

Computational Insight into the Nature and Strength of the π-Hole Type Chalcogen∙∙∙Chalcogen Interactions in the XO2∙∙∙CH3YCH3 Complexes (X = S, Se, Te; Y = O, S, Se, Te)

 ,
,  , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Molecular Electrostatic Surface Potential (MESP)
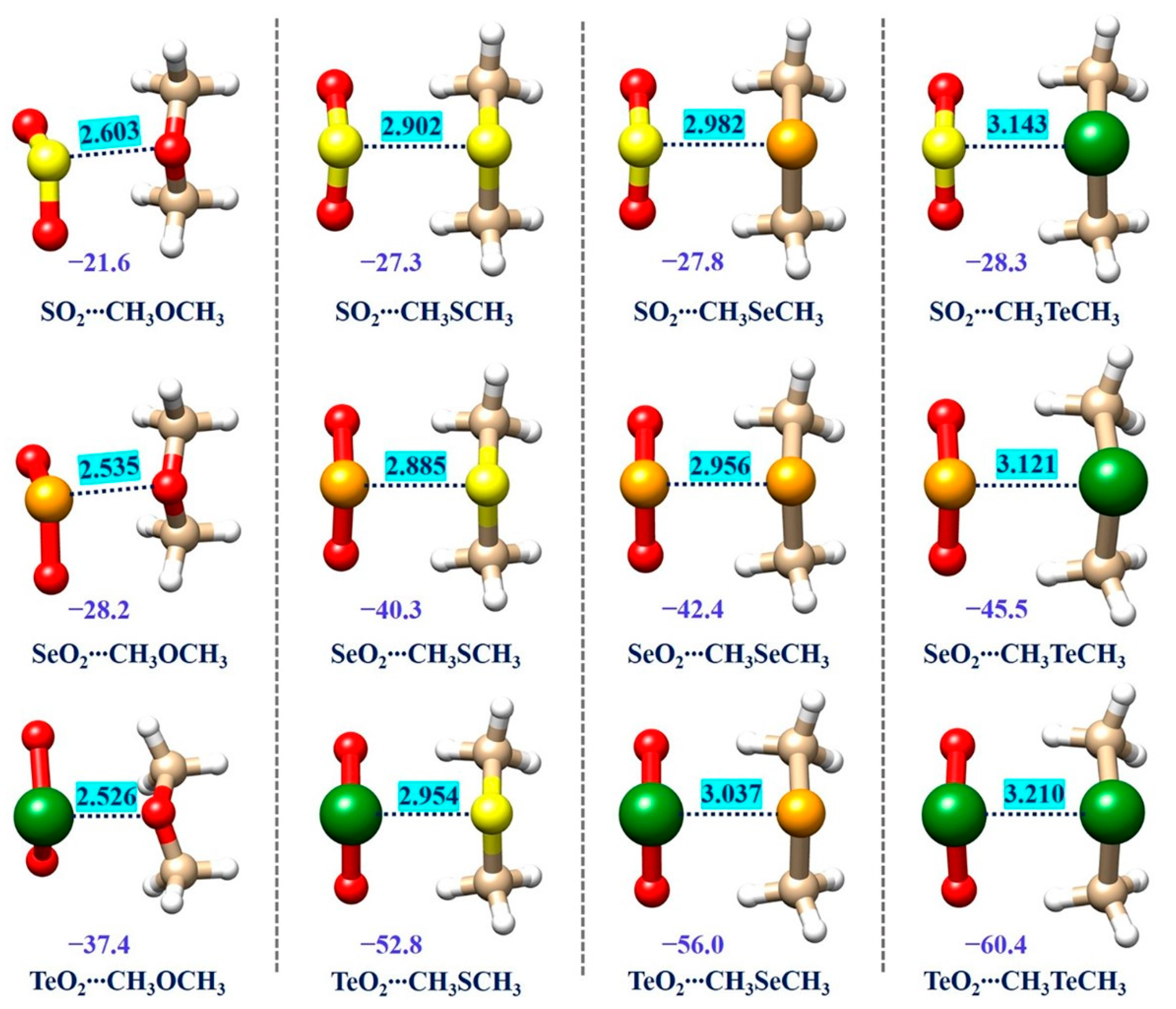
2.2. Geometrical Structures and Binding Energies of the Studied Complexes
2.3. Quantum Theory of Atoms in Molecules (QTAIM) Analysis
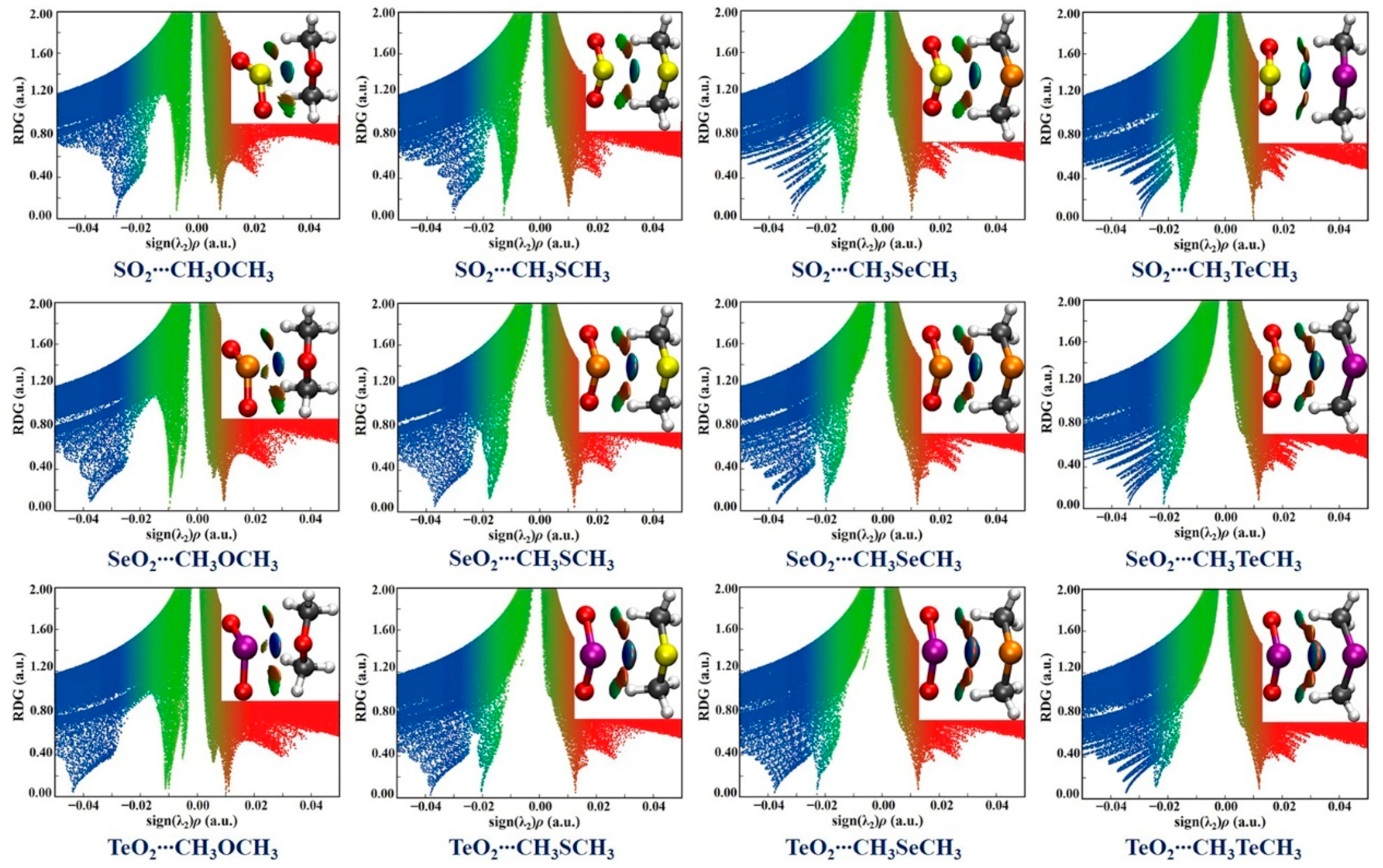
2.4. Non-Covalent Interaction Plot (NCIplot) Analysis
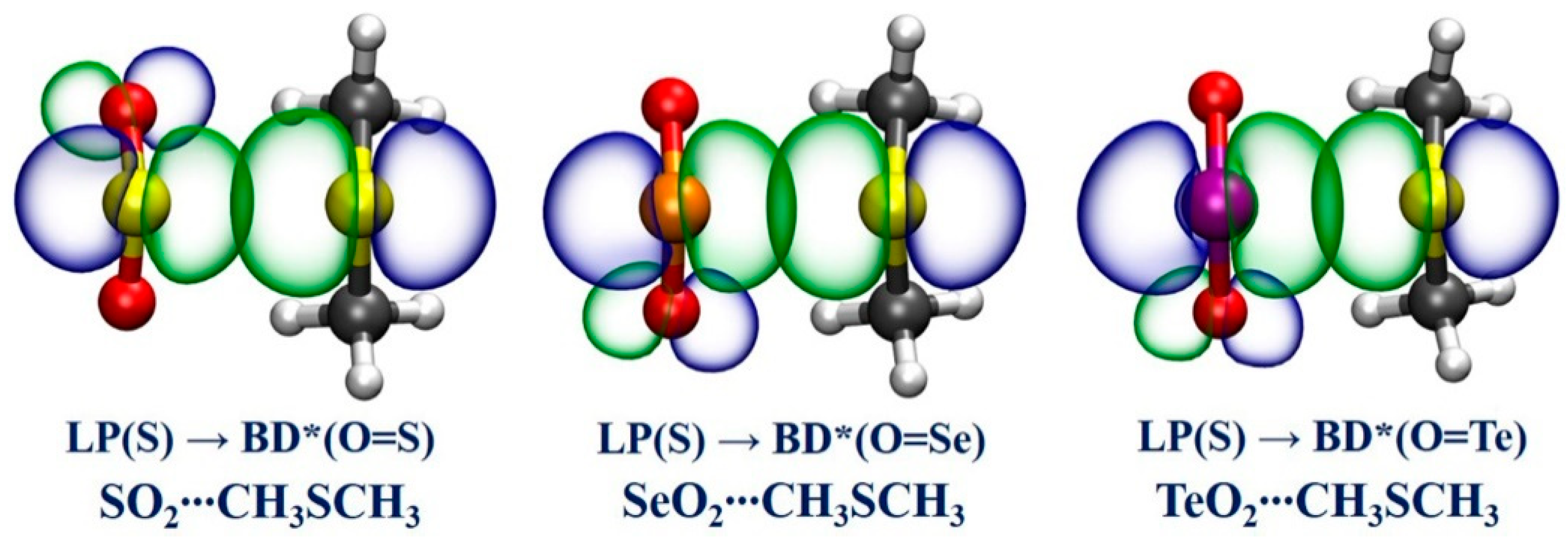
2.5. Natural Bond Orbital (NBO) Analysis
2.6. Symmetry-Adapted Perturbation Theory (SAPT) Analysis
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pascoe, D.J.; Ling, K.B.; Cockroft, S.L. The Origin of Chalcogen-Bonding Interactions. J. Am. Chem. Soc. 2017, 139, 15160–15167. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Bryce, D.I.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Vogel, L.; Wonner, P.; Huber, S.M. Chalcogen Bonding: An Overview. Angew. Chem. Int. Ed. 2018, 58, 1880–1891. [Google Scholar] [CrossRef]
- Scilabra, P.; Terraneo, G.; Resnati, G. The Chalcogen Bond in Crystalline Solids: A World Parallel to Halogen Bond. Acc. Chem. Res. 2019, 52, 1313–1324. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Clark, T.; Riley, K.E.; Politzer, P. σ-Holes, π-holes and electrostatically-driven interactions. J. Mol. Model. 2012, 18, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. The bright future of unconventional σ/π-hole interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole bond vs π-hole bond: A comparison based on halogen bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. An Overview of Strengths and Directionalities of Noncovalent Interactions: σ-Holes and π-Holes. Crystals 2019, 9, 165. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. σ-Hole Interactions: Perspectives and Misconceptions. Crystals 2017, 7, 212. [Google Scholar] [CrossRef]
- Frontera, A. σ- and π-Hole Interactions. Crystals 2020, 10, 721. [Google Scholar] [CrossRef]
- Scheiner, S. Dissection of the Origin of π-Holes and the Noncovalent Bonds in Which They Engage. J. Phys. Chem. A 2021, 125, 6514–6528. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Sato, N. Origin of attraction in chalcogen–nitrogen interaction of 1,2,5-chalcogenadiazole dimers. J. Phys. Chem. B 2013, 117, 6849–6855. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Gilli, G.; Gilli, P. The Nature of the Hydrogen Bond; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Adhikari, U.; Scheiner, S. Effects of charge and substituent on the S···N chalcogen bond. J. Phys. Chem. A 2014, 118, 3183–3192. [Google Scholar] [CrossRef] [PubMed]
- Nziko, V.d.P.N.; Scheiner, S. Chalcogen Bonding between Tetravalent SF4 and Amines. J. Phys. Chem. A 2014, 118, 10849–10856. [Google Scholar] [CrossRef]
- Geboes, Y.; De Vleeschouwer, F.; De Proft, F.; Herrebout, W.A. Exploiting the σ-hole concept: An Infrared and Raman-based characterization of the S···O chalcogen bond between 2,2,4,4-tetrafluoro-1,3-dithiethane and dimethyl ether. Chem. Eur. J. 2017, 23, 17384–17392. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Wysokiński, R.; Michalczyk, M.; Scheiner, S. Chalcogen bonding of two ligands to hypervalent YF4 (Y = S, Se, Te, Po). Phys. Chem. Chem. Phys. 2019, 21, 20829–20839. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Zheng, Y.; Gou, Q.; Hou, G.-L.; Feng, G. Rotational characterization of S···F chalcogen bonds in the complex of 2,2,4,4-tetrafluoro-1,3-dithietane and difluoromethane. Phys. Chem. Chem. Phys. 2019, 21, 24659–24665. [Google Scholar] [CrossRef]
- Santos, L.d.A.; Ramalho, T.C.; Hamlin, T.A.; Bickelhaupt, F.M. Chalcogen bonds: Hierarchical ab initio benchmark and density functional theory performance study. J. Comput. Chem. 2021, 42, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Esrafili, M.D.; Vakili, M.; Solimannejad, M. Cooperative interaction between π-hole and single-electron σ-hole interactions in O2S···NCX···CH3 and O2Se···NCX···CH3 complexes (X = F, Cl, Br and I). Mol. Phys. 2014, 112, 2078–2084. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mohammadian-Sabet, F. Homonuclear chalcogen–chalcogen bond interactions in complexes pairing YO3 and YHX molecules (Y = S, Se; X = H, Cl, Br, CCH, NC, OH, OCH3): Influence of substitution and cooperativity. Int. J. Quantum Chem. 2016, 116, 529–536. [Google Scholar] [CrossRef]
- Obenchain, D.A.; Spada, L.; Alessandrini, S.; Rampino, S.; Herbers, S.; Tasinato, N.; Mendolicchio, M.; Kraus, P.; Gauss, J.; Puzzarini, C.; et al. Unveiling the Sulfur–Sulfur Bridge: Accurate Structural and Energetic Characterization of a Homochalcogen Intermolecular Bond. Angew. Chem. Int. Ed. 2018, 57, 15822–15826. [Google Scholar] [CrossRef]
- Wang, W.; Zhu, H.; Liu, S.; Zhao, Z.; Zhang, L.; Hao, J.; Wang, Y. Chalcogen–Chalcogen Bonding Catalysis Enables Assembly of Discrete Molecules. J. Am. Chem. Soc. 2019, 141, 9175–9179. [Google Scholar] [CrossRef]
- Wang, W.; Zhu, H.; Feng, L.; Yu, Q.; Hao, J.; Zhu, R.; Wang, Y. Dual chalcogen–chalcogen bonding catalysis. J. Am. Chem. Soc. 2020, 142, 3117–3124. [Google Scholar] [CrossRef] [PubMed]
- Pale, P.; Mamane, V. Chalcogen Bonding Catalysis: Tellurium, the Last Frontier? Chem. Eur. J. 2023, 29, e202302755. [Google Scholar] [CrossRef]
- Zhao, Z.; Pang, Y.; Zhao, Z.; Zhou, P.-P.; Wang, Y. Supramolecular catalysis with ethers enabled by dual chalcogen bonding activation. Nat. Commun. 2023, 14, 6347. [Google Scholar] [CrossRef]
- Ho, P.C.; Wang, J.Z.; Meloni, F.; Vargas-Baca, I. Chalcogen bonding in materials chemistry. Coordin. Chem. Rev. 2020, 422, 213464. [Google Scholar] [CrossRef]
- Beno, B.R.; Yeung, K.-S.; Bartberger, M.D.; Pennington, L.D.; Meanwell, N.A. A Survey of the Role of Noncovalent Sulfur Interactions in Drug Design. J. Med. Chem. 2015, 58, 4383–4438. [Google Scholar] [CrossRef]
- Carugo, O.; Resnati, G.; Metrangolo, P. Chalcogen Bonds Involving Selenium in Protein Structures. ACS Chem. Biol. 2021, 16, 1622–1627. [Google Scholar] [CrossRef]
- Piña, M.d.L.N.; Frontera, A.; Bauza, A. Charge Assisted S/Se Chalcogen Bonds in SAM Riboswitches: A Combined PDB and ab Initio Study. ACS Chem. Biol. 2021, 16, 1701–1708. [Google Scholar] [CrossRef] [PubMed]
- Riveras, J.A.F.; Frontera, A.; Bauzá, A. Selenium chalcogen bonds are involved in protein–carbohydrate recognition: A combined PDB and theoretical study. Phys. Chem. Chem. Phys. 2021, 23, 17656–17662. [Google Scholar] [CrossRef]
- Biswal, H.S.; Sahu, A.K.; Galmés, B.; Frontera, A.; Chopra, D. Se···O/S and S···O chalcogen bonds in small molecules and proteins: A combined CSD and PDB study. ChemBioChem 2022, 23, e202100498. [Google Scholar] [CrossRef] [PubMed]
- Gleiter, R.; Haberhauer, G.; Werz, D.B.; Rominger, F.; Bleiholder, C. From Noncovalent Chalcogen–Chalcogen Interactions to Supramolecular Aggregates: Experiments and Calculations. Chem. Rev. 2018, 118, 2010–2041. [Google Scholar] [CrossRef]
- Fourmigué, M.; Dhaka, A. Chalcogen bonding in crystalline diselenides and selenocyanates: From molecules of pharmaceutical interest to conducting materials. Coordin. Chem. Rev. 2020, 403, 213084. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Gurbanov, A.V.; Aliyeva, V.A.; da Silva, M.F.C.G.; Resnati, G.; Pombeiro, A.J. Chalcogen bonding in coordination chemistry. Coordin. Chem. Rev. 2022, 464, 214556. [Google Scholar] [CrossRef]
- Lim, J.Y.; Beer, P.D. Sigma-Hole Interactions in Anion Recognition. Chem 2018, 4, 731–783. [Google Scholar] [CrossRef]
- Biot, N.; Bonifazi, D. Chalcogen-bond driven molecular recognition at work. Coordin. Chem. Rev. 2020, 413, 213243. [Google Scholar] [CrossRef]
- Bleiholder, C.; Werz, D.B.; Köppel, H.; Gleiter, R. Theoretical Investigations on Chalcogen−Chalcogen Interactions: What Makes These Nonbonded Interactions Bonding? J. Am. Chem. Soc. 2006, 128, 2666–2674. [Google Scholar] [CrossRef] [PubMed]
- Bleiholder, C.; Gleiter, R.; Werz, D.B.; Köppel, H. Theoretical Investigations on Heteronuclear Chalcogen—Chalcogen Interactions: On the Nature of Weak Bonds between Chalcogen Centers. Inorg. Chem. 2007, 46, 2249–2260. [Google Scholar] [CrossRef] [PubMed]
- Kříž, K.; Fanfrlík, J.; Lepšík, M. Chalcogen Bonding in Protein−Ligand Complexes: PDB Survey and Quantum Mechanical Calculations. Chemphyschem 2018, 19, 2540–2548. [Google Scholar] [CrossRef] [PubMed]
- Lundemba, A.S.; Bibelayi, D.D.; Tsalu, P.V.; Wood, P.A.; Cole, J.; Kayembe, J.S.; Yav, Z.G. Chalcogen Bonds in Small-Organic Molecule Compounds Derived from the Cambridge Structural Database (CSD). Cryst. Struct. Theory Appl. 2021, 10, 57–69. [Google Scholar] [CrossRef]
- Carugo, O.I. Chalcogen bonds formed by protein sulfur atoms in proteins. A survey of high-resolution structures deposited in the protein data bank. J. Biomol. Struct. Dyn. 2022, 91, 1–7. [Google Scholar] [CrossRef]
- Piña, M.d.L.N.; Bauzá, A. On the Importance of Halogen and Chalcogen Bonds in the Solid State of Nucleic Acids: A Combined Crystallographic and Theoretical Perspective. Int. J. Mol. Sci. 2023, 24, 13035. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Saeidi, N.; Baneshi, M.M. Chalcogen–chalcogen interactions in furan-YHX and thiophene-YHX complexes (X = F, Cl, Br; Y = S, Se): An ab initio study. Bull. Chem. Soc. Jpn. 2015, 88, 1683–1692. [Google Scholar] [CrossRef]
- Yan, N.; Huo, S.; Li, X.; Zeng, Y.; Meng, L. The chalcogen bond in F2P(S)N···SX2, F2PNS···SX2, F2PSN···SX2 (X = F, Cl, Br, OH, CH3, NH2) complexes. J. Mol. Model. 2019, 25, 19. [Google Scholar]
- Weiss, R.; Aubert, E.; Groslambert, L.; Pale, P.; Mamane, V. Evidence for and evaluation of fluorine–tellurium chalcogen bonding. Chem. Sci. 2023, 14, 7221–7229. [Google Scholar] [CrossRef]
- Matsumoto, K.; Gerken, M. Recent advances in sulfur tetrafluoride chemistry: Syntheses, structures, and applications. Dalton Trans. 2021, 50, 12791–12799. [Google Scholar] [CrossRef]
- Liang, J.; Shi, Y.; Lu, Y.; Xu, Z.; Liu, H. Square tetravalent chalcogen bonds in dimeric aggregates: A joint crystallographic survey and theoretical study. CrystEngComm 2022, 24, 975–986. [Google Scholar] [CrossRef]
- Franconetti, A.; Quiñonero, D.; Frontera, A.; Resnati, G. Unexpected chalcogen bonds in tetravalent sulfur compounds. Phys. Chem. Chem. Phys. 2019, 21, 11313–11319. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.A.; Saeed, R.R.A.; Shehata, M.N.I.; Moussa, N.A.M.; Tawfeek, A.M.; Ahmed, M.N.; El-Rahman, M.K.A.; Shoeib, T. Sigma-Hole and Lone-Pair-Hole Site-Based Interactions of Seesaw Tetravalent Chalcogen-Bearing Molecules with Lewis Bases. ACS Omega 2023, 8, 32828–32837. [Google Scholar] [CrossRef] [PubMed]
- Azofra, L.M.; Scheiner, S. Substituent effects in the noncovalent bonding of SO2 to molecules containing a carbonyl group. The dominating role of the chalcogen bond. J. Phys. Chem. A 2014, 118, 3835–3845. [Google Scholar] [CrossRef]
- Jin, Y.; Saragi, R.T.; Juanes, M.; Feng, G.; Lesarri, A. Interaction topologies of the S···O chalcogen bond: The conformational equilibrium of the cyclohexanol···SO2 cluster. Phys. Chem. Chem. Phys. 2021, 23, 10799–10806. [Google Scholar] [CrossRef]
- Tulsiyan, K.D.; Jena, S.; Dutta, J.; Biswal, H.S. Hydrogen bonding with polonium. Phys. Chem. Chem. Phys. 2022, 24, 17185–17194. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Charaya, H.; Chakraborty, S. An experimental exploration of C−H···X hydrogen bond in [CHCl3−X(CH3)2] complexes (X= O, S, and Se). ChemPhysChem 2023, 24, e202300124. [Google Scholar] [CrossRef]
- Beckmann, J.L.; Krieft, J.; Vishnevskiy, Y.V.; Neumann, B.; Stammler, H.-G.; Mitzel, N.W. A bidentate antimony pnictogen bonding host-system. Angew. Chem. Int. Ed. 2023, 62, e202310439. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Concha, M.C. σ-hole bonding between like atoms; a fallacy of atomic charges. J. Mol. Model. 2008, 14, 659–665. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital methods. WIREs Comput. Mol. Sci. 2011, 2, 1–42. [Google Scholar] [CrossRef]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of symmetry adapted perturbation theory (SAPT). I. Efficiency and performance for interaction energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [CrossRef] [PubMed]
- Mantina, M.; Chamberlin, A.C.; Valero, R.; Cramer, C.J.; Truhlar, D.G. Consistent van der Waals Radii for the Whole Main Group. J. Phys. Chem. A 2009, 113, 5806–5812. [Google Scholar] [CrossRef]
- Cordero, B.; Gómez, V.; Platero-Prats, A.E.; Revés, M.; Echeverría, J.; Cremades, E.; Barragán, F.; Alvarez, S. Covalent radii revisited. Dalton Trans. 2008, 21, 2832–2838. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. B 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. Systematically convergent basis sets with relativistic pseudopotentials. II. Small-core pseudopotentials and correlation consistent basis sets for the post-d group 16–18 elements. J. Chem. Phys. 2003, 119, 11113–11123. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.T.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F.D. The Calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Parrish, R.M.; Burns, L.A.; Smith, D.G.A.; Simmonett, A.C.; DePrince, A.E., III; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Di Remigio, R.; Richard, R.M.; et al. Psi4 1.1: An open-source electronic structure program emphasizing automation, advanced libraries, and interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complexes | EB | ChBs | RChB (Å) | Rsum,1 a (Å) | λ b | Rsum,2 c (Å) |
|---|---|---|---|---|---|---|
| SO2∙∙∙CH3OCH3 | −21.6 | S∙∙∙O | 2.603 | 3.32 (21.6%) d | 0.78 | 1.66 [56.8%] e |
| SO2∙∙∙CH3SCH3 | −27.3 | S∙∙∙S | 2.920 | 3.60 (18.9%) | 0.81 | 2.06 [41.7%] |
| SO2∙∙∙CH3SeCH3 | −27.8 | S∙∙∙Se | 2.982 | 3.70 (19.4%) | 0.81 | 2.19 [36.2%] |
| SO2∙∙∙CH3TeCH3 | −28.3 | S∙∙∙Te | 3.143 | 3.86 (18.6%) | 0.81 | 2.39 [31.5%] |
| SeO2∙∙∙CH3OCH3 | −28.2 | Se∙∙∙O | 2.535 | 3.42 (25.9%) | 0.74 | 1.79 [41.6%] |
| SeO2∙∙∙CH3SCH3 | −40.3 | Se∙∙∙S | 2.885 | 3.70 (22.0%) | 0.78 | 2.19 [31.7%] |
| SeO2∙∙∙CH3SeCH3 | −42.4 | Se∙∙∙Se | 2.956 | 3.80 (22.2%) | 0.78 | 2.32 [27.4%] |
| SeO2∙∙∙CH3TeCH3 | −45.5 | Se∙∙∙Te | 3.121 | 3.96 (21.2%) | 0.79 | 2.52 [23.8%] |
| TeO2∙∙∙CH3OCH3 | −37.4 | Te∙∙∙O | 2.526 | 3.58 (29.4%) | 0.71 | 1.99 [26.9%] |
| TeO2∙∙∙CH3SCH3 | −52.8 | Te∙∙∙S | 2.954 | 3.86 (23.5%) | 0.77 | 2.39 [23.6%] |
| TeO2∙∙∙CH3SeCH3 | −56.0 | Te∙∙∙Se | 3.037 | 3.96 (23.3%) | 0.77 | 2.52 [20.5%] |
| TeO2∙∙∙CH3TeCH3 | −60.4 | Te∙∙∙Te | 3.210 | 4.12 (22.1%) | 0.78 | 2.72 [18.0%] |
| Complexes | BCP | ρ(r) | ∇2ρ(r) | H(r) | |G(r)/V(r)| |
|---|---|---|---|---|---|
| SO2∙∙∙CH3OCH3 | S∙∙∙O | 0.0290 | 0.0831 | 0.0002 | 1.0087 |
| SO2∙∙∙CH3SCH3 | S∙∙∙S | 0.0307 | 0.0463 | −0.0031 | 0.8266 |
| SO2∙∙∙CH3SeCH3 | S∙∙∙Se | 0.0315 | 0.0395 | −0.0037 | 0.7864 |
| SO2∙∙∙CH3TeCH3 | S∙∙∙Te | 0.0292 | 0.0303 | −0.0034 | 0.7654 |
| SeO2∙∙∙CH3OCH3 | Se∙∙∙O | 0.0377 | 0.0911 | −0.0023 | 0.9172 |
| SeO2∙∙∙CH3SCH3 | Se∙∙∙S | 0.0370 | 0.0420 | −0.0053 | 0.7479 |
| SeO2∙∙∙CH3SeCH3 | Se∙∙∙Se | 0.0371 | 0.0345 | −0.0057 | 0.7153 |
| SeO2∙∙∙CH3TeCH3 | Se∙∙∙Te | 0.0340 | 0.0254 | −0.0051 | 0.6923 |
| TeO2∙∙∙CH3OCH3 | Te∙∙∙O | 0.0438 | 0.0893 | −0.0063 | 0.8202 |
| TeO2∙∙∙CH3SCH3 | Te∙∙∙S | 0.0383 | 0.0328 | −0.0069 | 0.6871 |
| TeO2∙∙∙CH3SeCH3 | Te∙∙∙Se | 0.0378 | 0.0255 | −0.0069 | 0.6576 |
| TeO2∙∙∙CH3TeCH3 | Te∙∙∙Te | 0.0346 | 0.0184 | −0.0058 | 0.6419 |
| Complexes | Donor | Acceptor | E(2) |
|---|---|---|---|
| SO2∙∙∙CH3OCH3 | LP(O) | BD*(O=S) | 7.7 |
| SO2∙∙∙CH3SCH3 | LP(S) | BD*(O=S) | 55.3 |
| SO2∙∙∙CH3SeCH3 | LP(Se) | BD*(O=S) | 66.2 |
| SO2∙∙∙CH3TeCH3 | LP(Te) | BD*(O=S) | 70.1 |
| SeO2∙∙∙CH3OCH3 | LP(O) | BD*(O=Se) | 44.0 |
| SeO2∙∙∙CH3SCH3 | LP(S) | BD*(O=Se) | 109.3 |
| SeO2∙∙∙CH3SeCH3 | LP(Se) | BD*(O=Se) | 131.5 |
| SeO2∙∙∙CH3TeCH3 | LP(Te) | BD*(O=Se) | NA b |
| TeO2∙∙∙CH3OCH3 | LP(O) | BD*(O=Te) | 74.1 |
| TeO2∙∙∙CH3SCH3 | LP(S) | BD*(O=Te) | 137.3 |
| TeO2∙∙∙CH3SeCH3 | LP(Se) | BD*(O=Te) | NA b |
| TeO2∙∙∙CH3TeCH3 | LP(Te) | BD*(O=Te) | NA b |
| Complexes | Eelec | Eind | Edisp | Eex-re | Etotal |
|---|---|---|---|---|---|
| SO2∙∙∙CH3OCH3 | −58.9(49%) b | −27.5(22%) | −35.0(29%) | 90.1 | −31.3 |
| SO2∙∙∙CH3SCH3 | −79.2(43%) | −55.3(30%) | −48.2(27%) | 139.6 | −43.2 |
| SO2∙∙∙CH3SeCH3 | −88.6(43%) | −65.4(32%) | −52.8(25%) | 160.3 | −46.4 |
| SO2∙∙∙CH3TeCH3 | −87.6(42%) | −65.5(31%) | −55.2(27%) | 160.3 | −48.0 |
| SeO2∙∙∙CH3OCH3 | −95.2(49%) | −53.4(27%) | −47.5(24%) | 147.2 | −48.9 |
| SeO2∙∙∙CH3SCH3 | −120.3(43%) | −95.8(34%) | −65.5(23%) | 206.6 | −75.0 |
| SeO2∙∙∙CH3SeCH3 | −134.4(43%) | −108.5(34%) | −71.0(23%) | 230.9 | −83.0 |
| SeO2∙∙∙CH3TeCH3 | −129.8(41%) | −114.4(36%) | −74.4(23%) | 228.7 | −89.9 |
| TeO2∙∙∙CH3OCH3 | −145.7(51%) | −80.3(28%) | −59.3(21%) | 204.6 | −80.7 |
| TeO2∙∙∙CH3SCH3 | −154.9(46%) | −104.4(31%) | −77.9(23%) | 228.7 | −108.5 |
| TeO2∙∙∙CH3SeCH3 | −166.1(45%) | −117.4(32%) | −83.6(23%) | 248.2 | −119.0 |
| TeO2∙∙∙CH3TeCH3 | −161.8(44%) | −119.5(32%) | −87.8(24%) | 239.2 | −129.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, F.; Liu, Q.; Zhong, Y.; Cui, X.; Yu, J.; Hu, Z.; Feng, G.; Zeng, Z.; Lu, T. Computational Insight into the Nature and Strength of the π-Hole Type Chalcogen∙∙∙Chalcogen Interactions in the XO2∙∙∙CH3YCH3 Complexes (X = S, Se, Te; Y = O, S, Se, Te). Int. J. Mol. Sci. 2023, 24, 16193. https://doi.org/10.3390/ijms242216193
Lei F, Liu Q, Zhong Y, Cui X, Yu J, Hu Z, Feng G, Zeng Z, Lu T. Computational Insight into the Nature and Strength of the π-Hole Type Chalcogen∙∙∙Chalcogen Interactions in the XO2∙∙∙CH3YCH3 Complexes (X = S, Se, Te; Y = O, S, Se, Te). International Journal of Molecular Sciences. 2023; 24(22):16193. https://doi.org/10.3390/ijms242216193
Chicago/Turabian StyleLei, Fengying, Qingyu Liu, Yeshuang Zhong, Xinai Cui, Jie Yu, Zuquan Hu, Gang Feng, Zhu Zeng, and Tao Lu. 2023. "Computational Insight into the Nature and Strength of the π-Hole Type Chalcogen∙∙∙Chalcogen Interactions in the XO2∙∙∙CH3YCH3 Complexes (X = S, Se, Te; Y = O, S, Se, Te)" International Journal of Molecular Sciences 24, no. 22: 16193. https://doi.org/10.3390/ijms242216193
APA StyleLei, F., Liu, Q., Zhong, Y., Cui, X., Yu, J., Hu, Z., Feng, G., Zeng, Z., & Lu, T. (2023). Computational Insight into the Nature and Strength of the π-Hole Type Chalcogen∙∙∙Chalcogen Interactions in the XO2∙∙∙CH3YCH3 Complexes (X = S, Se, Te; Y = O, S, Se, Te). International Journal of Molecular Sciences, 24(22), 16193. https://doi.org/10.3390/ijms242216193