
Roles of Oxidative Injury and Nitric Oxide System Derangements in Kawasaki Disease Pathogenesis: A Systematic Review

 , , , and
, , , and
Abstract
:1. Introduction
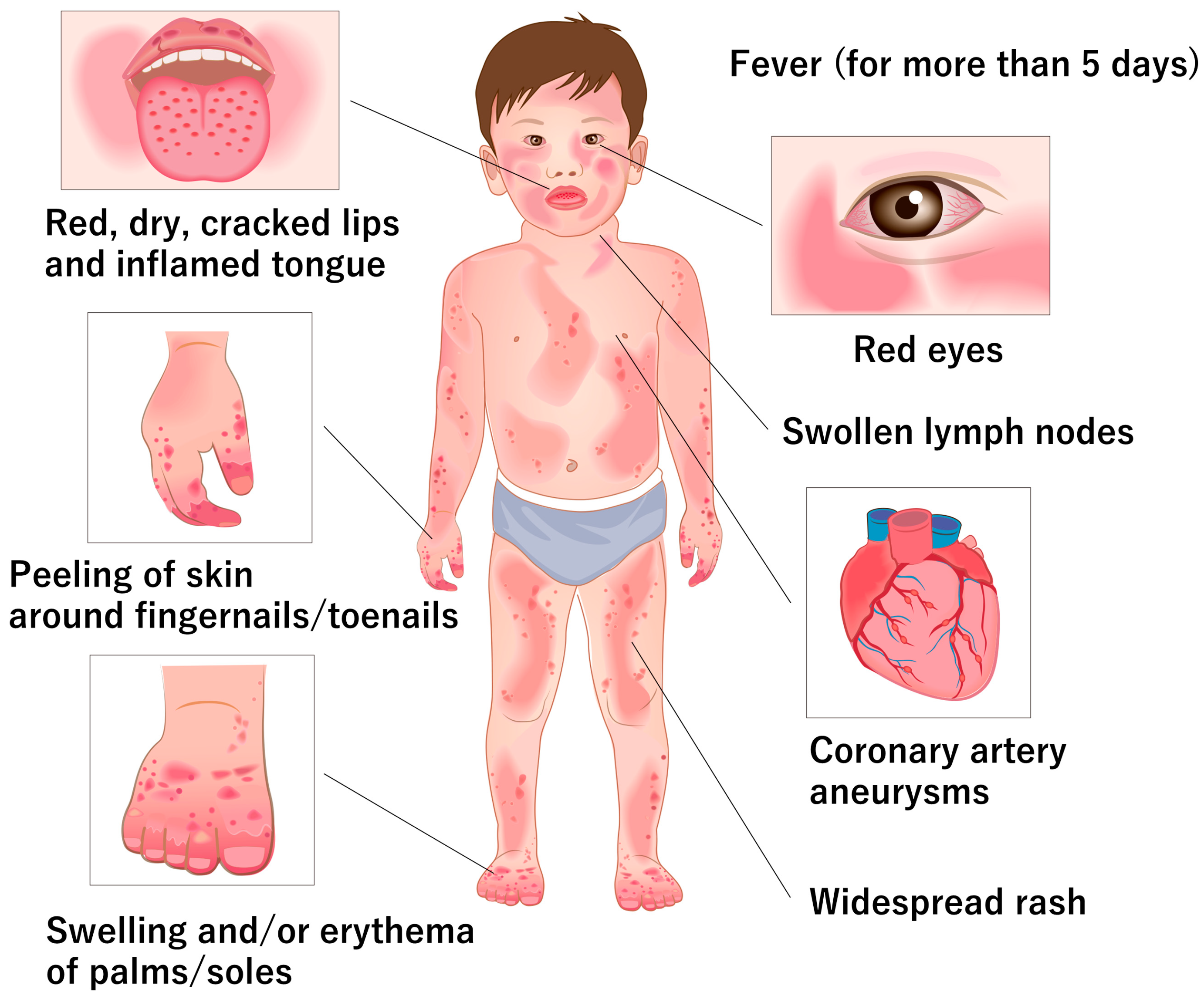
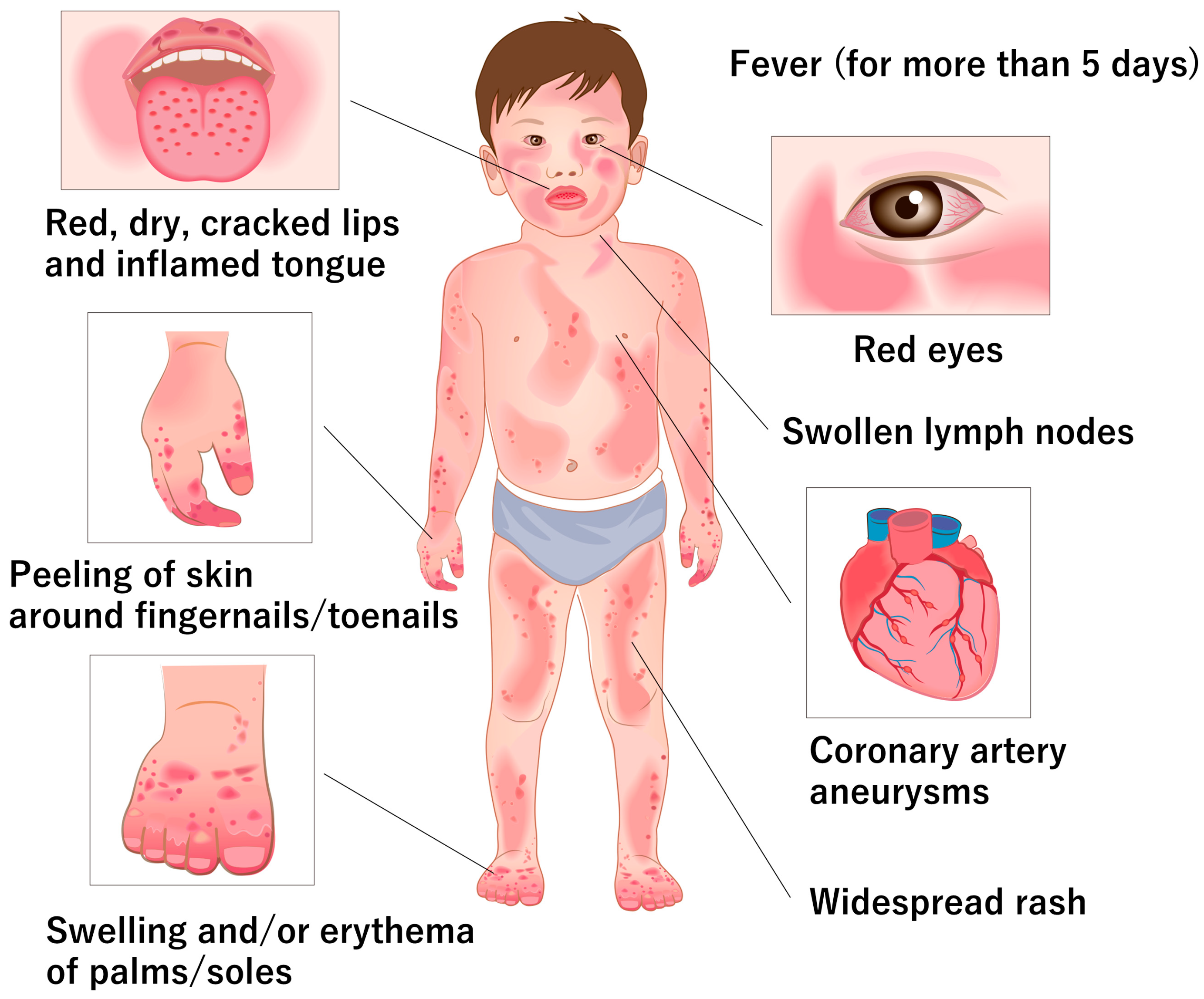
2. Clinical and Pathological Features of KD
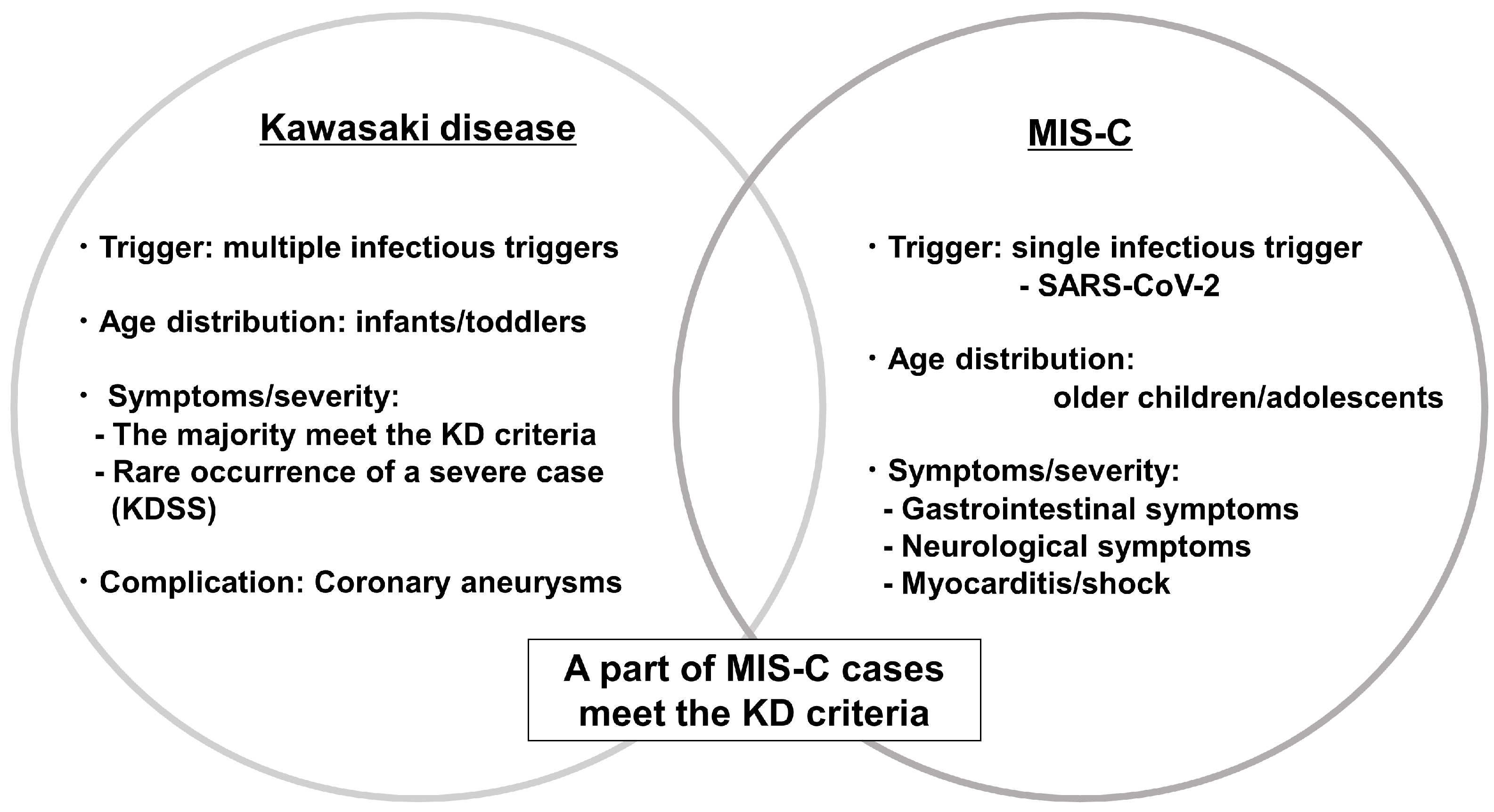
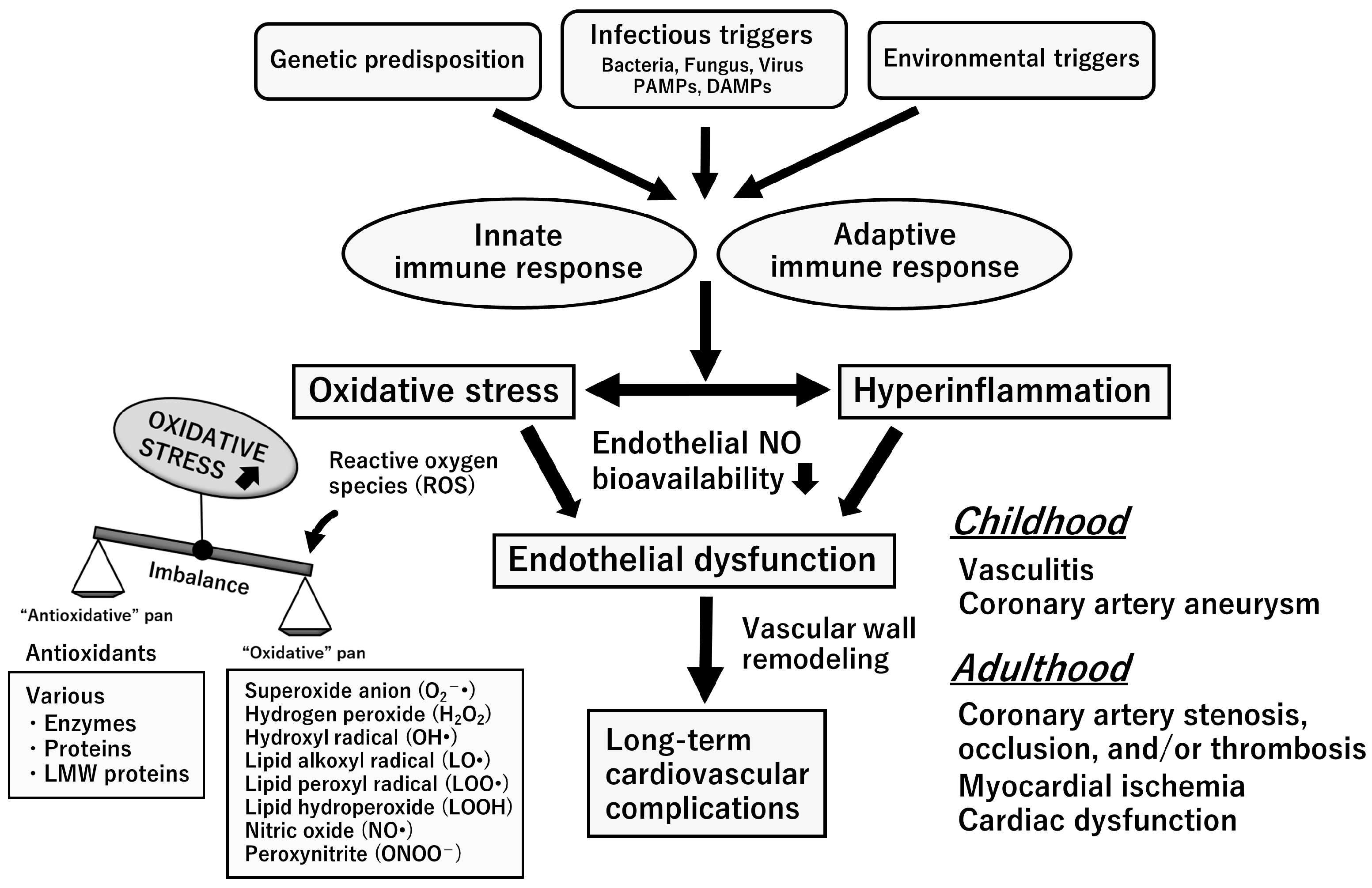
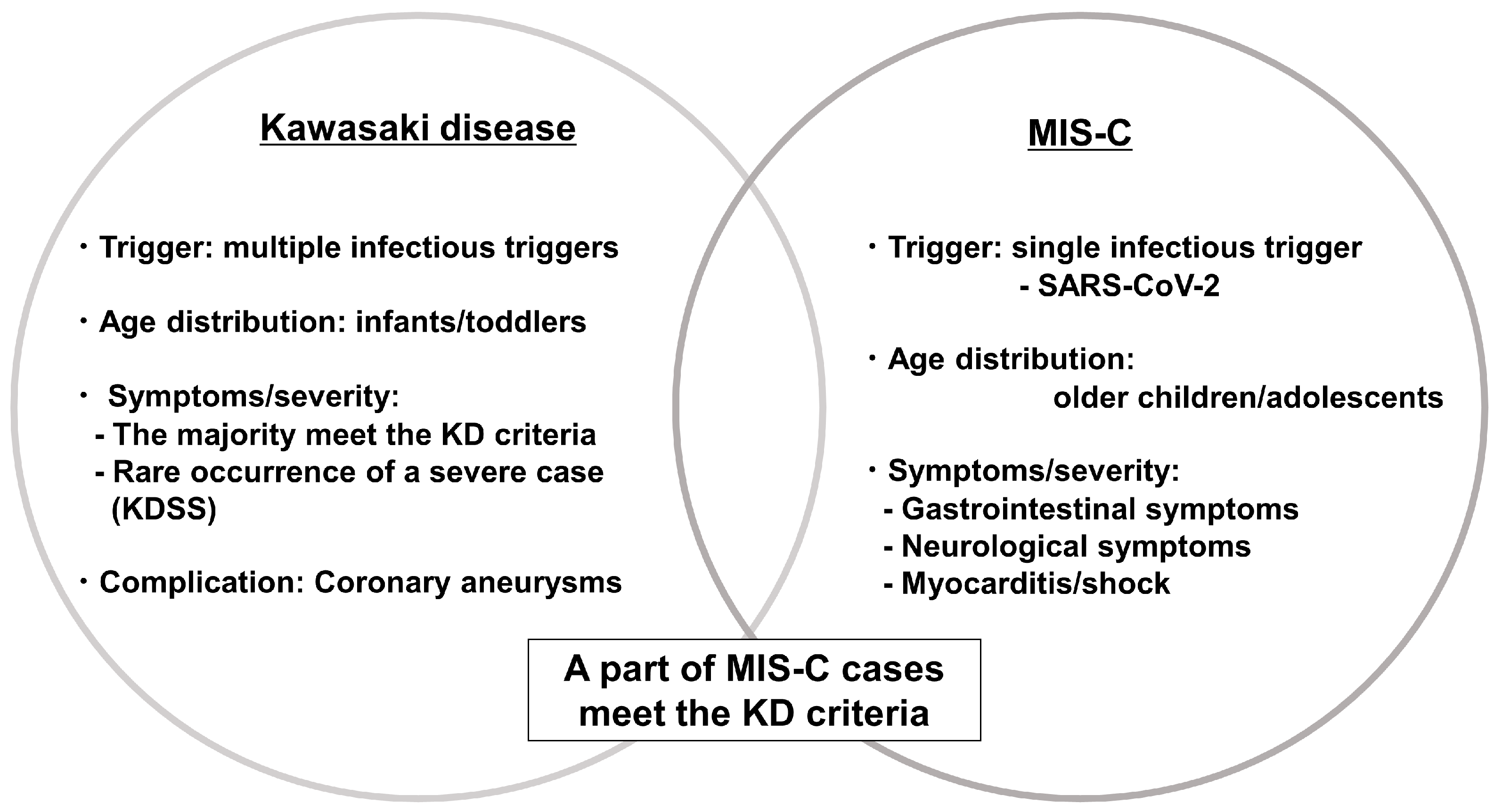
3. KD and Multisystem Inflammatory Syndrome in Children
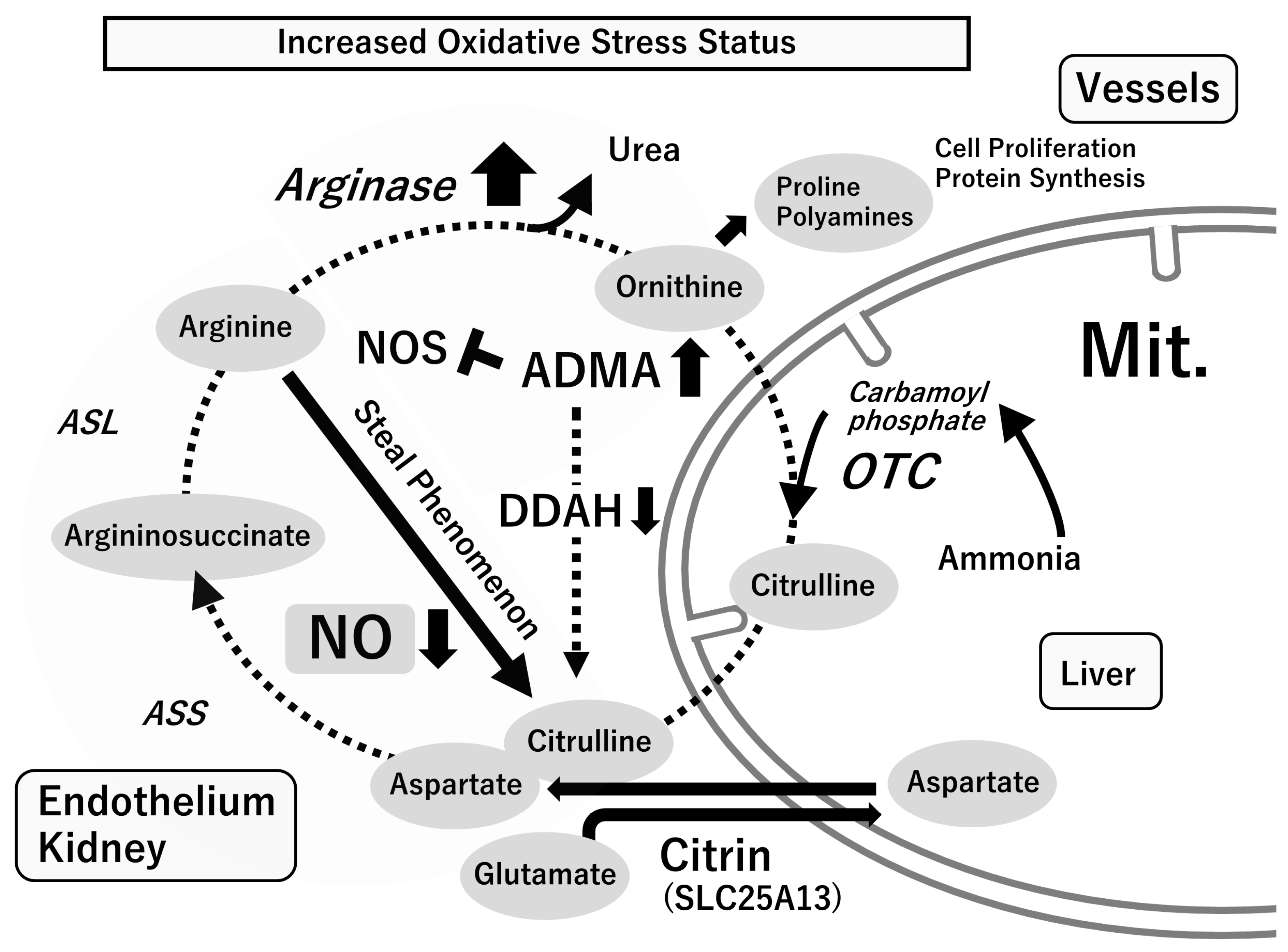
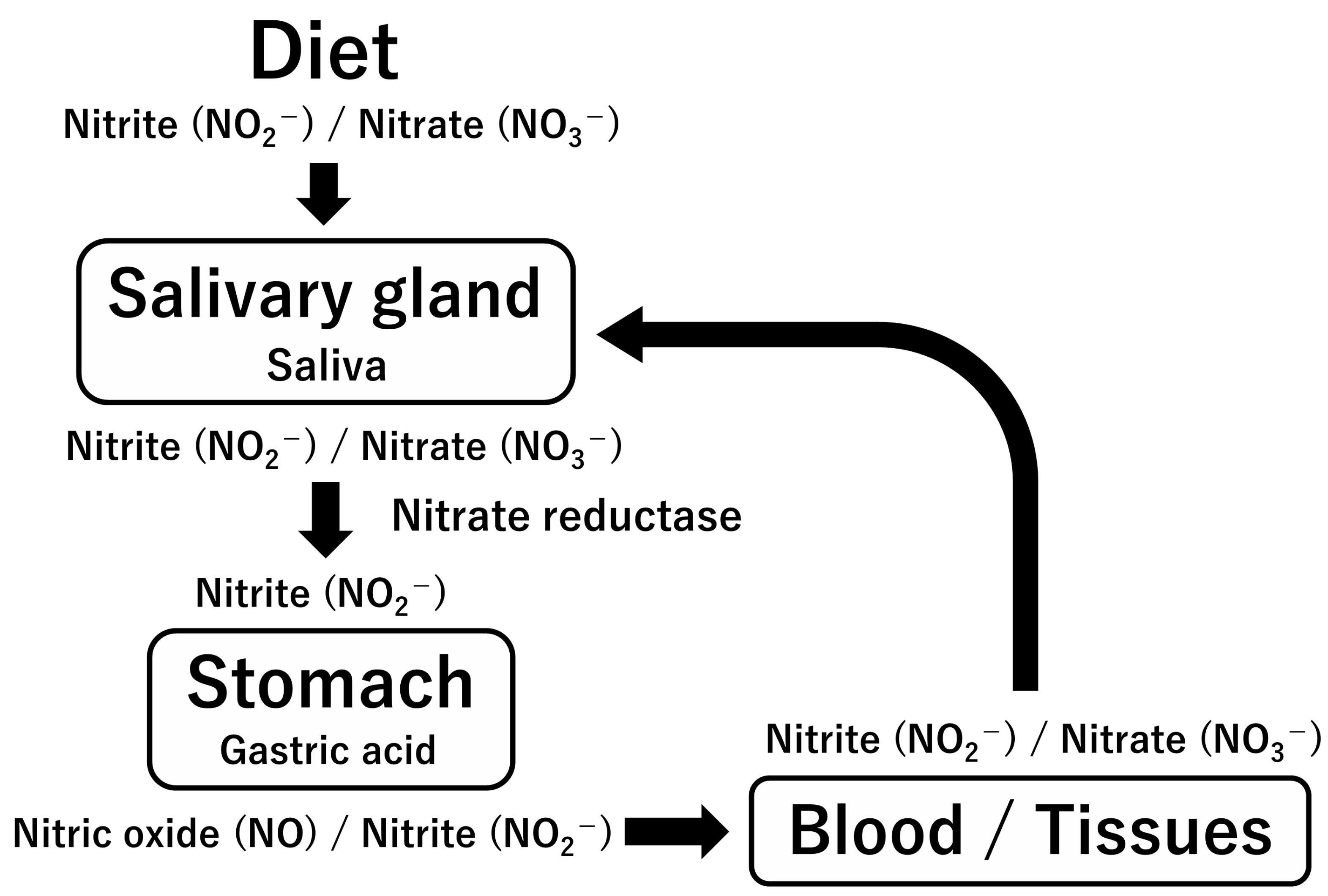
4. L-Arginine and Nitric Oxide Production in Physiological and Pathological Conditions
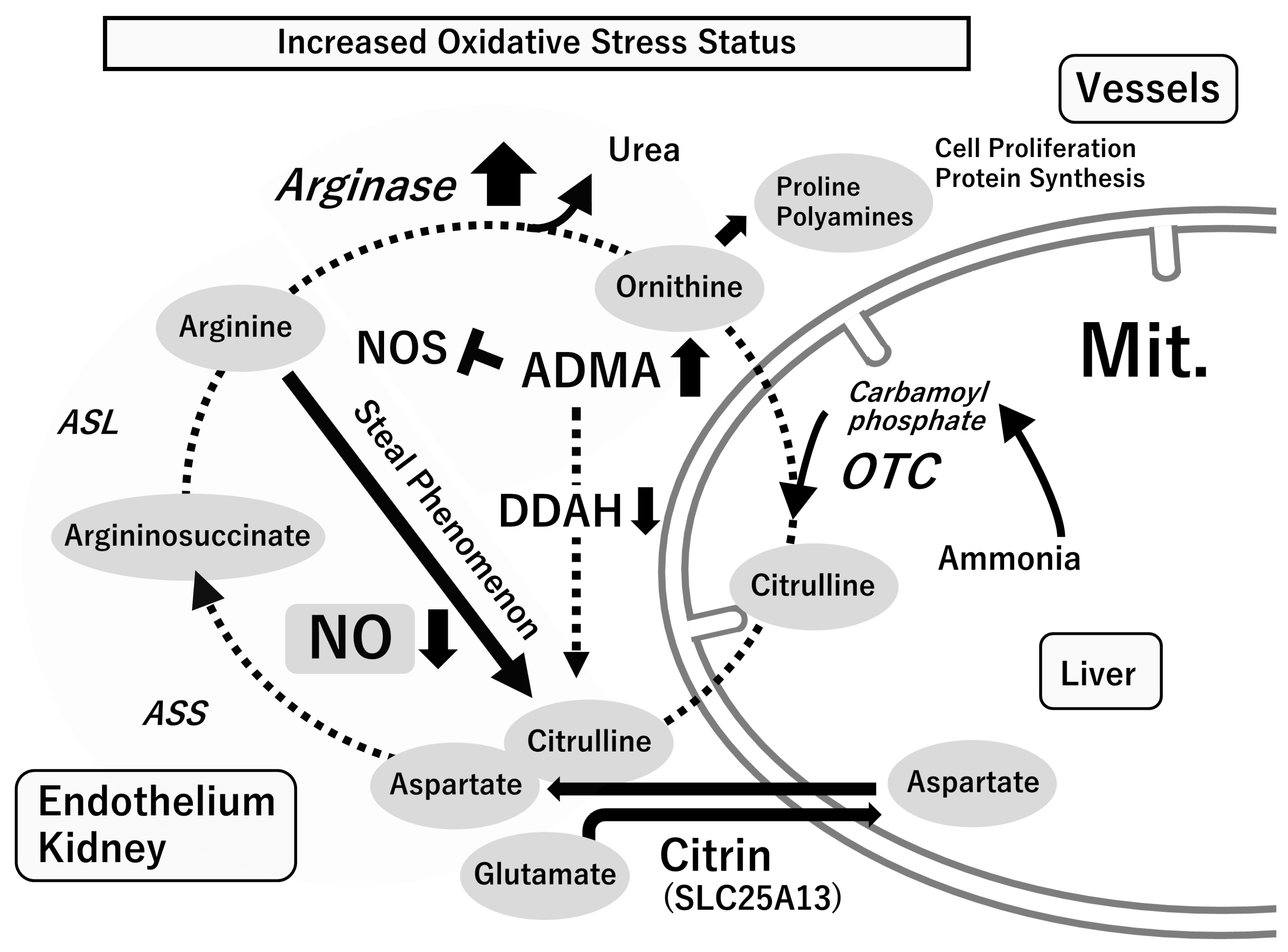
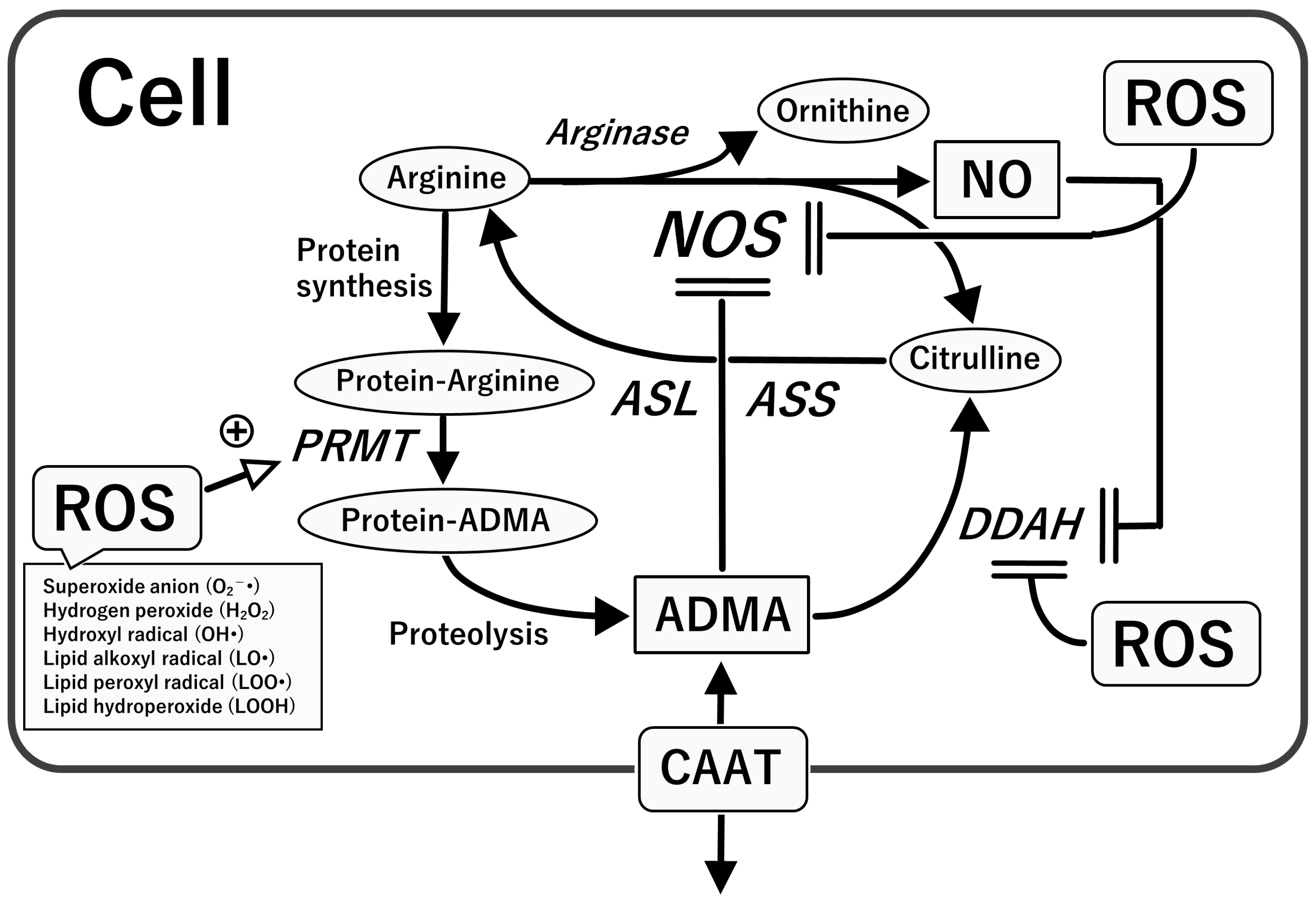
4.1. L-Arginine–Nitric Oxide (NO) System
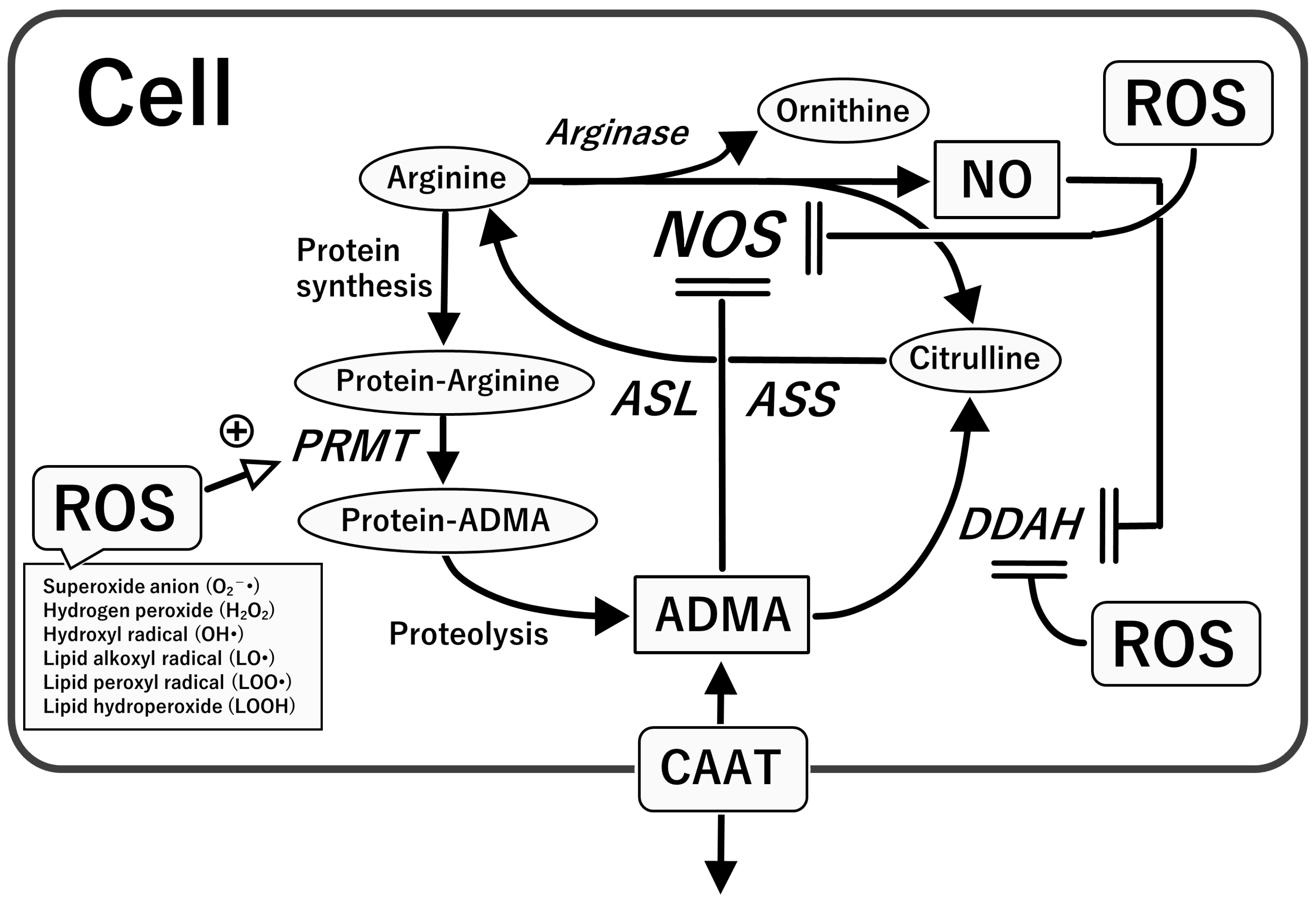
4.2. Asymmetric Dimethylarginine (ADMA), an Endogenous NOS Inhibitor
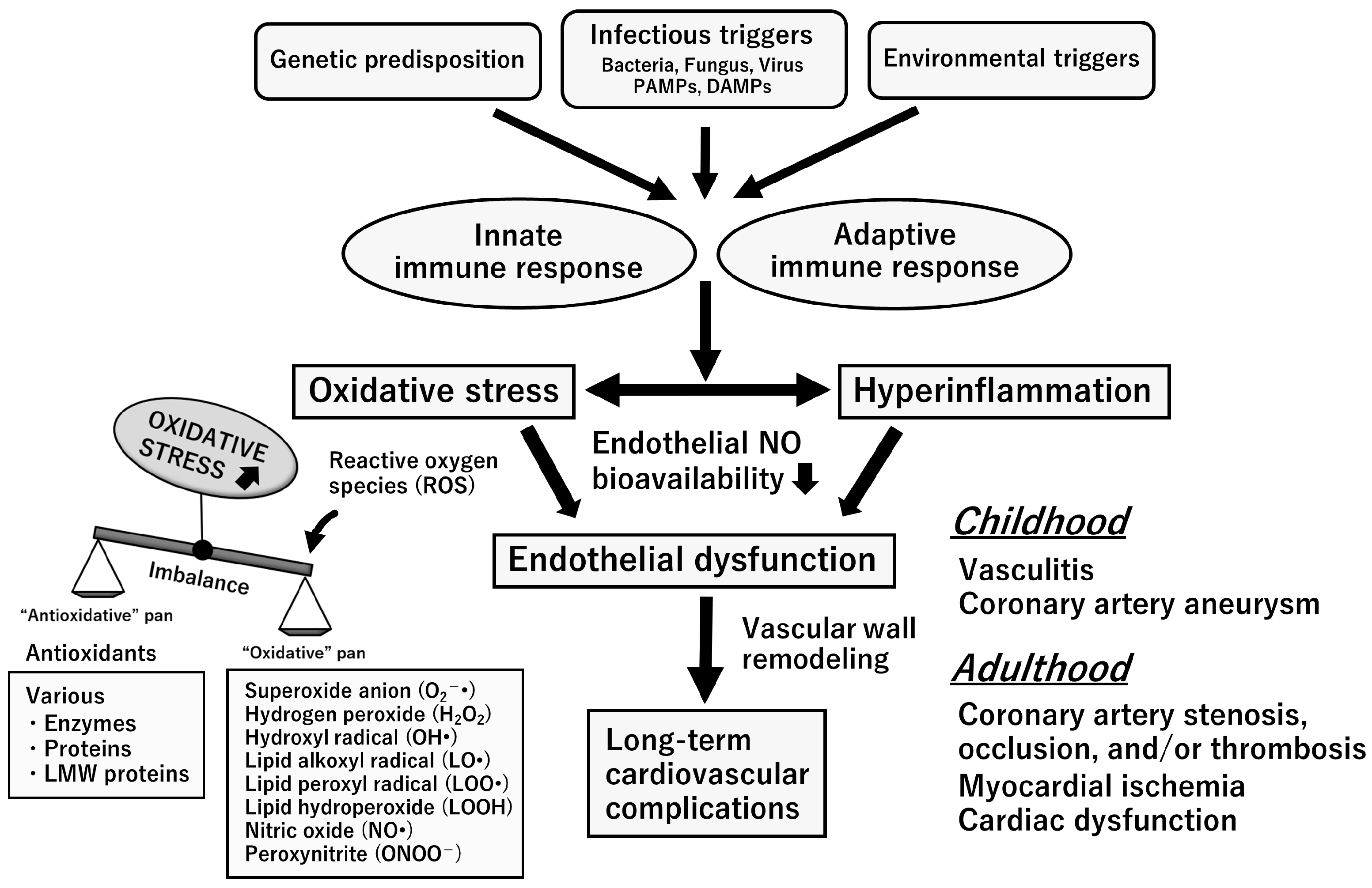
5. Oxidative Stress with Relevance to the L-Arginine-NOS-NO Pathway
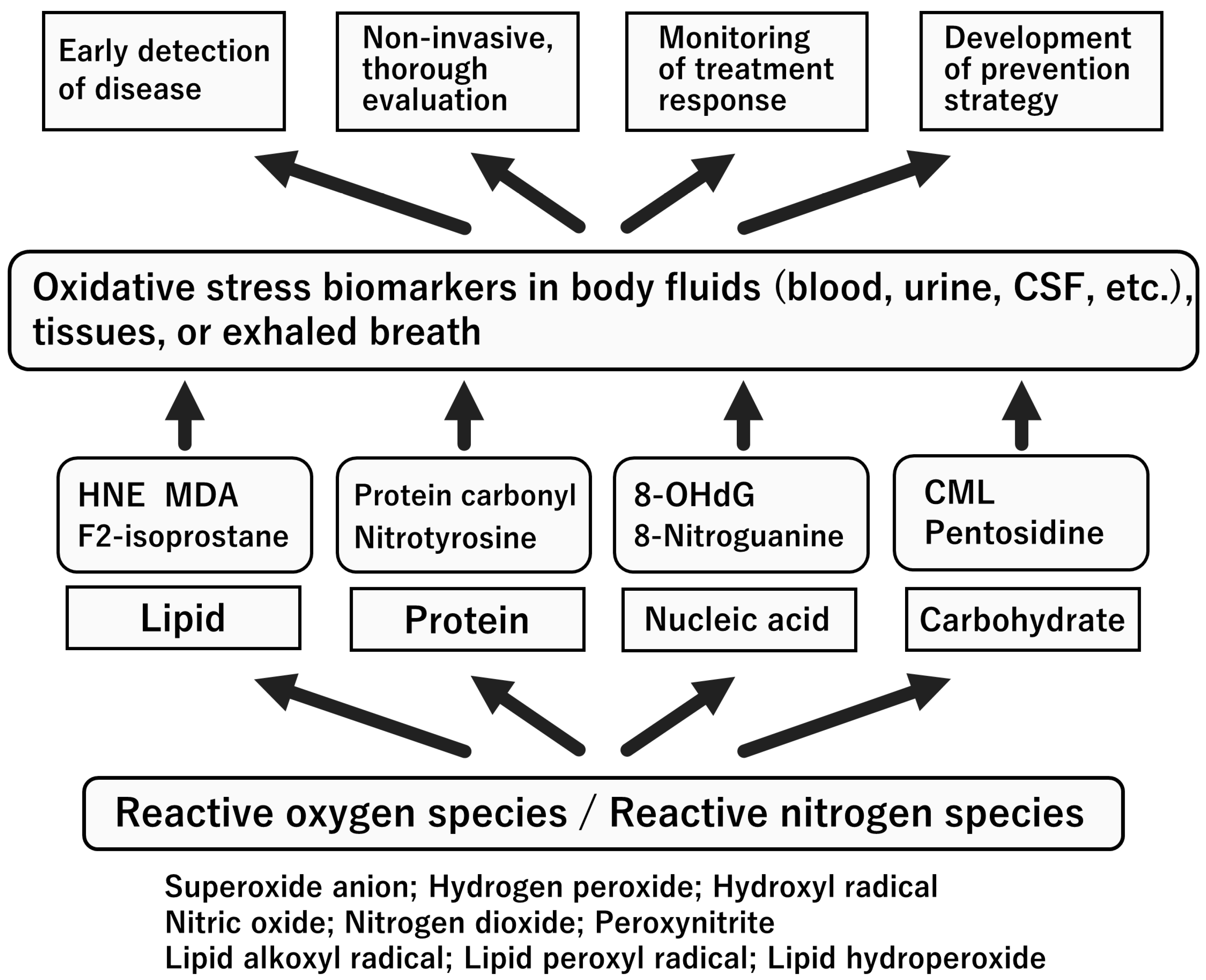
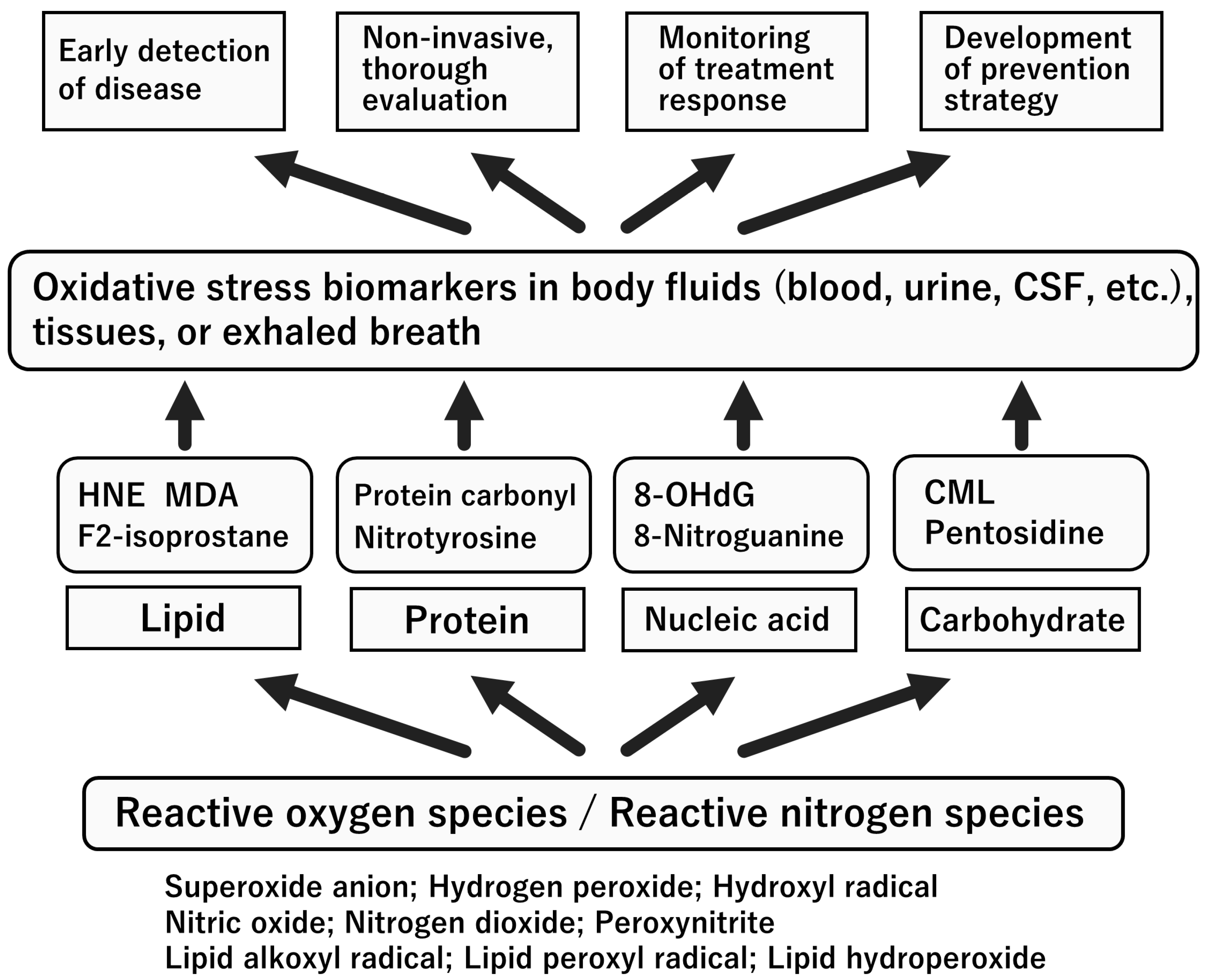
6. Biomarkers for Oxidative Stress and NO System
7. Oxidative Stress Status in the Acute Stage of KD
8. Nitrosative Stress Status in the Acute Stage of KD
9. Endothelial Dysfunction Syndrome in the Chronic Stage of KD
10. Environmental and Toxicological Factors in the Development of KD
10.1. Breast Feeding and Risk of KD
10.2. Redox Modulating Factors in Human Breast Milk
10.3. Ambient Pollution and Risk of KD
11. Adjunct Therapy for Vascular Endothelial Dysfunction in Patients with KD
12. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kawasaki, T.; Kosaki, F.; Okawa, S.; Shigematsu, I.; Yanagawa, H. A new infantile acute febrile mucocutaneous lymph node syndrome (MLNS) prevailing in Japan. Pediatrics 1974, 54, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Koike, S.; Yamamoto, M.; Ito, Y.; Yano, E. Coronary aneurysms in infants and young children with acute febrile mucocutaneous lymph node syndrome. J. Pediatr. 1975, 86, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Wu, M.H. The global epidemiology of Kawasaki disease: Review and future perspectives. Glob. Cardiol. Sci. Pract. 2017, 2017, e201720. [Google Scholar] [CrossRef] [PubMed]
- Makino, N.; Nakamura, Y.; Yashiro, M.; Kosami, K.; Matsubara, Y.; Ae, R.; Aoyama, Y.; Yanagawa, H. Nationwide epidemiologic survey of Kawasaki disease in Japan, 2015–2016. Pediatr. Int. 2019, 61, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.B.; Park, S.; Eun, L.Y.; Han, J.W.; Lee, S.Y.; Yoon, K.L.; Yu, J.J.; Choi, J.W.; Lee, K.Y. Epidemiology and Clinical Features of Kawasaki Disease in South Korea, 2012–2014. Pediatr. Infect. Dis. J. 2017, 36, 482–485. [Google Scholar] [CrossRef]
- McCrindle, B.W.; Rowley, A.H.; Newburger, J.W.; Burns, J.C.; Bolger, A.F.; Gewitz, M.; Baker, A.L.; Jackson, M.A.; Takahashi, M.; Shah, P.B.; et al. Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Scientific Statement for Health Professionals from the American Heart Association. Circulation 2017, 135, e927–e999. [Google Scholar] [CrossRef]
- Newburger, J.W.; Takahashi, M.; Burns, J.C. Kawasaki Disease. J. Am. Coll. Cardiol. 2016, 67, 1738–1749. [Google Scholar] [CrossRef]
- Hara, T.; Yamamura, K.; Sakai, Y. The up-to-date pathophysiology of Kawasaki disease. Clin. Transl. Immunol. 2021, 10, e1284. [Google Scholar] [CrossRef]
- Chang, L.; Yang, H.W.; Lin, T.Y.; Yang, K.D. Perspective of Immunopathogenesis and Immunotherapies for Kawasaki Disease. Front. Pediatr. 2021, 9, 697632. [Google Scholar] [CrossRef]
- Pilania, R.K.; Jindal, A.K.; Bhattarai, D.; Naganur, S.H.; Singh, S. Cardiovascular Involvement in Kawasaki Disease Is Much More Than Mere Coronary Arteritis. Front. Pediatr. 2020, 8, 526969. [Google Scholar] [CrossRef]
- Uehara, R.; Belay, E.D.; Maddox, R.A.; Holman, R.C.; Nakamura, Y.; Yashiro, M.; Oki, I.; Ogino, H.; Schonberger, L.B.; Yanagawa, H. Analysis of potential risk factors associated with nonresponse to initial intravenous immunoglobulin treatment among Kawasaki disease patients in Japan. Pediatr. Infect. Dis. J. 2008, 27, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Ogata, S.; Tremoulet, A.H.; Sato, Y.; Ueda, K.; Shimizu, C.; Sun, X.; Jain, S.; Silverstein, L.; Baker, A.L.; Tanaka, N.; et al. Coronary artery outcomes among children with Kawasaki disease in the United States and Japan. Int. J. Cardiol. 2013, 168, 3825–3828. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.Y.; Zhang, M.; Ko, S.; Chen, F. An Update on Cardiovascular Risk Factors after Kawasaki Disease. Front. Cardiovasc. Med. 2021, 8, 671198. [Google Scholar] [CrossRef] [PubMed]
- Ae, R.; Shibata, Y.; Kosami, K.; Nakamura, Y.; Hamada, H. Kawasaki Disease and Pediatric Infectious Diseases during the Coronavirus Disease 2019 Pandemic. J. Pediatr. 2021, 239, 50–58.e2. [Google Scholar] [CrossRef]
- Sharma, K.; Vignesh, P.; Srivastava, P.; Sharma, J.; Chaudhary, H.; Mondal, S.; Kaur, A.; Kaur, H.; Singh, S. Epigenetics in Kawasaki Disease. Front. Pediatr. 2021, 9, 673294. [Google Scholar] [CrossRef]
- Rowley, A.H. Is Kawasaki disease an infectious disorder? Int. J. Rheum. Dis. 2018, 21, 20–25. [Google Scholar] [CrossRef]
- Rhim, J.W.; Kang, H.M.; Han, J.W.; Lee, K.Y. A Presumed Etiology of Kawasaki Disease Based on Epidemiological Comparison with Infectious or Immune-Mediated Diseases. Front. Pediatr. 2019, 7, 202. [Google Scholar] [CrossRef]
- Chang, L.Y.; Lu, C.Y.; Shao, P.L.; Lee, P.I.; Lin, M.T.; Fan, T.Y.; Cheng, A.L.; Lee, W.L.; Hu, J.J.; Yeh, S.J.; et al. Viral infections associated with Kawasaki disease. J. Formos. Med. Assoc. 2014, 113, 148–154. [Google Scholar] [CrossRef]
- Noval Rivas, M.; Arditi, M. Kawasaki disease: Pathophysiology and insights from mouse models. Nat. Rev. Rheumatol. 2020, 16, 391–405. [Google Scholar] [CrossRef]
- Lo, M.S. A framework for understanding Kawasaki disease pathogenesis. Clin. Immunol. 2020, 214, 108385. [Google Scholar] [CrossRef]
- Lindquist, M.E.; Hicar, M.D. B Cells and Antibodies in Kawasaki Disease. Int. J. Mol. Sci. 2019, 20, 1834. [Google Scholar] [CrossRef]
- Takahashi, K.; Oharaseki, T.; Yokouchi, Y. Histopathological aspects of cardiovascular lesions in Kawasaki disease. Int. J. Rheum. Dis. 2018, 21, 31–35. [Google Scholar] [CrossRef]
- Harada, M.; Yokouchi, Y.; Oharaseki, T.; Matsui, K.; Tobayama, H.; Tanaka, N.; Akimoto, K.; Takahashi, K.; Kishiro, M.; Shimizu, T.; et al. Histopathological characteristics of myocarditis in acute-phase Kawasaki disease. Histopathology 2012, 61, 1156–1167. [Google Scholar] [CrossRef]
- Orenstein, J.M.; Shulman, S.T.; Fox, L.M.; Baker, S.C.; Takahashi, M.; Bhatti, T.R.; Russo, P.A.; Mierau, G.W.; de Chadarévian, J.P.; Perlman, E.J.; et al. Three linked vasculopathic processes characterize Kawasaki disease: A light and transmission electron microscopic study. PLoS ONE 2012, 7, e38998. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Oharaseki, T.; Naoe, S.; Wakayama, M.; Yokouchi, Y. Neutrophilic involvement in the damage to coronary arteries in acute stage of Kawasaki disease. Pediatr. Int. 2005, 47, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Riphagen, S.; Gomez, X.; Gonzalez-Martinez, C.; Wilkinson, N.; Theocharis, P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet 2020, 395, 1607–1608. [Google Scholar] [CrossRef]
- Ouldali, N.; Pouletty, M.; Mariani, P.; Beyler, C.; Blachier, A.; Bonacorsi, S.; Danis, K.; Chomton, M.; Maurice, L.; Le Bourgeois, F.; et al. Emergence of Kawasaki disease related to SARS-CoV-2 infection in an epicentre of the French COVID-19 epidemic: A time-series analysis. Lancet Child. Adolesc. Health 2020, 4, 662–668. [Google Scholar] [CrossRef]
- Feldstein, L.R.; Rose, E.B.; Horwitz, S.M.; Collins, J.P.; Newhams, M.M.; Son, M.B.F.; Newburger, J.W.; Kleinman, L.C.; Heidemann, S.M.; Martin, A.A.; et al. Multisystem Inflammatory Syndrome in U.S. Children and Adolescents. N. Engl. J. Med. 2020, 383, 334–346. [Google Scholar] [CrossRef]
- Uehara, R.; Belay, E.D. Epidemiology of Kawasaki disease in Asia, Europe, and the United States. J. Epidemiol. 2012, 22, 79–85. [Google Scholar] [CrossRef]
- Yan, F.; Pan, B.; Sun, H.; Tian, J.; Li, M. Risk Factors of Coronary Artery Abnormality in Children with Kawasaki Disease: A Systematic Review and Meta-Analysis. Front. Pediatr. 2019, 7, 374. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Harahsheh, A.S.; Raghuveer, G.; Jain, S.; Choueiter, N.F.; Garrido-Garcia, L.M.; Dahdah, N.; Portman, M.A.; Misra, N.; Khoury, M.; et al. Emerging Insights into the Pathophysiology of Multisystem Inflammatory Syndrome Associated with COVID-19 in Children. Can. J. Cardiol. 2023, 39, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Molloy, M.J.; Auger, K.A.; Hall, M.; Shah, S.S.; Schondelmeyer, A.C.; Parikh, K.; Kazmier, K.M.; Katragadda, H.; Jacob, S.A.; Jerardi, K.E.; et al. Epidemiology and Severity of Illness of MIS-C and Kawasaki Disease during the COVID-19 Pandemic. Pediatrics 2023, e2023062101. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, B.J.; Cho, K.S.; Rhim, J.W.; Lee, S.Y.; Jeong, D.C. Similarities and Differences between Multisystem Inflammatory Syndrome in Children (MIS-C) and Kawasaki Disease Shock Syndrome. Children 2023, 10, 1527. [Google Scholar] [CrossRef]
- Godfred-Cato, S.; Abrams, J.Y.; Balachandran, N.; Jaggi, P.; Jones, K.; Rostad, C.A.; Lu, A.T.; Fan, L.; Jabbar, A.; Anderson, E.J.; et al. Distinguishing Multisystem Inflammatory Syndrome in Children from COVID-19, Kawasaki Disease and Toxic Shock Syndrome. Pediatr. Infect. Dis. J. 2022, 41, 315–323. [Google Scholar] [CrossRef]
- Abrams, J.Y.; Oster, M.E.; Godfred-Cato, S.E.; Bryant, B.; Datta, S.D.; Campbell, A.P.; Leung, J.W.; Tsang, C.A.; Pierce, T.J.; Kennedy, J.L.; et al. Factors linked to severe outcomes in multisystem inflammatory syndrome in children (MIS-C) in the USA: A retrospective surveillance study. Lancet Child. Adolesc. Health 2021, 5, 323–331. [Google Scholar] [CrossRef]
- Ae, R.; Makino, N.; Kuwabara, M.; Matsubara, Y.; Kosami, K.; Sasahara, T.; Nakamura, Y. Incidence of Kawasaki Disease before and after the COVID-19 Pandemic in Japan: Results of the 26th Nationwide Survey, 2019 to 2020. JAMA Pediatr. 2022, 176, 1217–1224. [Google Scholar] [CrossRef]
- Burney, J.A.; Roberts, S.C.; DeHaan, L.L.; Shimizu, C.; Bainto, E.V.; Newburger, J.W.; Dominguez, S.; Jone, P.N.; Jaggi, P.; Szmuszkovicz, J.R.; et al. Epidemiological and Clinical Features of Kawasaki Disease during the COVID-19 Pandemic in the United States. JAMA Netw. Open 2022, 5, e2217436. [Google Scholar] [CrossRef]
- Shahbaz, F.F.; Martins, R.S.; Umair, A.; Ukrani, R.D.; Jabeen, K.; Sohail, M.R.; Khan, E. A Review of Coronaviruses Associated with Kawasaki Disease: Possible Implications for Pathogenesis of the Multisystem Inflammatory Syndrome Associated with COVID-19. Clin. Med. Insights Pediatr. 2022, 16, 11795565221075319. [Google Scholar] [CrossRef]
- Esper, F.; Shapiro, E.D.; Weibel, C.; Ferguson, D.; Landry, M.L.; Kahn, J.S. Association between a novel human coronavirus and Kawasaki disease. J. Infect. Dis. 2005, 191, 499–502. [Google Scholar] [CrossRef]
- Moncada, S.; Higgs, A. The L-arginine-nitric oxide pathway. N. Engl. J. Med. 1993, 329, 2002–2012. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol. Rev. 2019, 99, 311–379. [Google Scholar] [CrossRef]
- Förstermann, U.; Münzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef]
- Tsukahara, H. Oxidative Stress Biomarkers: Current Status and Future Perspective. In Studies on Pediatric Disorders; Tsukahara, H., Kaneko, K., Eds.; Springer: New York, NY, USA, 2014; pp. 87–113. [Google Scholar]
- Taniguchi, A.; Tsuge, M.; Miyahara, N.; Tsukahara, H. Reactive Oxygen Species and Antioxidative Defense in Chronic Obstructive Pulmonary Disease. Antioxidants 2021, 10, 1537. [Google Scholar] [CrossRef] [PubMed]
- O’Riordan, E.; Chen, J.; Brodsky, S.V.; Smirnova, I.; Li, H.; Goligorsky, M.S. Endothelial cell dysfunction: The syndrome in making. Kidney Int. 2005, 67, 1654–1658. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, J.; Khondkar, W.; Morelli, M.B.; Wang, X.; Santulli, G.; Trimarco, V. Arginine and Endothelial Function. Biomedicines 2020, 8, 277. [Google Scholar] [CrossRef]
- Morris, C.R.; Kato, G.J.; Poljakovic, M.; Wang, X.; Blackwelder, W.C.; Sachdev, V.; Hazen, S.L.; Vichinsky, E.P.; Morris, S.M., Jr.; Gladwin, M.T. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA 2005, 294, 81–90. [Google Scholar] [CrossRef]
- Pernow, J.; Jung, C. Arginase as a potential target in the treatment of cardiovascular disease: Reversal of arginine steal? Cardiovasc. Res. 2013, 98, 334–343. [Google Scholar] [CrossRef]
- Pope, A.J.; Karuppiah, K.; Cardounel, A.J. Role of the PRMT-DDAH-ADMA axis in the regulation of endothelial nitric oxide production. Pharmacol. Res. 2009, 60, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Tain, Y.L. Asymmetric Dimethylarginine (ADMA) in Pediatric Renal Diseases: From Pathophysiological Phenomenon to Clinical Biomarker and Beyond. Children 2021, 8, 837. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.G.; Tresini, M. Oxidative stress and gene regulation. Free Radic. Biol. Med. 2000, 28, 463–499. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Toyokuni, S. Reactive oxygen species-induced molecular damage and its application in pathology. Pathol. Int. 1999, 49, 91–102. [Google Scholar] [CrossRef]
- Auten, R.L.; Davis, J.M. Oxygen toxicity and reactive oxygen species: The devil is in the details. Pediatr. Res. 2009, 66, 121–127. [Google Scholar] [CrossRef]
- Tsuge, M.; Ikeda, M.; Matsumoto, N.; Yorifuji, T.; Tsukahara, H. Current Insights into Atopic March. Children 2021, 8, 1067. [Google Scholar] [CrossRef]
- Ibáñez-Cabellos, J.S.; Pallardó, F.V.; García-Giménez, J.L.; Seco-Cervera, M. Oxidative Stress and Epigenetics: miRNA Involvement in Rare Autoimmune Diseases. Antioxidants 2023, 12, 800. [Google Scholar] [CrossRef]
- Zatz, R.; Baylis, C. Chronic nitric oxide inhibition model six years on. Hypertension 1998, 32, 958–964. [Google Scholar] [CrossRef]
- Usui, M.; Egashira, K.; Kitamoto, S.; Koyanagi, M.; Katoh, M.; Kataoka, C.; Shimokawa, H.; Takeshita, A. Pathogenic role of oxidative stress in vascular angiotensin-converting enzyme activation in long-term blockade of nitric oxide synthesis in rats. Hypertension 1999, 34, 546–551. [Google Scholar] [CrossRef]
- Tsukahara, H.; Hiraoka, M.; Kobata, R.; Hata, I.; Ohshima, Y.; Jiang, M.Z.; Noiri, E.; Mayumi, M. Increased oxidative stress in rats with chronic nitric oxide depletion: Measurement of urinary 8-hydroxy-2′-deoxyguanosine excretion. Redox Rep. 2000, 5, 23–28. [Google Scholar] [CrossRef]
- Oktem, F.; Kirbas, A.; Armagan, A.; Kuybulu, A.E.; Yilmaz, H.R.; Ozguner, F.; Uz, E. Lisinopril attenuates renal oxidative injury in L-NAME-induced hypertensive rats. Mol. Cell Biochem. 2011, 352, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, K.; Ohtake, K.; Uchida, H.; Ito, J.; Uchida, M.; Natsume, H.; Tamada, H.; Kobayashi, J. Dietary nitrite supplementation attenuates cardiac remodeling in l-NAME-induced hypertensive rats. Nitric. Oxide 2017, 67, 1–9. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Mozos, I.; Luca, C.T. Crosstalk between Oxidative and Nitrosative Stress and Arterial Stiffness. Curr. Vasc. Pharmacol. 2017, 15, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Noiri, E.; Tsukahara, H. Parameters for measurement of oxidative stress in diabetes mellitus: Applicability of enzyme-linked immunosorbent assay for clinical evaluation. J. Investig. Med. 2005, 53, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K. Rapid Diagnostic Tests for Oxidative Stress Status. In Studies on Pediatric Disorders; Tsukahara, H., Kaneko, K., Eds.; Springer: New York, NY, USA, 2014; pp. 137–148. [Google Scholar]
- Yahata, T.; Hamaoka, K. Oxidative stress and Kawasaki disease: How is oxidative stress involved from the acute stage to the chronic stage? Rheumatology 2017, 56, 6–13. [Google Scholar] [CrossRef]
- Uchida, N.; Asayama, K.; Dobashi, K.; Hayashibe, H.; Kato, K. Antioxidant enzymes and lipoperoxide in blood in patients with Kawasaki disease. Comparison with the changes in acute infections. Acta Paediatr. Jpn. 1990, 32, 242–248. [Google Scholar] [CrossRef]
- Lebranchu, Y.; Malvy, D.; Richard, M.J.; Arnaud, J.; Favier, A.; Bardos, P. Kawasaki disease and oxidative metabolism. Clin. Chim. Acta 1990, 187, 193–198. [Google Scholar] [CrossRef]
- Yahata, T.; Suzuki, C.; Hamaoka, A.; Fujii, M.; Hamaoka, K. Dynamics of reactive oxygen metabolites and biological antioxidant potential in the acute stage of Kawasaki disease. Circ. J. 2011, 75, 2453–2459. [Google Scholar] [CrossRef]
- Kaneko, K.; Takahashi, M.; Yoshimura, K.; Kitao, T.; Yamanouchi, S.; Kimata, T.; Tsuji, S. Intravenous immunoglobulin counteracts oxidative stress in Kawasaki disease. Pediatr. Cardiol. 2012, 33, 1086–1088. [Google Scholar] [CrossRef]
- Takatsuki, S.; Ito, Y.; Takeuchi, D.; Hoshida, H.; Nakayama, T.; Matsuura, H.; Saji, T. IVIG reduced vascular oxidative stress in patients with Kawasaki disease. Circ. J. 2009, 73, 1315–1318. [Google Scholar] [CrossRef]
- Yachie, A.; Toma, T.; Mizuno, K.; Okamoto, H.; Shimura, S.; Ohta, K.; Kasahara, Y.; Koizumi, S. Heme oxygenase-1 production by peripheral blood monocytes during acute inflammatory illnesses of children. Exp. Biol. Med. 2003, 228, 550–556. [Google Scholar] [CrossRef] [PubMed]
- He, Y.E.; Qiu, H.X.; Wu, R.Z.; Rong, X.; Xu, H.T.; Xiang, R.L.; Chu, M.P. Oxidised Low-Density Lipoprotein and Its Receptor-Mediated Endothelial Dysfunction Are Associated with Coronary Artery Lesions in Kawasaki Disease. J. Cardiovasc. Transl. Res. 2020, 13, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, H.; Hata, I.; Sudo, M. Urinary nitrite versus nitrate excretion in children. J. Pediatr. 1997, 130, 161. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, T.; Oishi, K.; Sasaki, M.; Hatanaka, Y.; Minatogawa, Y.; Uemura, S.; Koike, M. Nitric oxide and aneurysm formation in Kawasaki disease. Acta Paediatr. 1997, 86, 470–473. [Google Scholar] [CrossRef]
- Tsukahara, H.; Kikuchi, K.; Matsuda, M.; Saito, M.; Hata, I.; Tsuchida, S.; Sudo, M. Endogenous nitric oxide production in Kawasaki disease. Scand. J. Clin. Lab. Investig. 1997, 57, 43–47. [Google Scholar] [CrossRef]
- Ikemoto, Y.; Teraguchi, M.; Ono, A.; Kino, M.; Yoshimura, K.; Kobayashi, Y. Serial changes of plasma nitrate in the acute phase of Kawasaki disease. Pediatr. Int. 2003, 45, 421–425. [Google Scholar] [CrossRef]
- Wang, C.L.; Wu, Y.T.; Lee, C.J.; Liu, H.C.; Huang, L.T.; Yang, K.D. Decreased nitric oxide production after intravenous immunoglobulin treatment in patients with Kawasaki disease. J. Pediatr. 2002, 141, 560–565. [Google Scholar] [CrossRef]
- Yu, X.; Hirono, K.I.; Ichida, F.; Uese, K.; Rui, C.; Watanabe, S.; Watanabe, K.; Hashimoto, I.; Kumada, T.; Okada, E.; et al. Enhanced iNOS expression in leukocytes and circulating endothelial cells is associated with the progression of coronary artery lesions in acute Kawasaki disease. Pediatr. Res. 2004, 55, 688–694. [Google Scholar] [CrossRef]
- Straface, E.; Marchesi, A.; Gambardella, L.; Metere, A.; Tarissi de Jacobis, I.; Viora, M.; Giordani, L.; Villani, A.; Del Principe, D.; Malorni, W.; et al. Does oxidative stress play a critical role in cardiovascular complications of Kawasaki disease? Antioxid. Redox Signal 2012, 17, 1441–1446. [Google Scholar] [CrossRef]
- Straface, E.; Gambardella, L.; Metere, A.; Marchesi, A.; Palumbo, G.; Cortis, E.; Villani, A.; Pietraforte, D.; Viora, M.; Malorni, W.; et al. Oxidative stress and defective platelet apoptosis in naïve patients with Kawasaki disease. Biochem. Biophys. Res. Commun. 2010, 392, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, K.; Tatsumi, K.; Iharada, A.; Tsuji, S.; Tateiwa, A.; Teraguchi, M.; Ogino, H.; Kaneko, K. Increased nitric oxide production by neutrophils in early stage of Kawasaki disease. Eur. J. Pediatr. 2009, 168, 1037–1041. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Tain, Y.L.; Lee, C.P.; Kuo, H.C. Asymmetric and symmetric dimethylarginine are associated with coronary artery lesions in Kawasaki disease. J. Pediatr. 2014, 165, 295–299. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N. Toxic Dimethylarginines: Asymmetric Dimethylarginine (ADMA) and Symmetric Dimethylarginine (SDMA). Toxins 2017, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Graciano-Machuca, O.; Villegas-Rivera, G.; López-Pérez, I.; Macías-Barragán, J.; Sifuentes-Franco, S. Multisystem Inflammatory Syndrome in Children (MIS-C) Following SARS-CoV-2 Infection: Role of Oxidative Stress. Front. Immunol. 2021, 12, 723654. [Google Scholar] [CrossRef] [PubMed]
- Perrone, S.; Cannavò, L.; Manti, S.; Rullo, I.; Buonocore, G.; Esposito, S.M.R.; Gitto, E. Pediatric Multisystem Syndrome Associated with SARS-CoV-2 (MIS-C): The Interplay of Oxidative Stress and Inflammation. Int. J. Mol. Sci. 2022, 23, 12836. [Google Scholar] [CrossRef] [PubMed]
- Durante, W. Targeting Arginine in COVID-19-Induced Immunopathology and Vasculopathy. Metabolites 2022, 12, 240. [Google Scholar] [CrossRef]
- Kato, H.; Sugimura, T.; Akagi, T.; Sato, N.; Hashino, K.; Maeno, Y.; Kazue, T.; Eto, G.; Yamakawa, R. Long-term consequences of Kawasaki disease. A 10- to 21-year follow-up study of 594 patients. Circulation 1996, 94, 1379–1385. [Google Scholar] [CrossRef]
- Senzaki, H. Long-term outcome of Kawasaki disease. Circulation 2008, 118, 2763–2772. [Google Scholar] [CrossRef]
- Sato, S.; Tsukahara, H.; Ohta, N.; Todoroki, Y.; Nishida, K.; Mayumi, M. Endothelial dysfunction in Kawasaki disease: Focus on nitric oxide. Pediatr. Int. 2004, 46, 114. [Google Scholar] [CrossRef]
- Higashi, Y.; Noma, K.; Yoshizumi, M.; Kihara, Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ. J. 2009, 73, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Noto, N.; Okada, T.; Yamasuge, M.; Taniguchi, K.; Karasawa, K.; Ayusawa, M.; Sumitomo, N.; Harada, K. Noninvasive assessment of the early progression of atherosclerosis in adolescents with Kawasaki disease and coronary artery lesions. Pediatrics 2001, 107, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.B.; Li, T.L.; Xiang, H.J.; Chang, Q.; Li, C.L. Impaired endothelial function in the brachial artery after Kawasaki disease and the effects of intravenous administration of vitamin C. Pediatr. Infect. Dis. J. 2003, 22, 34–39. [Google Scholar] [CrossRef]
- Ikemoto, Y.; Ogino, H.; Teraguchi, M.; Kobayashi, Y. Evaluation of preclinical atherosclerosis by flow-mediated dilatation of the brachial artery and carotid artery analysis in patients with a history of Kawasaki disease. Pediatr. Cardiol. 2005, 26, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Y.F.; O, K.; Woo, C.W.; Armstrong, S.; Siow, Y.L.; Chow, P.C.; Cheung, E.W. Oxidative stress in children late after Kawasaki disease: Relationship with carotid atherosclerosis and stiffness. BMC Pediatr. 2008, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Niboshi, A.; Hamaoka, K.; Sakata, K.; Yamaguchi, N. Endothelial dysfunction in adult patients with a history of Kawasaki disease. Eur. J. Pediatr. 2008, 167, 189–196. [Google Scholar] [CrossRef]
- Hamaoka, A.; Hamaoka, K.; Yahata, T.; Fujii, M.; Ozawa, S.; Toiyama, K.; Nishida, M.; Itoi, T. Effects of HMG-CoA reductase inhibitors on continuous post-inflammatory vascular remodeling late after Kawasaki disease. J. Cardiol. 2010, 56, 245–253. [Google Scholar] [CrossRef]
- AlHuzaimi, A.; Al Mashham, Y.; Potts, J.E.; De Souza, A.M.; Sandor, G.G. Echo-Doppler assessment of arterial stiffness in pediatric patients with Kawasaki disease. J. Am. Soc. Echocardiogr. 2013, 26, 1084–1089. [Google Scholar] [CrossRef]
- Ishikawa, T.; Iwashima, S. Endothelial dysfunction in children within 5 years after onset of Kawasaki disease. J. Pediatr. 2013, 163, 1117–1121. [Google Scholar] [CrossRef]
- Ishikawa, T.; Seki, K. The association between oxidative stress and endothelial dysfunction in early childhood patients with Kawasaki disease. BMC Cardiovasc. Disord. 2018, 18, 30. [Google Scholar] [CrossRef]
- Motoji, Y.; Fukazawa, R.; Matsui, R.; Abe, Y.; Uehara, I.; Watanabe, M.; Hashimoto, Y.; Miyagi, Y.; Nagi-Miura, N.; Tanaka, N.; et al. Statins Show Anti-Atherosclerotic Effects by Improving Endothelial Cell Function in a Kawasaki Disease-like Vasculitis Mouse Model. Int. J. Mol. Sci. 2022, 23, 16108. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, H. Redox modulatory factors of human breast milk. In Handbook of Dietary and Nutritional Aspects of Human Breast Milk; Human Health Handbooks; Wageningen Academic Publishers: Wageningen, The Netherlands, 2013; Volume 5, pp. 599–614. [Google Scholar]
- Cacho, N.T.; Lawrence, R.M. Innate Immunity and Breast Milk. Front. Immunol. 2017, 8, 584. [Google Scholar] [CrossRef] [PubMed]
- Nolan, L.S.; Parks, O.B.; Good, M. A Review of the Immunomodulating Components of Maternal Breast Milk and Protection against Necrotizing Enterocolitis. Nutrients 2019, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Shoji, H.; Shimizu, T. Effect of human breast milk on biological metabolism in infants. Pediatr. Int. 2019, 61, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, N.; Yorifuji, T.; Nakamura, K.; Ikeda, M.; Tsukahara, H.; Doi, H. Breastfeeding and risk of food allergy: A nationwide birth cohort in Japan. Allergol. Int. 2020, 69, 91–97. [Google Scholar] [CrossRef]
- Yorifuji, T.; Tsukahara, H.; Doi, H. Breastfeeding and Risk of Kawasaki Disease: A Nationwide Longitudinal Survey in Japan. Pediatrics 2016, 137, e20153919. [Google Scholar] [CrossRef]
- Fouhy, F.; Watkins, C.; Hill, C.J.; O’Shea, C.A.; Nagle, B.; Dempsey, E.M.; O’Toole, P.W.; Ross, R.P.; Ryan, C.A.; Stanton, C. Perinatal factors affect the gut microbiota up to four years after birth. Nat. Commun. 2019, 10, 1517. [Google Scholar] [CrossRef]
- Kaneko, K.; Akagawa, S.; Akagawa, Y.; Kimata, T.; Tsuji, S. Our Evolving Understanding of Kawasaki Disease Pathogenesis: Role of the Gut Microbiota. Front. Immunol. 2020, 11, 1616. [Google Scholar] [CrossRef]
- Takeuchi, A.; Namba, T.; Matsumoto, N.; Tamai, K.; Nakamura, K.; Nakamura, M.; Kageyama, M.; Kubo, T.; Tsukahara, H.; Yorifuji, T. Preterm birth and Kawasaki disease: A nationwide Japanese population-based study. Pediatr. Res. 2022, 92, 557–562. [Google Scholar] [CrossRef]
- Nakamura, K.; Matsumoto, N.; Nakamura, M.; Takeuchi, A.; Kageyama, M.; Yorifuji, T. Exclusively Breastfeeding Modifies the Adverse Association of Late Preterm Birth and Gastrointestinal Infection: A Nationwide Birth Cohort Study. Breastfeed. Med. 2020, 15, 509–515. [Google Scholar] [CrossRef]
- Isayama, T.; Lewis-Mikhael, A.M.; O’Reilly, D.; Beyene, J.; McDonald, S.D. Health Services Use by Late Preterm and Term Infants from Infancy to Adulthood: A Meta-analysis. Pediatrics 2017, 140, e20170266. [Google Scholar] [CrossRef]
- Kobayashi, J. Nitrite in breast milk: Roles in neonatal pathophysiology. Pediatr. Res. 2021, 90, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Shoji, H.; Oguchi, S.; Shimizu, T.; Yamashiro, Y. Effect of human breast milk on urinary 8-hydroxy-2′-deoxyguanosine excretion in infants. Pediatr. Res. 2003, 53, 850–852. [Google Scholar] [CrossRef] [PubMed]
- Shoji, H.; Shimizu, T.; Shinohara, K.; Oguchi, S.; Shiga, S.; Yamashiro, Y. Suppressive effects of breast milk on oxidative DNA damage in very low birthweight infants. Arch. Dis. Child.-Fetal Neonatal Ed. 2004, 89, F136. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, T.; Sasaki, M.; Oishi, K.; Uemura, S.; Koike, M.; Shinozaki, M. Non-enzymatic nitric oxide generation in the stomachs of breastfed neonates. Acta Paediatr. 1999, 88, 1053–1055. [Google Scholar] [CrossRef]
- Akçay, F.; Aksoy, H.; Memisoğullari, R. Effect of breast-feeding on concentration of nitric oxide in breast milk. Ann. Clin. Biochem. 2002, 39, 68–69. [Google Scholar] [CrossRef]
- Ohta, N.; Tsukahara, H.; Ohshima, Y.; Nishii, M.; Ogawa, Y.; Sekine, K.; Kasuga, K.; Mayumi, M. Nitric oxide metabolites and adrenomedullin in human breast milk. Early Hum. Dev. 2004, 78, 61–65. [Google Scholar] [CrossRef]
- Hord, N.G.; Ghannam, J.S.; Garg, H.K.; Berens, P.D.; Bryan, N.S. Nitrate and nitrite content of human, formula, bovine, and soy milks: Implications for dietary nitrite and nitrate recommendations. Breastfeed. Med. 2011, 6, 393–399. [Google Scholar] [CrossRef]
- Fernandes, J.O.; Tella, S.O.C.; Ferraz, I.S.; Ciampo, L.A.D.; Tanus-Santos, J.E. Assessment of nitric oxide metabolites concentrations in plasma, saliva, and breast milk and their relationship in lactating women. Mol. Cell Biochem. 2021, 476, 1293–1302. [Google Scholar] [CrossRef]
- Todoroki, Y.; Tsukahara, H.; Ohshima, Y.; Shukunami, K.; Nishijima, K.; Kotsuji, F.; Hata, A.; Kasuga, K.; Sekine, K.; Nakamura, H.; et al. Concentrations of thioredoxin, a redox-regulating protein, in umbilical cord blood and breast milk. Free Radic. Res. 2005, 39, 291–297. [Google Scholar] [CrossRef]
- Zarban, A.; Taheri, F.; Chahkandi, T.; Sharifzadeh, G.; Khorashadizadeh, M. Antioxidant and radical scavenging activity of human colostrum, transitional and mature milk. J. Clin. Biochem. Nutr. 2009, 45, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Ohtake, K.; Uchida, H. NO-Rich Diet for Lifestyle-Related Diseases. Nutrients 2015, 7, 4911–4937. [Google Scholar] [CrossRef] [PubMed]
- DeMartino, A.W.; Kim-Shapiro, D.B.; Patel, R.P.; Gladwin, M.T. Nitrite and nitrate chemical biology and signalling. Br. J. Pharmacol. 2019, 176, 228–245. [Google Scholar] [CrossRef]
- Burke, H.; Leonardi-Bee, J.; Hashim, A.; Pine-Abata, H.; Chen, Y.; Cook, D.G.; Britton, J.R.; McKeever, T.M. Prenatal and passive smoke exposure and incidence of asthma and wheeze: Systematic review and meta-analysis. Pediatrics 2012, 129, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, M.; Franchi, S.; Pistorio, A.; Petecchia, L.; Rusconi, F. Smoke exposure, wheezing, and asthma development: A systematic review and meta-analysis in unselected birth cohorts. Pediatr. Pulmonol. 2015, 50, 353–362. [Google Scholar] [CrossRef]
- Yorifuji, T.; Tsukahara, H.; Kashima, S.; Doi, H. Intrauterine and Early Postnatal Exposure to Particulate Air Pollution and Kawasaki Disease: A Nationwide Longitudinal Survey in Japan. J. Pediatr. 2018, 193, 147–154.e2. [Google Scholar] [CrossRef]
- Yorifuji, T.; Tsukahara, H.; Doi, H. Early childhood exposure to maternal smoking and Kawasaki Disease: A longitudinal survey in Japan. Sci. Total Environ. 2019, 655, 141–146. [Google Scholar] [CrossRef]
- Takeuchi, A.; Sugino, N.; Namba, T.; Tamai, K.; Nakamura, K.; Nakamura, M.; Kageyama, M.; Yorifuji, T.; Bonno, M. Neonatal sepsis and Kawasaki disease. Eur. J. Pediatr. 2022, 181, 2927–2933. [Google Scholar] [CrossRef]
- Cetinkaya, F.; Uslu, H.S.; Nuhoğlu, A. Effect of neonatal sepsis on the development of allergies and asthma in later childhood. Int. Arch. Allergy Immunol. 2007, 142, 145–150. [Google Scholar] [CrossRef]
- Mantzarlis, K.; Tsolaki, V.; Zakynthinos, E. Role of Oxidative Stress and Mitochondrial Dysfunction in Sepsis and Potential Therapies. Oxid. Med. Cell Longev. 2017, 2017, 5985209. [Google Scholar] [CrossRef]
- Norooznezhad, A.H.; Mansouri, K. Endothelial cell dysfunction, coagulation, and angiogenesis in coronavirus disease 2019 (COVID-19). Microvasc. Res. 2021, 137, 104188. [Google Scholar] [CrossRef]
- Kidde, J.; Gorabi, A.M.; Jamialahmadi, T.; Sahebkar, A. COVID-19 Is an Endothelial Disease: Implications of Nitric Oxide. Adv. Exp. Med. Biol. 2021, 1321, 109–113. [Google Scholar] [CrossRef]
- Margaritis, M.; Channon, K.M.; Antoniades, C. Statins as regulators of redox state in the vascular endothelium: Beyond lipid lowering. Antioxid. Redox Signal 2014, 20, 1198–1215. [Google Scholar] [CrossRef]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic Effects of Statins on the Cardiovascular System. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Tremoulet, A.H.; Jain, S.; Jone, P.N.; Best, B.M.; Duxbury, E.H.; Franco, A.; Printz, B.; Dominguez, S.R.; Heizer, H.; Anderson, M.S.; et al. Phase I/IIa Trial of Atorvastatin in Patients with Acute Kawasaki Disease with Coronary Artery Aneurysm. J. Pediatr. 2019, 215, 107–117.e12. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, C.; Kim, J.; He, M.; Tremoulet, A.H.; Hoffman, H.M.; Shyy, J.Y.; Burns, J.C. RNA Sequencing Reveals Beneficial Effects of Atorvastatin on Endothelial Cells in Acute Kawasaki Disease. J. Am. Heart Assoc. 2022, 11, e025408. [Google Scholar] [CrossRef] [PubMed]
- Song, S.L.; Hays, S.B.; Panton, C.E.; Mylona, E.K.; Kalligeros, M.; Shehadeh, F.; Mylonakis, E. Statin Use Is Associated with Decreased Risk of Invasive Mechanical Ventilation in COVID-19 Patients: A Preliminary Study. Pathogens 2020, 9, 759. [Google Scholar] [CrossRef]
- Torres-Peña, J.D.; Pérez-Belmonte, L.M.; Fuentes-Jiménez, F.; López Carmona, M.D.; Pérez-Martinez, P.; López-Miranda, J.; Carrasco Sánchez, F.J.; Vargas Núñez, J.A.; Del Corral Beamonte, E.; Magallanes Gamboa, J.O.; et al. Prior Treatment with Statins is Associated with Improved Outcomes of Patients with COVID-19: Data from the SEMI-COVID-19 Registry. Drugs 2021, 81, 685–695. [Google Scholar] [CrossRef]
- Daniels, L.B.; Sitapati, A.M.; Zhang, J.; Zou, J.; Bui, Q.M.; Ren, J.; Longhurst, C.A.; Criqui, M.H.; Messer, K. Relation of Statin Use Prior to Admission to Severity and Recovery among COVID-19 Inpatients. Am. J. Cardiol. 2020, 136, 149–155. [Google Scholar] [CrossRef]
- Chacko, S.R.; DeJoy, R., 3rd; Lo, K.B.; Albano, J.; Peterson, E.; Bhargav, R.; Gu, F.; Salacup, G.; Pelayo, J.; Azmaiparashvili, Z.; et al. Association of Pre-Admission Statin Use with Reduced In-Hospital Mortality in COVID-19. Am. J. Med. Sci. 2021, 361, 725–730. [Google Scholar] [CrossRef]
- Gupta, A.; Madhavan, M.V.; Poterucha, T.J.; DeFilippis, E.M.; Hennessey, J.A.; Redfors, B.; Eckhardt, C.; Bikdeli, B.; Platt, J.; Nalbandian, A.; et al. Association between antecedent statin use and decreased mortality in hospitalized patients with COVID-19. Nat. Commun. 2021, 12, 1325. [Google Scholar] [CrossRef] [PubMed]
- Ashor, A.W.; Lara, J.; Mathers, J.C.; Siervo, M. Effect of vitamin C on endothelial function in health and disease: A systematic review and meta-analysis of randomised controlled trials. Atherosclerosis 2014, 235, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Rashid, J.; Kumar, S.S.; Job, K.M.; Liu, X.; Fike, C.D.; Sherwin, C.M.T. Therapeutic Potential of Citrulline as an Arginine Supplement: A Clinical Pharmacology Review. Paediatr. Drugs 2020, 22, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.M.; Townsend, J.R.; Pinzone, A.G.; Hoffman, J.R. Supplementation with Nitric Oxide Precursors for Strength Performance: A Review of the Current Literature. Nutrients 2023, 15, 660. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formation of Modified Molecules by Reactive Oxygen Species: | Kawasaki Disease vs. Controls |
|---|---|
| Lipid peroxides (plasma) | ↑ [69] |
| Malonyldialdehyde/organic hydroperoxides (plasma) | ↑ [70] |
| Total hydroperoxides (blood) | ↑ [71] a |
| Total hydroperoxides (serum) | ↑ [72] a |
| 8-isoprostane (urine) | ↑ [73] |
| Oxidized low-density lipoprotein (plasma) | ↑ [75] |
| Lectin-like oxidized low-density lipoprotein receptor-1 (mononuclear cells) | ↑ [75] |
| Antioxidative molecules and enzymes: | |
| Manganese superoxide dismutase (leukocytes) | ↑ [69] |
| Glutathione peroxidase/catalase (erythrocytes) | ↑ [69] |
| Copper/zinc superoxide dismutase (leukocytes/erythrocytes) | → [69] |
| Heme oxygenase-1 (monocytes) | ↑ [74] |
| Others: | |
| Zinc (plasma) | ↓ [70] |
| Biological antioxidative potential (blood) | → [71] (IVIG-responding) a ↓ [71] (IVIG-nonresponding) a |
| Biological antioxidative potential (serum) | ↓ [72] a |
| Biomarkers of L-Arginine–Nitric Oxide System | Kawasaki Disease vs. Controls |
|---|---|
| Nitrite/nitrate (urine) | ↑ [77] a |
| Nitrite/nitrate (urine) | ↑ [78] |
| Nitrate (plasma) | ↑ [79] |
| Nitrite/nitrate (plasma/urine) | ↑ [80] |
| Inducible nitric oxide synthase (NOS2) (mononuclear cells) | ↑ [80] a |
| Inducible nitric oxide synthase (NOS2) (neutrophils/circulating endothelial cells) | ↑ [81] |
| Inducible nitric oxide synthase (NOS2) (monocytes) | → [81] b |
| Reactive oxygen- and nitrogen-derived species (e.g., peroxynitrite) (whole blood) | ↑ [82] c |
| Asymmetric dimethylarginine (plasma) | ↓ [82] |
| Nitric oxide (neutrophils) | ↑ [84] d |
| L-arginine/asymmetric dimethylarginine/symmetric dimethylarginine (plasma) | ↓ [85] |
| Stage | Nitrate (in 100 mL) | Nitrite (in 100 mL) | Nitrate (in 750 mL) | Nitrite (in 750 mL) |
|---|---|---|---|---|
| Colostrum | 0.19 ± 0.03 (mg) 3.1 ± 0.5 (µmol) | 0.08 ± 0.02 (mg) 1.7 ± 0.4 (µmol) | 1.43 ± 0.24 (mg) 23.1 ± 3.9 (µmol) | 0.60 ± 0.15 (mg) 13.0 ± 3.3 (µmol) |
| Transition | 0.52 ± 0.10 (mg) 8.4 ± 1.6 (µmol) | 0.001 ± 0.001 (mg) 0.02 ± 0.02 (µmol) | 3.90 ± 0.75 (mg) 62.9 ± 12.1 (µmol) | 0.008 ± 0.008 (mg) 0.2 ± 0.2 (µmol) |
| Mature | 0.31 ± 0.02 (mg) 5.0 ± 0.3 (µmol) | 0.001 ± 0.001 (mg) 0.02 ± 0.02 (µmol) | 2.32 ± 0.21 (mg) 37.4 ± 3.4 (µmol) | 0.008 ± 0.008 (mg) 0.2 ± 0.2 (µmol) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsuge, M.; Uda, K.; Eitoku, T.; Matsumoto, N.; Yorifuji, T.; Tsukahara, H. Roles of Oxidative Injury and Nitric Oxide System Derangements in Kawasaki Disease Pathogenesis: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 15450. https://doi.org/10.3390/ijms242015450
Tsuge M, Uda K, Eitoku T, Matsumoto N, Yorifuji T, Tsukahara H. Roles of Oxidative Injury and Nitric Oxide System Derangements in Kawasaki Disease Pathogenesis: A Systematic Review. International Journal of Molecular Sciences. 2023; 24(20):15450. https://doi.org/10.3390/ijms242015450
Chicago/Turabian StyleTsuge, Mitsuru, Kazuhiro Uda, Takahiro Eitoku, Naomi Matsumoto, Takashi Yorifuji, and Hirokazu Tsukahara. 2023. "Roles of Oxidative Injury and Nitric Oxide System Derangements in Kawasaki Disease Pathogenesis: A Systematic Review" International Journal of Molecular Sciences 24, no. 20: 15450. https://doi.org/10.3390/ijms242015450
APA StyleTsuge, M., Uda, K., Eitoku, T., Matsumoto, N., Yorifuji, T., & Tsukahara, H. (2023). Roles of Oxidative Injury and Nitric Oxide System Derangements in Kawasaki Disease Pathogenesis: A Systematic Review. International Journal of Molecular Sciences, 24(20), 15450. https://doi.org/10.3390/ijms242015450