bachelot, T.; Juric, D.; et al. Alpelisib (ALP) + Fulvestrant (FUL) in Patients (Pts) with PIK3CA-Mutated (Mut) Hormone Receptor-Positive (HR+), Human Epidermal Growth Factor Receptor 2-Negative (HER2–) Advanced Breast Cancer (ABC) Previously Treated with Cyclin-Dependent Kinase 4/6 Inhibitor (CDKi) + Aromatase Inhibitor (AI): BYLieve Study Results. J. Clin. Oncol. 2020, 38, 1006. [Google Scholar] [CrossRef]
- Juric, D.; Rugo, H.S.; Chia, S.K.L.; Lerebours, F.; Ruiz-Borrego, M.; Drullinsky, P.; Ciruelos, E.M.; Neven, P.; Park, Y.H.; Arce, C.H.; et al. Alpelisib (ALP) + Endocrine Therapy (ET) in Patients (Pts) with Hormone Receptor-Positive (HR+), Human Epidermal Growth Factor Receptor 2-Negative (HER2–), PIK3CA-Mutated (Mut) Advanced Breast Cancer (ABC): Baseline Biomarker Analysis and Progression-Free Survival (PFS) by Duration of Prior Cyclin-Dependent Kinase 4/6 Inhibitor (CDK4/6i) Therapy in the BYLieve Study. J. Clin. Oncol. 2022, 40, 1018. [Google Scholar] [CrossRef]
- A Study of LOXO-783 in Patients with Breast Cancer/Other Solid Tumors. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT05307705 (accessed on 27 July 2023).
- First-in-Human Study of Mutant-Selective PI3Kα Inhibitor, RLY-2608, as a Single Agent in Advanced Solid Tumor Patients and in Combination with Fulvestrant in Patients with Advanced Breast Cancer. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT05216432 (accessed on 11 July 2023).
- Piccart, M.; Hortobagyi, G.N.; Campone, M.; Pritchard, K.I.; Lebrun, F.; Ito, Y.; Noguchi, S.; Perez, A.; Rugo, H.S.; Deleu, I.; et al. Everolimus plus Exemestane for Hormone-Receptor-Positive, Human Epidermal Growth Factor Receptor-2-Negative Advanced Breast Cancer: Overall Survival Results from BOLERO-2. Ann. Oncol. 2014, 25, 2357–2362. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Hurvitz, S.A.; DeMichele, A.; Clark, A.S.; Zelnak, A.B.; Yardley, D.A.; Karuturi, M.S.; Sanft, T.B.; Blau, S.; Hart, L.L.; et al. Triplet Therapy (Continuous Ribociclib, Everolimus, Exemestane) in HR+/HER2− Advanced Breast Cancer Postprogression of a CDK4/6 Inhibitor (TRINITI-1): Efficacy, Safety, and Biomarker Results. J. Clin. Oncol. 2019, 37, 1016. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S.A.; DeMichele, A.; Clark, A.S.; Zelnak, A.; Yardley, D.A.; Karuturi, M.; Sanft, T.; Blau, S.; Hart, L.; et al. Phase I/II Trial of Exemestane, Ribociclib, and Everolimus in Women with HR+/HER2− Advanced Breast Cancer after Progression on CDK4/6 Inhibitors (TRINITI-1). Clin. Cancer Res. 2021, 27, 4177–4185. [Google Scholar] [CrossRef]
- A Study Evaluating the Efficacy and Safety of Giredestrant Plus Everolimus Compared with the Physician’s Choice of Endocrine Therapy Plus Everolimus in Participants with Estrogen Receptor-Positive, HER2-Negative, Locally Advanced or Metastatic Breast Cancer (EvERA Breast Cancer). Available online: https://classic.clinicaltrials.gov/ct2/show/NCT05306340 (accessed on 11 July 2023).
- Korotchkina, L.G.; Leontieva, O.V.; Bukreeva, E.I.; Demidenko, Z.N.; Gudkov, A.V.; Blagosklonny, M.V. The Choice between P53-Induced Senescence and Quiescence Is Determined in Part by the MTOR Pathway. Aging 2010, 2, 344–352. [Google Scholar] [CrossRef]
- Palbociclib with Everolimus + Exemestane in BC. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT02871791 (accessed on 11 July 2023).
- CDK 4/6 Inhibitor, Ribociclib, with Adjuvant Endocrine Therapy for ER-Positive Breast Cancer. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03285412 (accessed on 11 July 2023).
- This Study in Patients with Different Types of Cancer (Solid Tumours) Aims to Find a Safe Dose of Xentuzumab in Combination with Abemaciclib with or without Hormonal Therapies. The Study Also Tests How Effective These Medicines Are in Patients with Lung and Breast Cancer. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03099174 (accessed on 11 July 2023).
- A Study to Assess the Tolerability and Clinical Activity of Gedatolisib in Combination with Palbociclib/Letrozole or Palbociclib/Fulvestrant in Women with Metastatic Breast Cancer. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT02684032 (accessed on 11 July 2023).
- Fulvestrant, Palbociclib and Erdafitinib in ER+/HER2−/FGFR-Amplified Metastatic Breast Cancer. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03238196 (accessed on 11 July 2023).
- Wander, S.A.; Juric, D.; Supko, J.G.; Micalizzi, D.S.; Spring, L.; Vidula, N.; Beeler, M.; Habin, K.R.; Viscosi, E.; Fitzgerald, D.M.; et al. Phase Ib Trial to Evaluate Safety and Anti-Tumor Activity of the AKT Inhibitor, Ipatasertib, in Combination with Endocrine Therapy and a CDK4/6 Inhibitor for Patients with Hormone Receptor Positive (HR+)/HER2 Negative Metastatic Breast Cancer (MBC) (TAKTIC). J. Clin. Oncol. 2020, 38, 1066. [Google Scholar] [CrossRef]
- Turner, N.C.; Oliveira, M.; Howell, S.J.; Dalenc, F.; Cortes, J.; Gomez Moreno, H.L.; Hu, X.; Jhaveri, K.; Krivorotko, P.; Loibl, S.; et al. Capivasertib in Hormone Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2023, 388, 2058–2070. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Li, W.; Hu, X.; Zhang, Q.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.; et al. Tucidinostat plus Exemestane for Postmenopausal Patients with Advanced, Hormone Receptor-Positive Breast Cancer (ACE): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2019, 20, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Connolly, R.M.; Zhao, F.; Miller, K.D.; Lee, M.J.; Piekarz, R.L.; Smith, K.L.; Brown-Glaberman, U.A.; Winn, J.S.; Faller, B.A.; Onitilo, A.A.; et al. E2112: Randomized Phase III Trial of Endocrine Therapy Plus Entinostat or Placebo in Hormone Receptor-Positive Advanced Breast Cancer. A Trial of the ECOG-ACRIN Cancer Research Group. J. Clin. Oncol. 2021, 39, 3171–3181. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.A.; Dowsett, M. BCL-2: A New Therapeutic Target in Estrogen Receptor-Positive Breast Cancer? Cancer Cell 2013, 24, 7–9. [Google Scholar] [CrossRef]
- Lok, S.W.; Whittle, J.R.; Vaillant, F.; Teh, C.E.; Lo, L.L.; Policheni, A.N.; Bergin, A.R.T.; Desai, J.; Ftouni, S.; Gandolfo, L.C.; et al. A Phase Ib Dose-Escalation and Expansion Study of the BCL2 Inhibitor Venetoclax Combined with Tamoxifen in ER and BCL2-Positive Metastatic Breast Cancer. Cancer Discov. 2019, 9, 354–369. [Google Scholar] [CrossRef]
- Lindeman, G.J.; Bowen, R.; Jerzak, K.J.; Song, X.; Decker, T.; Boyle, F.M.; McCune, S.L.; Armstrong, A.; Shannon, C.M.; Bertelli, G.; et al. Results from VERONICA: A Randomized, Phase II Study of Second-/Third-Line Venetoclax (VEN) + Fulvestrant (F) versus F Alone in Estrogen Receptor (ER)-Positive, HER2-Negative, Locally Advanced, or Metastatic Breast Cancer (LA/MBC). J. Clin. Oncol. 2021, 39, 1004. [Google Scholar] [CrossRef]
- Muttiah, C.; Whittle, J.R.; Oakman, C.; Lindeman, G.J. PALVEN: Phase Ib Trial of Palbociclib, Letrozole and Venetoclax in Estrogen Receptor- and BCL2-Positive Advanced Breast Cancer. Future Oncol. 2022, 18, 1805–1816. [Google Scholar] [CrossRef]
- Goel, S.; Decristo, M.J.; Watt, A.C.; Brinjones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 Inhibition Triggers Anti-Tumour Immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Litton, J.K.; Hurvitz, S.A.; Mina, L.A.; Rugo, H.S.; Lee, K.H.; Gonçalves, A.; Diab, S.; Woodward, N.; Goodwin, A.; Yerushalmi, R.; et al. Talazoparib versus Chemotherapy in Patients with Germline BRCA1/2-Mutated HER2-Negative Advanced Breast Cancer: Final Overall Survival Results from the EMBRACA Trial. Ann. Oncol. 2020, 31, 1526–1535. [Google Scholar] [CrossRef]
- Sammons, S.; Shastry, M.; Dent, S.; Anders, C.; Hamilton, E. Practical Treatment Strategies and Future Directions After Progression While Receiving CDK4/6 Inhibition and Endocrine Therapy in Advanced HR+/HER2− Breast Cancer. Clin. Breast Cancer 2020, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ogata, R.; Kishino, E.; Saitoh, W.; Koike, Y.; Kurebayashi, J. Resistance to Cyclin-Dependent Kinase (CDK) 4/6 Inhibitors Confers Cross-Resistance to Other CDK Inhibitors but Not to Chemotherapeutic Agents in Breast Cancer Cells. Breast Cancer 2021, 28, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Kolyadina, I.V.; Bolotina, L.; Zhukova, L.; Vladimirova, L.Y.; Sultanbaev, A.; Karabina, E.; Ganshina, I.; Ovchinnikova, E.; Kolyadina, I.V.; Antonova, G.; et al. The Effectiveness and Safety of Eribulin Therapy in HR-Positive HER2-Negative Metastatic Breast Cancer Post-CDK4/6 Inhibitor Therapy in Russian Clinical Practice. J. Clin. Oncol. 2021, 39, e13035. [Google Scholar] [CrossRef]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef]
- Modi, S.; Niikura, N.; Yamashita, T.; Jacot, W.; Sohn, J.; Tokunaga, E.; Vidal, M.J.; Park, Y.H.; Lee, K.S.; Chae, Y.; et al. Trastuzumab Deruxtecan (T-DXd) vs. Treatment of Physician’s Choice (TPC) in Patients (Pts) with HER2-Low, Hormone Receptor-Positive (HR+) Unresectable and/or Metastatic Breast Cancer (MBC): Exploratory Biomarker Analysis of DESTINY-Breast04. J. Clin. Oncol. 2023, 41, 1020. [Google Scholar] [CrossRef]
- Lloyd, M.R.; Spring, L.M.; Bardia, A.; Wander, S.A. Mechanisms of Resistance to CDK4/6 Blockade in Advanced Hormone Receptor-Positive, HER2-Negative Breast Cancer and Emerging Therapeutic Opportunities. Clin. Cancer Res. 2022, 28, 821–830. [Google Scholar] [CrossRef]
- Rugo, H.S.; Bardia, A.; Marmé, F.; Cortés, J.; Schmid, P.; Loirat, D.; Tredan, O.; Ciruelos, E.M.; Dalenc, F.; Gomez Pardo, P.; et al. LBA76 Overall Survival (OS) Results from the Phase III TROPiCS-02 Study of Sacituzumab Govitecan (SG) vs Treatment of Physician’s Choice (TPC) in Patients (Pts) with HR+/HER2− Metastatic Breast Cancer (MBC). Ann. Oncol. 2022, 33, S1386. [Google Scholar] [CrossRef]
- Pembrolizumab + Paclitaxel in Hormone Receptor-Positive (HR+)/Human Epidermal Growth Factor Receptor 2-Negative (HER2−) Non-Luminal (by PAM50) Advanced Breast Cancer After Cyclin-Dependent Kinase 4/6 (CDK4/6) Inhibitors Progression. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT04251169 (accessed on 27 July 2023).
- Study of ASTX727 Plus Talazoparib in Patients with Triple Negative or Hormone Resistant/HER2-Negative Metastatic Breast Cancer. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT04134884 (accessed on 27 July 2023).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villa, F.; Crippa, A.; Pelizzoni, D.; Ardizzoia, A.; Scartabellati, G.; Corbetta, C.; Cipriani, E.; Lavitrano, M.; Ardizzoia, A. Progression after First-Line Cyclin-Dependent Kinase 4/6 Inhibitor Treatment: Analysis of Molecular Mechanisms and Clinical Data. Int. J. Mol. Sci. 2023, 24, 14427. https://doi.org/10.3390/ijms241914427
Villa F, Crippa A, Pelizzoni D, Ardizzoia A, Scartabellati G, Corbetta C, Cipriani E, Lavitrano M, Ardizzoia A. Progression after First-Line Cyclin-Dependent Kinase 4/6 Inhibitor Treatment: Analysis of Molecular Mechanisms and Clinical Data. International Journal of Molecular Sciences. 2023; 24(19):14427. https://doi.org/10.3390/ijms241914427
Chicago/Turabian StyleVilla, Federica, Alessandra Crippa, Davide Pelizzoni, Alessandra Ardizzoia, Giulia Scartabellati, Cristina Corbetta, Eleonora Cipriani, Marialuisa Lavitrano, and Antonio Ardizzoia. 2023. "Progression after First-Line Cyclin-Dependent Kinase 4/6 Inhibitor Treatment: Analysis of Molecular Mechanisms and Clinical Data" International Journal of Molecular Sciences 24, no. 19: 14427. https://doi.org/10.3390/ijms241914427
APA StyleVilla, F., Crippa, A., Pelizzoni, D., Ardizzoia, A., Scartabellati, G., Corbetta, C., Cipriani, E., Lavitrano, M., & Ardizzoia, A. (2023). Progression after First-Line Cyclin-Dependent Kinase 4/6 Inhibitor Treatment: Analysis of Molecular Mechanisms and Clinical Data. International Journal of Molecular Sciences, 24(19), 14427. https://doi.org/10.3390/ijms241914427