
ACP-BC: A Model for Accurate Identification of Anticancer Peptides Based on Fusion Features of Bidirectional Long Short-Term Memory and Chemically Derived Information

Abstract
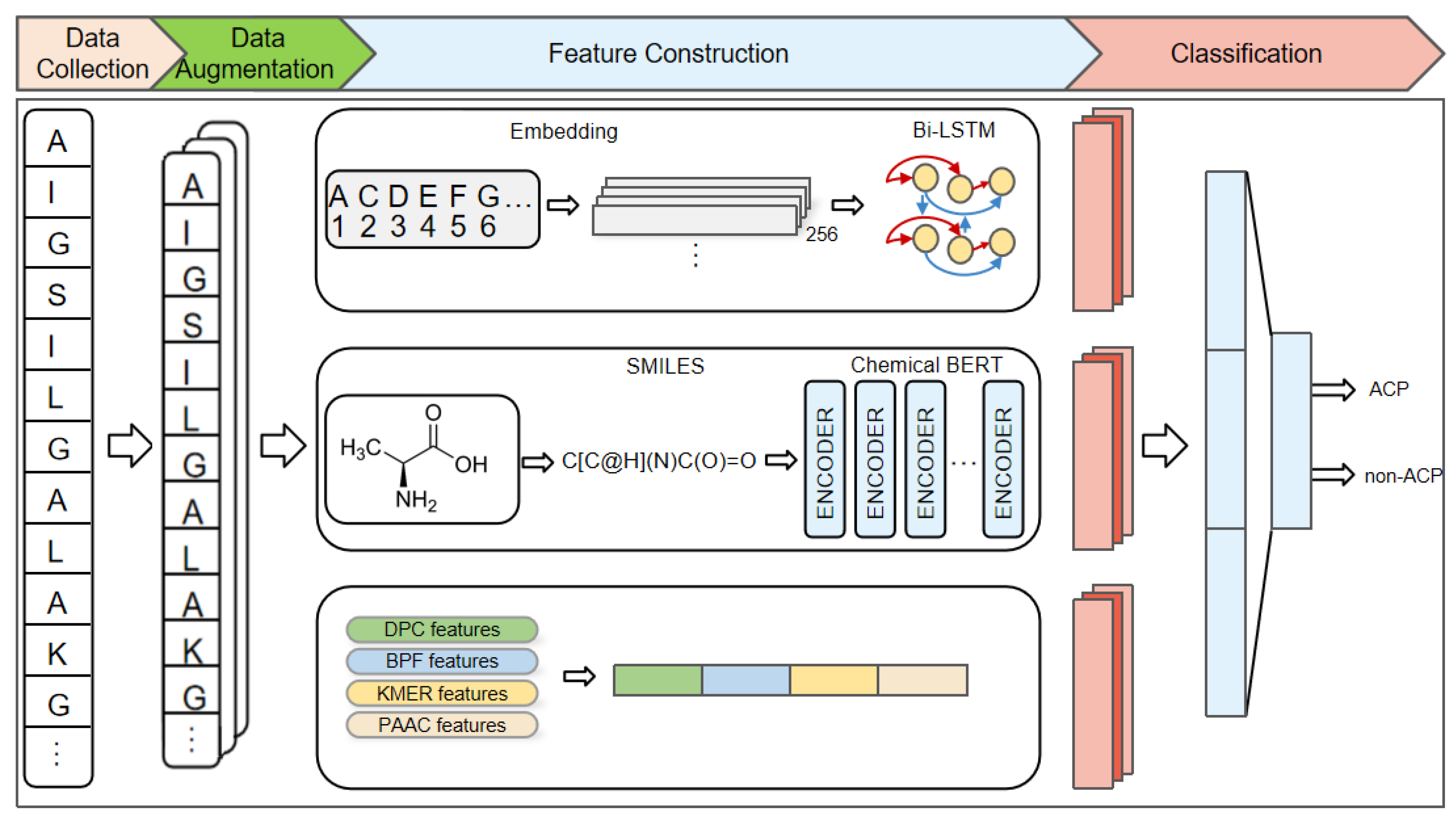
:1. Introduction
2. Results
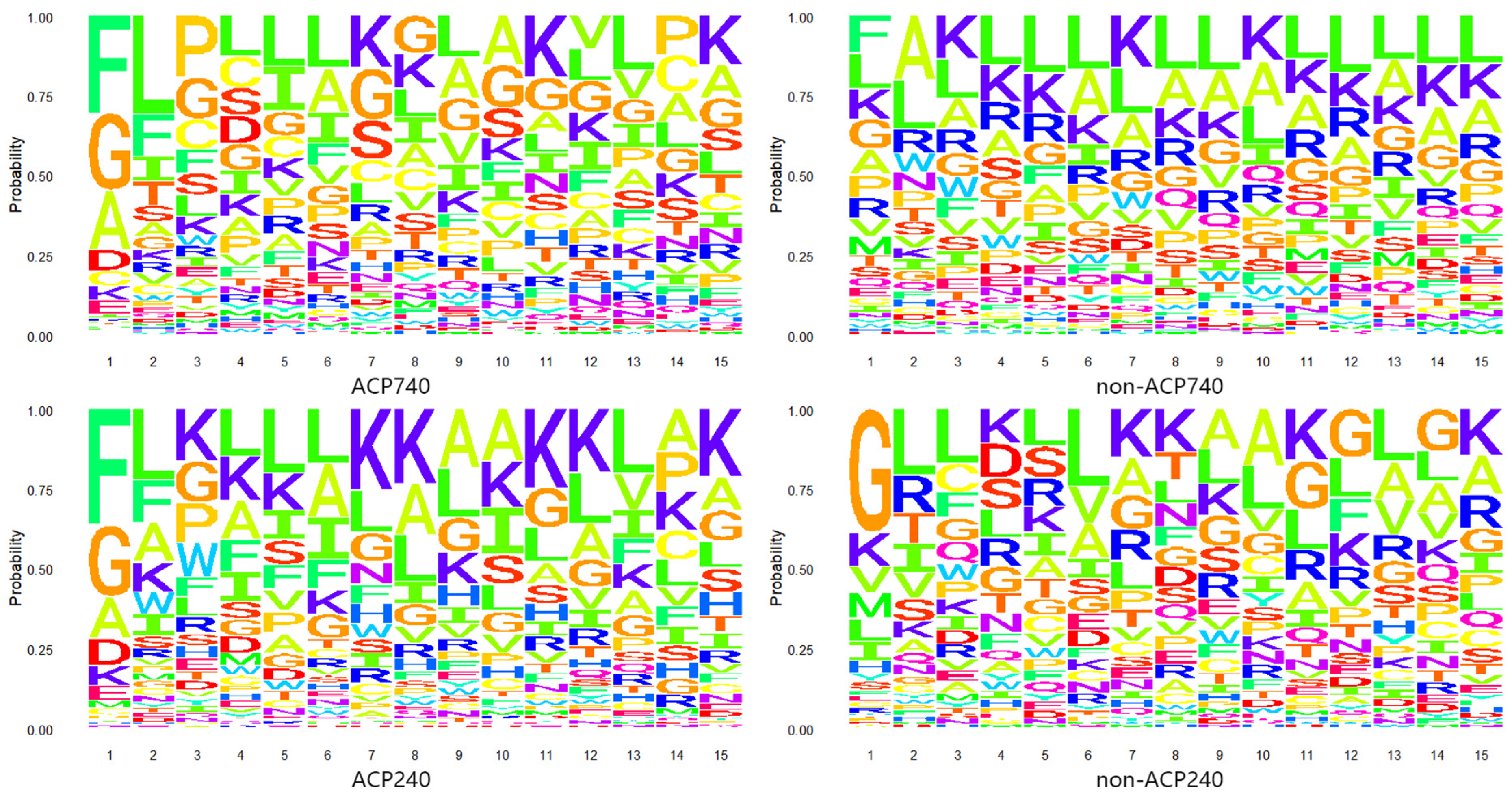
2.1. Analysis of Amino Acid Composition
2.2. Parameters of ACP-BC
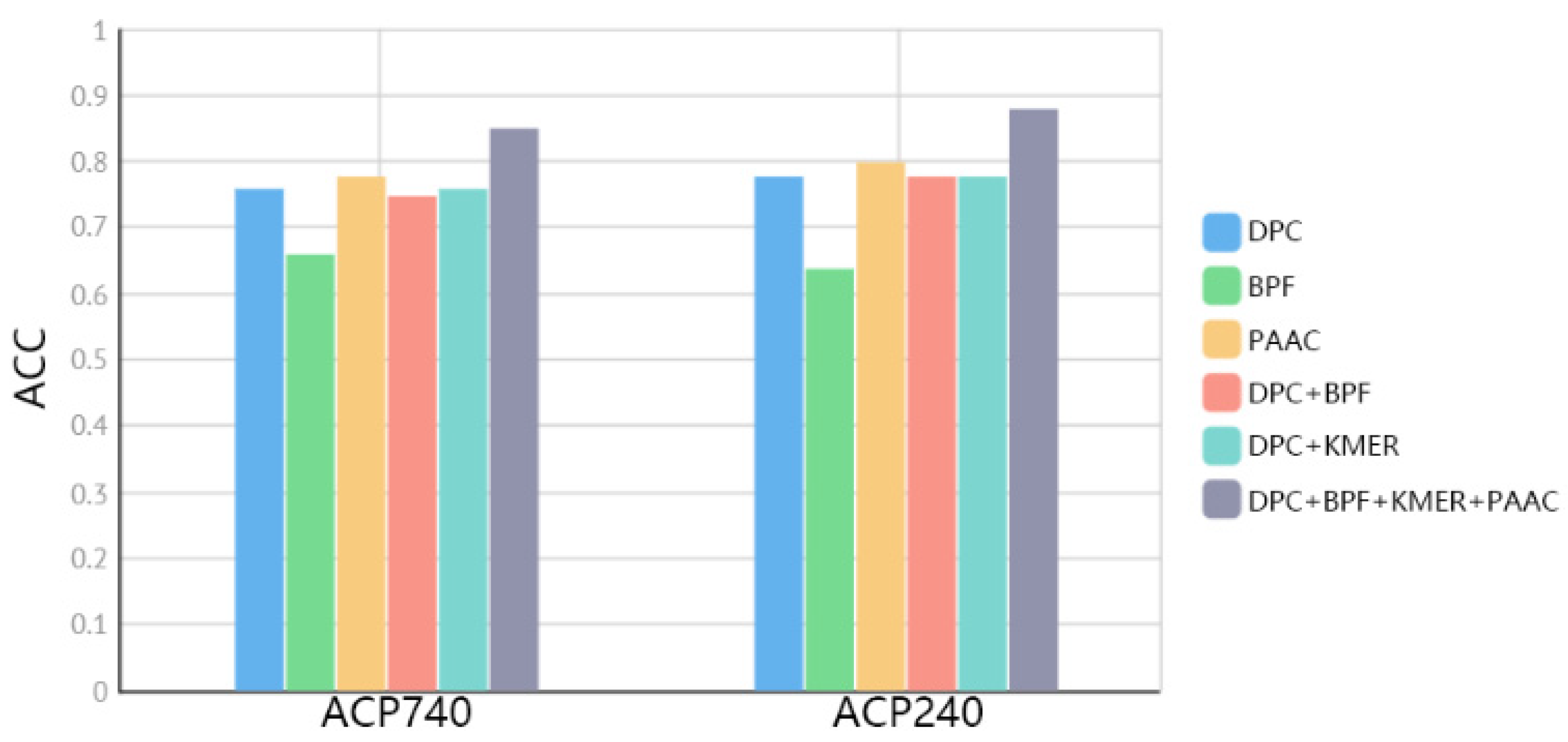
2.3. Comparison of Fused Features
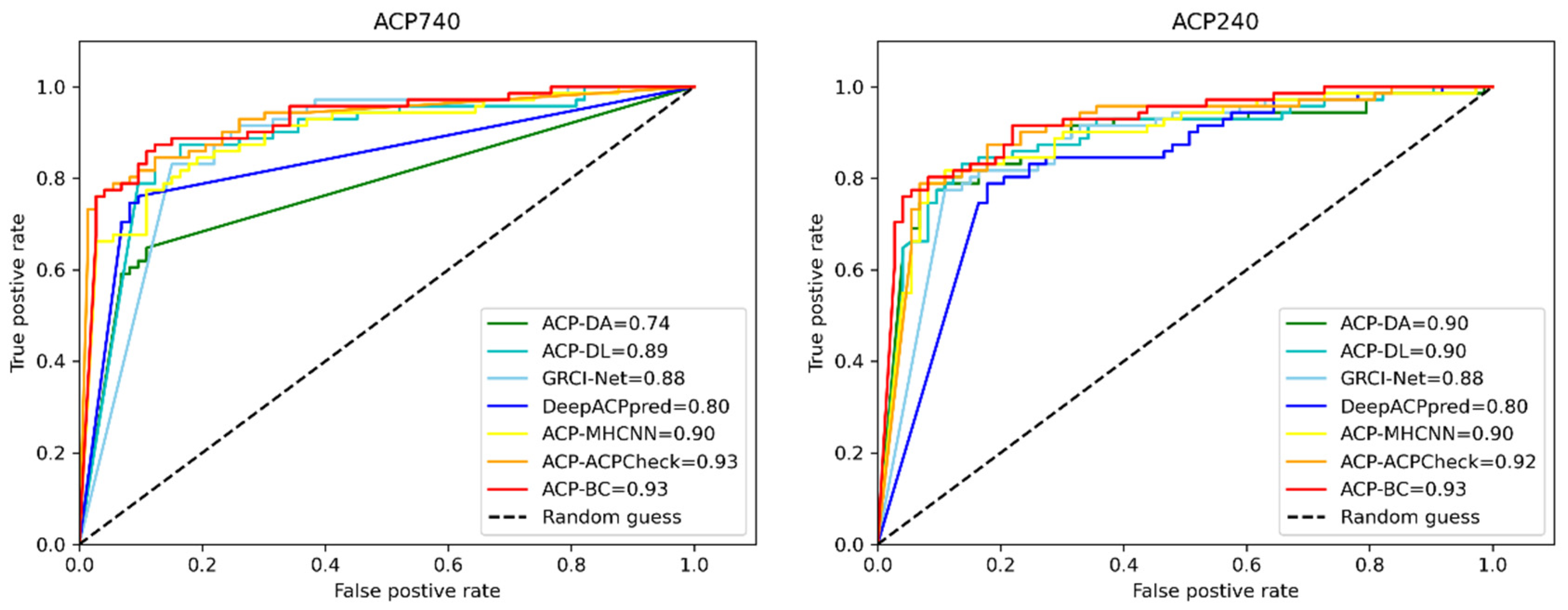
2.4. Comparison of Existing Methods
2.5. Independent Validation
2.5.1. Independently Validating on the ACPred-Fuse Dataset
2.5.2. Independently Validating on the ACP20 Dataset
3. Discussion
4. Materials and Methods
4.1. Dataset
4.1.1. Dataset ACP740
4.1.2. Dataset ACP240
4.1.3. Independent Validation Dataset ACP164
4.1.4. Independent Validation Dataset ACPred-Fuse
4.1.5. Independent Validation DatasetACP20
4.2. Data Augmentation
4.2.1. Replacement
4.2.2. Local Random Shuffling
4.2.3. Sequence Reversion
4.2.4. Combining Augmentations
4.3. Encoding and Embedding Representations of Amino Acid Sequences
4.3.1. BPF
4.3.2. DPC
4.3.3. PAAC
4.3.4. K-mer Sparse Matrix
4.4. Bi-LSTM
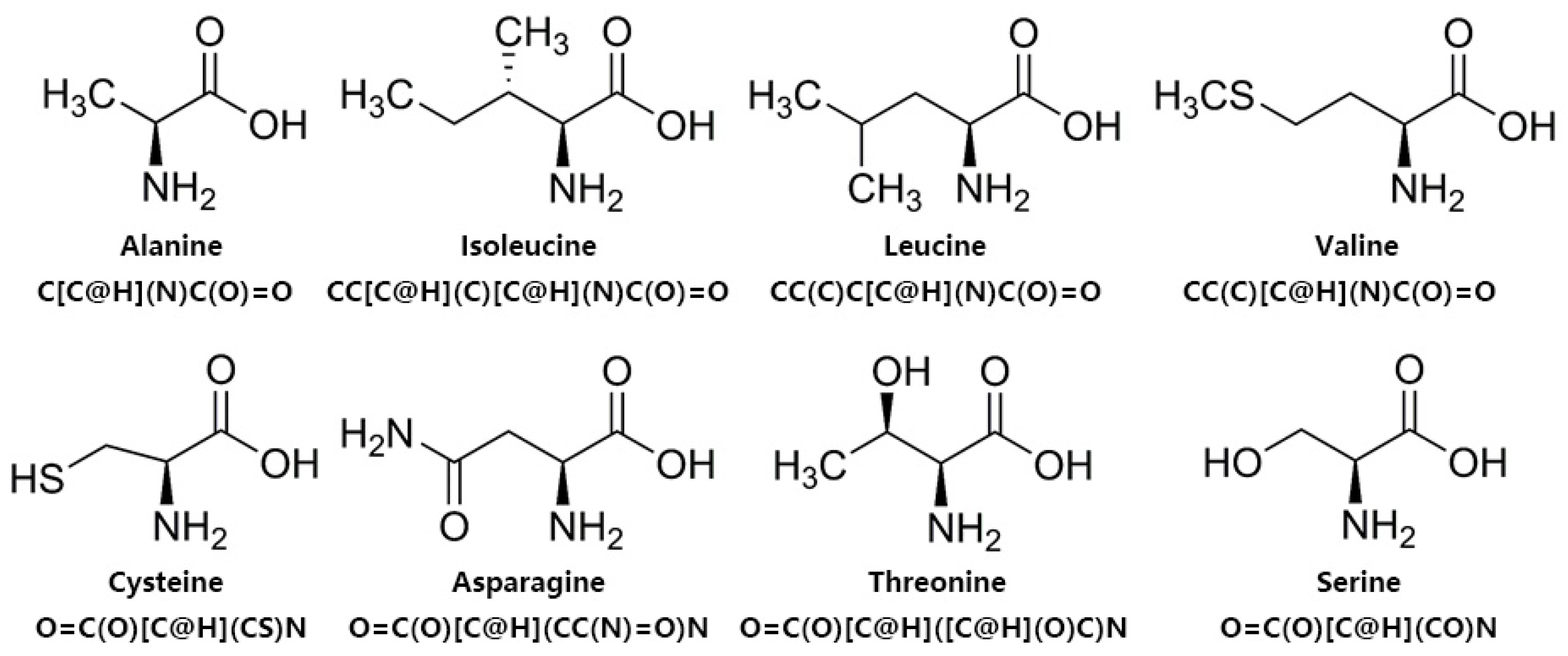
4.5. Chemical BERT
4.6. Performance Evaluation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACPs | Anticancer peptides | |
| non-ACPs | Non-Anticancer peptides | |
| AAC | amino acid composition | |
| DPC | dipeptide composition | |
| BPF | binary profiles feature | |
| PAAC | pseudo-amino acid composition | |
| g-gap DPC | g-gap dipeptide composition | |
| RBF | radial basis function | |
| ATC | atomic composition | |
| RAAAC | reduced amino acid alphabet composition | |
| CTD | composition-transition-distribution | |
| QSO | quasi-sequence-order | |
| AAIF | amino acid index | |
| GAAC | grouped amino acid composition | |
| Am-PAAC | amphiphilic pseudo amino acid composition | |
| OPF | overlap property feature | |
| TOBF | twenty-one-bit feature | |
| AKDC | daptive skip dipeptide composition | |
| CNN | convolutional neural networks | |
| GCN | graph convolutional networks | |
| SVM | support vector machine | |
| RF | Random Forest | |
| PNN | probability neural network | |
| GRNN | generalized regression neural network | |
| KNN | k-nearest neighbors | |
| RNN | recurrent neural network | |
| LSTM | long short-term memory | |
| CKSAAGP | k-spaced amino acid group pairs | |
| Bi-LSTM | bidirectional long short-term memory network | |
| NLP | natural language processing | |
| SMILES | Simplified Molecular Input Line Entry System | |
| BERT | bidirectional encoder representation transformer | |
| RoBerta | robustly optimized BERT pre-training approach | |
| BPE | byte pair encoding | |
| ChemBerta + ST | ChemBERTa with a SMILES tokenizer | |
| ChemBerta + BT | ChemBERTa with a BPE’s tokenizer | |
| TP | True Positive | |
| FP | False Positive | |
| TN | True Negative | |
| FN | False Negative | |
| ACC | accuracy | |
| MCC | Matthews correlation | |
| SE | sensitivity | |
| SP | specificity | |
| AUC | area under curve | |
| ROC | receiver operating characteristic |
References
- Arnold, M.; Karim-Kos, H.E.; Coebergh, J.W.; Byrnes, G.; Antilla, A.; Ferlay, J.; Renehan, A.G.; Forman, D.; Soerjomataram, I. Recent trends in incidence of five common cancers in 26 European countries since 1988: Analysis of the European Cancer Observatory. Eur. J. Cancer 2013, 51, 1164–1187. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Basith, S.; Shin, T.H.; Choi, S.; Kim, M.O.; Lee, G. MLACP: Machine-learning-based prediction of anticancer peptides. Oncotarget 2017, 8, 77121–77136. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Yaghoubi, A.; Khazaei, M.; Avan, A.; Hasanian, S.M.; Cho, W.C.; Soleimanpour, S. p28 bacterial peptide, as an anticancer agent. Front. Oncol. 2020, 10, 1303. [Google Scholar] [CrossRef]
- Wang, J.-J.; Lei, K.-F.; Han, F. Tumor microenvironment: Recent advances in various cancer treatments. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3855–3864. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Awasthi, R.; Malviya, R. Bioinspired microrobots: Opportunities and challenges in targeted cancer therapy. J. Control. Release 2023, 354, 439–452. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Singh, M.; Kumar, V.; Sikka, K.; Thakur, R.; Harioudh, M.K.; Mishra, D.P.; Ghosh, J.K.; Siddiqi, M.I. Computational Design of Biologically Active Anticancer Peptides and Their Interactions with Heterogeneous POPC/POPS Lipid Membranes. J. Chem. Inf. Model. 2019, 60, 332–341. [Google Scholar] [CrossRef]
- Tyagi, A.; Kapoor, P.; Kumar, R.; Chaudhary, K.; Gautam, A.; Raghava, G.P.S. In Silico Models for Designing and Discovering Novel Anticancer Peptides. Sci. Rep. 2013, 3, srep02984. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Peelle, B.; Lorens, J.; Li, W.; Bogenberger, J.; Payan, D.G.; Anderson, D. Intracellular protein scaffold-mediated display of random peptide libraries for phenotypic screens in mammalian cells. Chem. Biol. 2001, 8, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Norman, T.C.; Smith, D.L.; Sorger, P.K.; Drees, B.L.; O’Rourke, S.M.; Hughes, T.R.; Roberts, C.J.; Friend, S.H.; Fields, S.; Murray, A.W. Genetic Selection of Peptide Inhibitors of Biological Pathways. Science 1999, 285, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, S.M.H.; Chen, W.; Liu, Y.; Awal, A.; Ahmed, K.; Rahman, H.; Moni, M.A. PreDTIs: Prediction of drug–target interactions based on multiple feature information using gradient boosting framework with data balancing and feature selection techniques. Brief. Bioinform. 2021, 22, bbab046. [Google Scholar] [CrossRef]
- Shoombuatong, W.; Schaduangrat, N.; Pratiwi, R.; Nantasenamat, C. THPep: A machine learning-based approach for predicting tumor homing peptides. Comput. Biol. Chem. 2019, 80, 441–451. [Google Scholar] [CrossRef]
- Chen, J.; Cheong, H.H.; Siu, S.W.I. xDeep-AcPEP: Deep Learning Method for Anticancer Peptide Activity Prediction Based on Convolutional Neural Network and Multitask Learning. J. Chem. Inf. Model. 2021, 61, 3789–3803. [Google Scholar] [CrossRef]
- Han, G.-S.; Yu, Z.-G.; Anh, V. A two-stage SVM method to predict membrane protein types by incorporating amino acid classifications and physicochemical properties into a general form of Chou’s PseAAC. J. Theor. Biol. 2014, 344, 31–39. [Google Scholar] [CrossRef]
- Wang, G.; Vaisman, I.I.; van Hoek, M.L. Machine Learning Prediction of Antimicrobial Peptides. Methods Mol. Biol. 2022, 2405, 1–37. [Google Scholar] [CrossRef]
- Grisoni, F.; Neuhaus, C.S.; Gabernet, G.; Müller, A.T.; Hiss, J.A.; Schneider, G. Designing Anticancer Peptides by Constructive Machine Learning. ChemMedChem 2018, 13, 1300–1302. [Google Scholar] [CrossRef]
- Bhasin, M.; Raghava, G.P.S. Classification of Nuclear Receptors Based on Amino Acid Composition and Dipeptide Composition. J. Biol. Chem. 2004, 279, 23262–23266. [Google Scholar] [CrossRef]
- Saravanan, V.; Gautham, N. Harnessing Computational Biology for Exact Linear B-Cell Epitope Prediction: A Novel Amino Acid Composition-Based Feature Descriptor. OMICS J. Integr. Biol. 2015, 19, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Gautam, A.; Chaudhary, K.; Kumar, R.; Sharma, A.; Kapoor, P.; Tyagi, A. In silico approaches for designing highly effective cell penetrating peptides. J. Transl. Med. 2013, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- Hajisharifi, Z.; Piryaiee, M.; Beigi, M.M.; Behbahani, M.; Mohabatkar, H. Predicting anticancer peptides with Chou′s pseudo amino acid composition and investigating their mutagenicity via Ames test. J. Theor. Biol. 2014, 341, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Amanat, S.; Ashraf, A.; Hussain, W.; Rasool, N.; Khan, Y.D. Identification of lysine carboxylation sites in proteins by integrating statistical moments and position relative features via general PAAC. Curr. Bioinform. 2020, 15, 396–407. [Google Scholar] [CrossRef]
- Hasan, A.M.; Ben Islam, K.; Rahman, J.; Ahmad, S. Citrullination Site Prediction by Incorporating Sequence Coupled Effects into PseAAC and Resolving Data Imbalance Issue. Curr. Bioinform. 2020, 15, 235–245. [Google Scholar] [CrossRef]
- Naseer, S.; Hussain, W.; Khan, Y.D.; Rasool, N. Sequence-based Identification of Arginine Amidation Sites in Proteins Using Deep Representations of Proteins and PseAAC. Curr. Bioinform. 2021, 15, 937–948. [Google Scholar] [CrossRef]
- Shen, H.-B.; Chou, K.-C. PseAAC: A flexible web server for generating various kinds of protein pseudo amino acid composition. Anal. Biochem. 2008, 373, 386–388. [Google Scholar] [CrossRef]
- Chou, K.-C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins Struct. Funct. Bioinform. 2001, 43, 246–255. [Google Scholar] [CrossRef]
- Vijayakumar, S.; Ptv, L. ACPP: A Web Server for Prediction and Design of Anti-cancer Peptides. Int. J. Pept. Res. Ther. 2014, 21, 99–106. [Google Scholar] [CrossRef]
- Chen, W.; Ding, H.; Feng, P.; Lin, H.; Chou, K.-C. iACP: A sequence-based tool for identifying anticancer peptides. Oncotarget 2016, 7, 16895–16909. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Akbar, S.; Hayat, M.; Iqbal, M.; Jan, M.A. iACP-GAEnsC: Evolutionary genetic algorithm based ensemble classification of anticancer peptides by utilizing hybrid feature space. Artif. Intell. Med. 2017, 79, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Zhou, C.; Su, R.; Zou, Q. PEPred-Suite: Improved and robust prediction of therapeutic peptides using adaptive feature representation learning. Bioinformatics 2019, 35, 4272–4280. [Google Scholar] [CrossRef] [PubMed]
- Boopathi, V.; Subramaniyam, S.; Malik, A.; Lee, G.; Manavalan, B.; Yang, D.-C. mACPpred: A Support Vector Machine-Based Meta-Predictor for Identification of Anticancer Peptides. Int. J. Mol. Sci. 2019, 20, 1964. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, W.; Wang, D.; Wang, S.; Li, Q. Prediction of Anticancer Peptides Using a Low-Dimensional Feature Model. Front. Bioeng. Biotechnol. 2020, 8, 892. [Google Scholar] [CrossRef]
- Xu, L.; Liang, G.; Wang, L.; Liao, C. A Novel Hybrid Sequence-Based Model for Identifying Anticancer Peptides. Genes 2018, 9, 158. [Google Scholar] [CrossRef]
- Schaduangrat, N.; Nantasenamat, C.; Prachayasittikul, V.; Shoombuatong, W. ACPred: A Computational Tool for the Prediction and Analysis of Anticancer Peptides. Molecules 2019, 24, 1973. [Google Scholar] [CrossRef]
- Wei, L.; Zhou, C.; Chen, H.; Song, J.; Su, R. ACPred-FL: A sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics 2018, 34, 4007–4016. [Google Scholar] [CrossRef]
- Liang, X.; Li, F.; Chen, J.; Li, J.; Wu, H.; Li, S.; Song, J.; Liu, Q. Large-scale comparative review and assessment of computational methods for anti-cancer peptide identification. Briefings Bioinform. 2020, 22, bbaa312. [Google Scholar] [CrossRef]
- Ahmed, S.; Muhammod, R.; Khan, Z.H.; Adilina, S.; Sharma, A.; Shatabda, S.; Dehzangi, A. ACP-MHCNN: An accurate multi-headed deep-convolutional neural network to predict anticancer peptides. Sci. Rep. 2021, 11, 23676. [Google Scholar] [CrossRef]
- Wu, C.; Gao, R.; Zhang, Y.; De Marinis, Y. PTPD: Predicting therapeutic peptides by deep learning and word2vec. BMC Bioinform. 2019, 20, 456. [Google Scholar] [CrossRef] [PubMed]
- You, Z.-H.; Zhou, M.; Luo, X.; Li, S. Highly Efficient Framework for Predicting Interactions Between Proteins. IEEE Trans. Cybern. 2016, 47, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.-C.; You, Z.-H.; Zhou, X.; Cheng, L.; Li, X.; Jiang, T.-H.; Chen, Z.-H. ACP-DL: A Deep Learning Long Short-Term Memory Model to Predict Anticancer Peptides Using High-Efficiency Feature Representation. Mol. Ther. Nucleic Acids 2019, 17, 1–9. [Google Scholar] [CrossRef]
- Cao, R.; Wang, M.; Bin, Y.; Zheng, C. DLFF-ACP: Prediction of ACPs based on deep learning and multi-view features fusion. PeerJ 2021, 9, e11906. [Google Scholar] [CrossRef]
- Sun, M.; Yang, S.; Hu, X.; Zhou, Y. ACPNet: A Deep Learning Network to Identify Anticancer Peptides by Hybrid Sequence Information. Molecules 2022, 27, 1544. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, J.; Zhao, H.; Li, H.; Wang, J. CL-ACP: A parallel combination of CNN and LSTM anticancer peptide recognition model. BMC Bioinform. 2021, 22, 512. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-G.; Zhang, W.; Yang, X.; Li, C.; Chen, H. ACP-DA: Improving the Prediction of Anticancer Peptides Using Data Augmentation. Front. Genet. 2021, 12, 698477. [Google Scholar] [CrossRef]
- Rao, B.; Zhang, L.; Zhang, G. ACP-GCN: The Identification of Anticancer Peptides Based on Graph Convolution Networks. IEEE Access 2020, 8, 176005–176011. [Google Scholar] [CrossRef]
- Zhu, L.; Ye, C.; Hu, X.; Yang, S.; Zhu, C. ACP-check: An anticancer peptide prediction model based on bidirectional long short-term memory and multi-features fusion strategy. Comput. Biol. Med. 2022, 148, 105868. [Google Scholar] [CrossRef]
- You, H.; Yu, L.; Tian, S.; Ma, X.; Xing, Y.; Song, J.; Wu, W. Anti-cancer Peptide Recognition Based on Grouped Sequence and Spatial Dimension Integrated Networks. Interdiscip. Sci. Comput. Life Sci. 2021, 14, 196–208. [Google Scholar] [CrossRef]
- Lane, N.; Kahanda, I. DeepACPpred: A novel hybrid CNN-RNN architecture for predicting anti-cancer peptides. In Practical Applications of Computational Biology & Bioinformatics, 14th International Conference (PACBB 2020) 14; Springer International Publishing: Berlin/Heidelberg, Germany, 2021. [Google Scholar]
- Yu, L.; Jing, R.; Liu, F.; Luo, J.; Li, Y. DeepACP: A Novel Computational Approach for Accurate Identification of Anticancer Peptides by Deep Learning Algorithm. Mol. Ther. Nucleic Acids 2020, 22, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yang, Z.; Yang, J. 4mCBERT: A computing tool for the identification of DNA N4-methylcytosine sites by sequence- and chemical-derived information based on ensemble learning strategies. Int. J. Biol. Macromol. 2023, 231, 123180. [Google Scholar] [CrossRef] [PubMed]
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K. Bert: Pre-training of deep bidirectional transformers for language understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Hirohara, M.; Saito, Y.; Koda, Y.; Sato, K.; Sakakibara, Y. Convolutional neural network based on SMILES representation of compounds for detecting chemical motif. BMC Bioinform. 2018, 19, 526. [Google Scholar] [CrossRef]
- Quirós, M.; Gražulis, S.; Girdzijauskaitė, S.; Merkys, A.; Vaitkus, A. Using SMILES strings for the description of chemical connectivity in the Crystallography Open Database. J. Cheminform. 2018, 10, 23. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Tareen, A.; Kinney, J.B. Logomaker: Beautiful sequence logos in Python. Bioinformatics 2019, 36, 2272–2274. [Google Scholar] [CrossRef]
- Chithrananda, S.; Grand, G.; Ramsundar, B. ChemBERTa: Large-Scale Self-Supervised Pretraining for Molecular Property Prediction. arXiv 2020, arXiv:2010.09885. [Google Scholar] [CrossRef]
- Liu, Y.; Ott, M.; Goyal, N.; Du, J.; Joshi, M.; Chen, D.; Levy, O.; Lewis, M.; Zettlemoyer, L.; Stoyanov, V. Roberta: A robustly optimized BERT pretraining approach. arXiv 2019, arXiv:1907.11692. [Google Scholar]
- Radford, A.; Kim, J.W.; Xu, T.; Brockman, G.; McLeavey, C.; Sutskever, I. Robust speech recognition via large-scale weak supervision. arXiv 2022, arXiv:2212.04356. [Google Scholar]
- Rao, B.; Zhou, C.; Zhang, G.; Su, R.; Wei, L. ACPred-Fuse: Fusing multi-view information improves the prediction of anticancer peptides. Briefings Bioinform. 2019, 21, 1846–1855. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Tuknait, A.; Anand, P.; Gupta, S.; Sharma, M.; Mathur, D.; Joshi, A.; Singh, S.; Gautam, A.; Raghava, G.P.S. CancerPPD: A database of anticancer peptides and proteins. Nucleic Acids Res. 2015, 43, D837–D843. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Price, L.C.; Bahadori, T.; Seeger, F. Improving generalizability of protein sequence models with data augmentations. bioRxiv 2021. [Google Scholar] [CrossRef]
- Le, N.Q.K.; Ho, Q.-T.; Nguyen, V.-N.; Chang, J.-S. BERT-Promoter: An improved sequence-based predictor of DNA promoter using BERT pre-trained model and SHAP feature selection. Comput. Biol. Chem. 2022, 99, 107732. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Combination | R | C | D | ACC | MCC | SE | SP |
|---|---|---|---|---|---|---|---|
| c1 | 1.0 | 128 | 256 | 0.72 | 0.44 | 0.75 | 0.70 |
| c2 | 1.0 | 256 | 256 | 0.80 | 0.62 | 0.88 | 0.73 |
| c3 | 1.0 | 256 | 512 | 0.81 | 0.62 | 0.84 | 0.78 |
| c4 | 1.0 | 512 | 512 | 0.80 | 0.59 | 0.79 | 0.80 |
| c5 | 2.0 | 128 | 256 | 0.76 | 0.52 | 0.77 | 0.79 |
| c6 | 2.0 | 256 | 512 | 0.80 | 0.60 | 0.83 | 0.76 |
| c7 | 2.0 | 512 | 512 | 0.75 | 0.50 | 0.76 | 0.74 |
| Dataset | BERT | ACC | MCC | SE | SP |
|---|---|---|---|---|---|
| ACP740 | ChemBerta + ST | 0.87 | 0.75 | 0.87 | 0.88 |
| ChemBerta + BT | 0.85 | 0.69 | 0.85 | 0.85 | |
| BERT-base | 0.86 | 0.70 | 0.85 | 0.86 | |
| ACP240 | ChemBerta + ST | 0.90 | 0.90 | 0.90 | 0.89 |
| ChemBerta + BT | 0.86 | 0.72 | 0.87 | 0.85 | |
| BERT-base | 0.89 | 0.76 | 0.88 | 0.88 |
| Dataset | Methods | ACC | MCC | SE | SP | AUC |
|---|---|---|---|---|---|---|
| ACP740 | ACP-DA | 0.81 | 0.58 | 0.80 | 0.82 | 0.74 |
| ACP-DL | 0.81 | 0.62 | 0.81 | 0.80 | 0.89 | |
| GRCI-Net | 0.82 | 0.65 | 0.84 | 0.82 | 0.88 | |
| DeepACPpred | 0.85 | 0.71 | 0.85 | 0.85 | 0.80 | |
| ACP-MHCNN | 0.86 | 0.72 | 0.89 | 0.83 | 0.90 | |
| ACPCheck | 0.87 | 0.75 | 0.86 | 0.88 | 0.93 | |
| ACP-BC (ours) | 0.87 | 0.75 | 0.87 | 0.88 | 0.93 | |
| ACP240 | ACP-DA | 0.89 | 0.78 | 0.88 | 0.89 | 0.90 |
| ACP-DL | 0.84 | 0.68 | 0.88 | 0.78 | 0.90 | |
| GRCI-Net | 0.88 | 0.75 | 0.89 | 0.88 | 0.88 | |
| DeepACPpred | 0.86 | 0.72 | 0.88 | 0.84 | 0.80 | |
| ACP-MHCNN | 0.83 | 0.67 | 0.90 | 0.76 | 0.90 | |
| ACPCheck | 0.89 | 0.77 | 0.91 | 0.85 | 0.92 | |
| ACP-BC (ours) | 0.90 | 0.78 | 0.90 | 0.89 | 0.93 |
| Dataset | Methods | ACC | MCC | SE | SP | AUC |
|---|---|---|---|---|---|---|
| AntiCP_ACC | 0.88 | 0.29 | 0.68 | 0.89 | 0.85 | |
| AntiCP_DC | 0.82 | 0.22 | 0.68 | 0.83 | 0.83 | |
| iACP | 0.88 | 0.23 | 0.55 | 0.89 | 0.76 | |
| ACPred-Fuse | ACPred-FL | 0.85 | 0.26 | 0.70 | 0.86 | 0.85 |
| DeepACP | 0.86 | 0.31 | 0.78 | 0.86 | 0.88 | |
| DLFF-ACP | 0.86 | 0.32 | 0.83 | 0.86 | 0.90 | |
| ACP-BC (ours) | 0.91 | 0.40 | 0.81 | 0.91 | 0.92 |
| Sequence | Score | Lable |
|---|---|---|
| KLWKKIEKLIKKLLTSIR | 0.85 | ACPs |
| YIWARAERVWLWWGKFLSL | 0.88 | ACPs |
| DLFKQLQRLFLGILYCLYKIW | 0.82 | ACPs |
| AIKKFGPLAKIVAKV | 0.68 | ACPs |
| RWNGRIIKGFYNLVKIWKDLKG | 0.93 | ACPs |
| KVWKIKKNIRRLLHGIKRGWKG | 0.73 | ACPs |
| GFWARIGKVFAAVKNL | 0.78 | ACPs |
| AFLYRLTRQIRPWWRWLYKW | 0.78 | ACPs |
| RIWGKHSRYIKIVKRLIQ | 0.92 | ACPs |
| QIWHKIRKLWQIIKDGF | 0.67 | ACPs |
| CGESCVWIPCVTSIFNCKCKENKVCYHDKIP | 0.16 | non-ACPs |
| SDEKASPDKHHRFSLSRYAKLANRLANPKLLETFLSKWIGDRGNRSV | 0.18 | non-ACPs |
| DVKGMKKAIKGILDCVIEKGYDKLAAKLKKVIQQLWE | 0.10 | non-ACPs |
| AGWGSIFKHIFKAGKFIHGAIQAHND | 0.03 | non-ACPs |
| ATCDLASGFGVGSSLCAAHCIARRYRGGYCNSKAVCVCRN | 0.05 | non-ACPs |
| GWKIGKKLEHHGQNIRDGLISAGPAVFAVGQAATIYAAAK | 0.36 | non-ACPs |
| FLGALIKGAIHGGRFIHGMIQNHH | 0.25 | non-ACPs |
| FLPAIAGILSQLF | 0.40 | non-ACPs |
| ALWMTLLKKVLKAAAKALNAVLVGANA | 0.12 | non-ACPs |
| EGGGPQWAVGHFM | 0.29 | non-ACPs |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, M.; Hu, H.; Pang, W.; Zhou, Y. ACP-BC: A Model for Accurate Identification of Anticancer Peptides Based on Fusion Features of Bidirectional Long Short-Term Memory and Chemically Derived Information. Int. J. Mol. Sci. 2023, 24, 15447. https://doi.org/10.3390/ijms242015447
Sun M, Hu H, Pang W, Zhou Y. ACP-BC: A Model for Accurate Identification of Anticancer Peptides Based on Fusion Features of Bidirectional Long Short-Term Memory and Chemically Derived Information. International Journal of Molecular Sciences. 2023; 24(20):15447. https://doi.org/10.3390/ijms242015447
Chicago/Turabian StyleSun, Mingwei, Haoyuan Hu, Wei Pang, and You Zhou. 2023. "ACP-BC: A Model for Accurate Identification of Anticancer Peptides Based on Fusion Features of Bidirectional Long Short-Term Memory and Chemically Derived Information" International Journal of Molecular Sciences 24, no. 20: 15447. https://doi.org/10.3390/ijms242015447
APA StyleSun, M., Hu, H., Pang, W., & Zhou, Y. (2023). ACP-BC: A Model for Accurate Identification of Anticancer Peptides Based on Fusion Features of Bidirectional Long Short-Term Memory and Chemically Derived Information. International Journal of Molecular Sciences, 24(20), 15447. https://doi.org/10.3390/ijms242015447