
Thrombin-Induced COX-2 Expression and PGE2 Synthesis in Human Tracheal Smooth Muscle Cells: Role of PKCδ/Pyk2-Dependent AP-1 Pathway Modulation
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
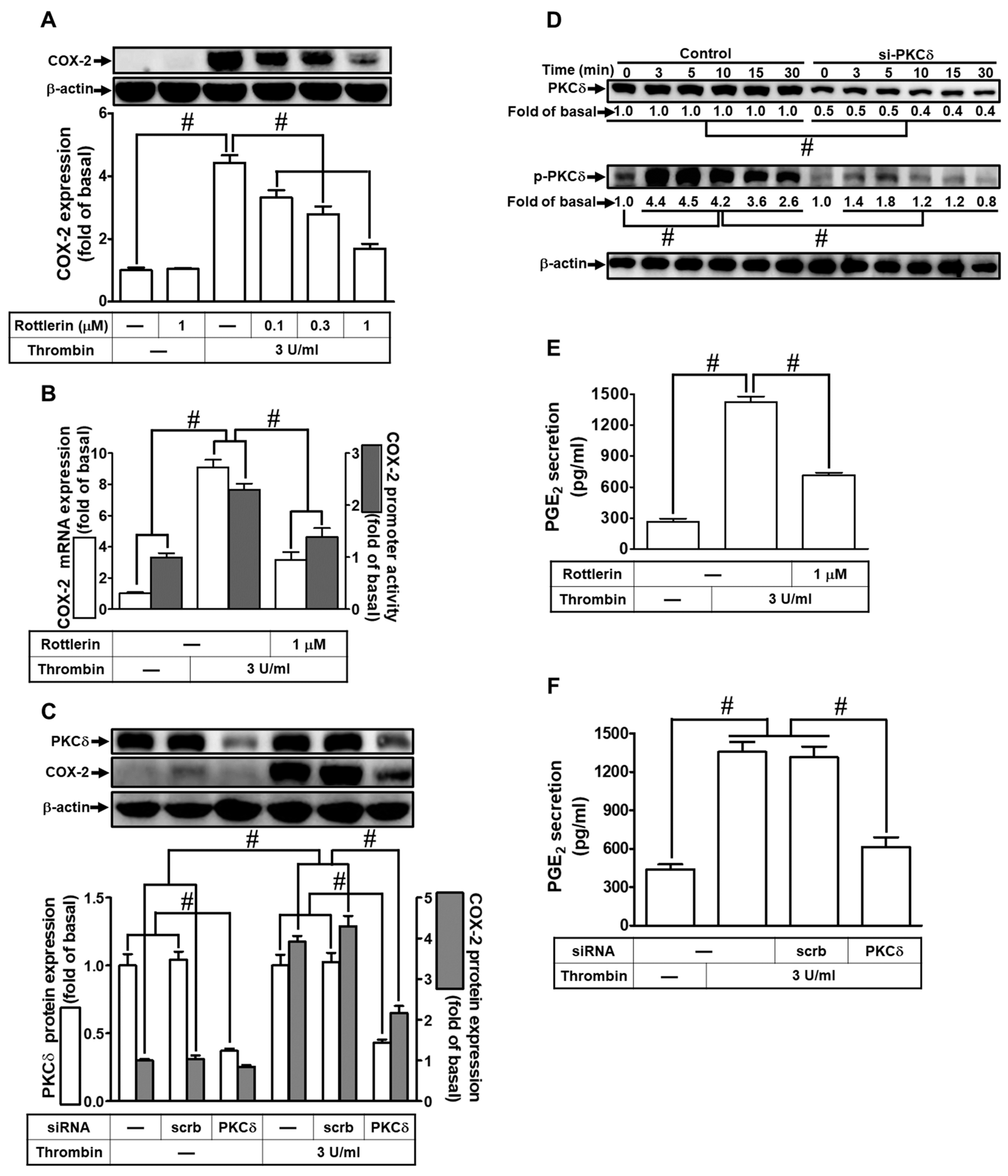
2.1. Thrombin Induces COX-2 Expression and PGE2 Release via PKCδ
2.2. Thrombin Enhances COX-2 Expression via PKCδ-Dependent Pyk2 Activation
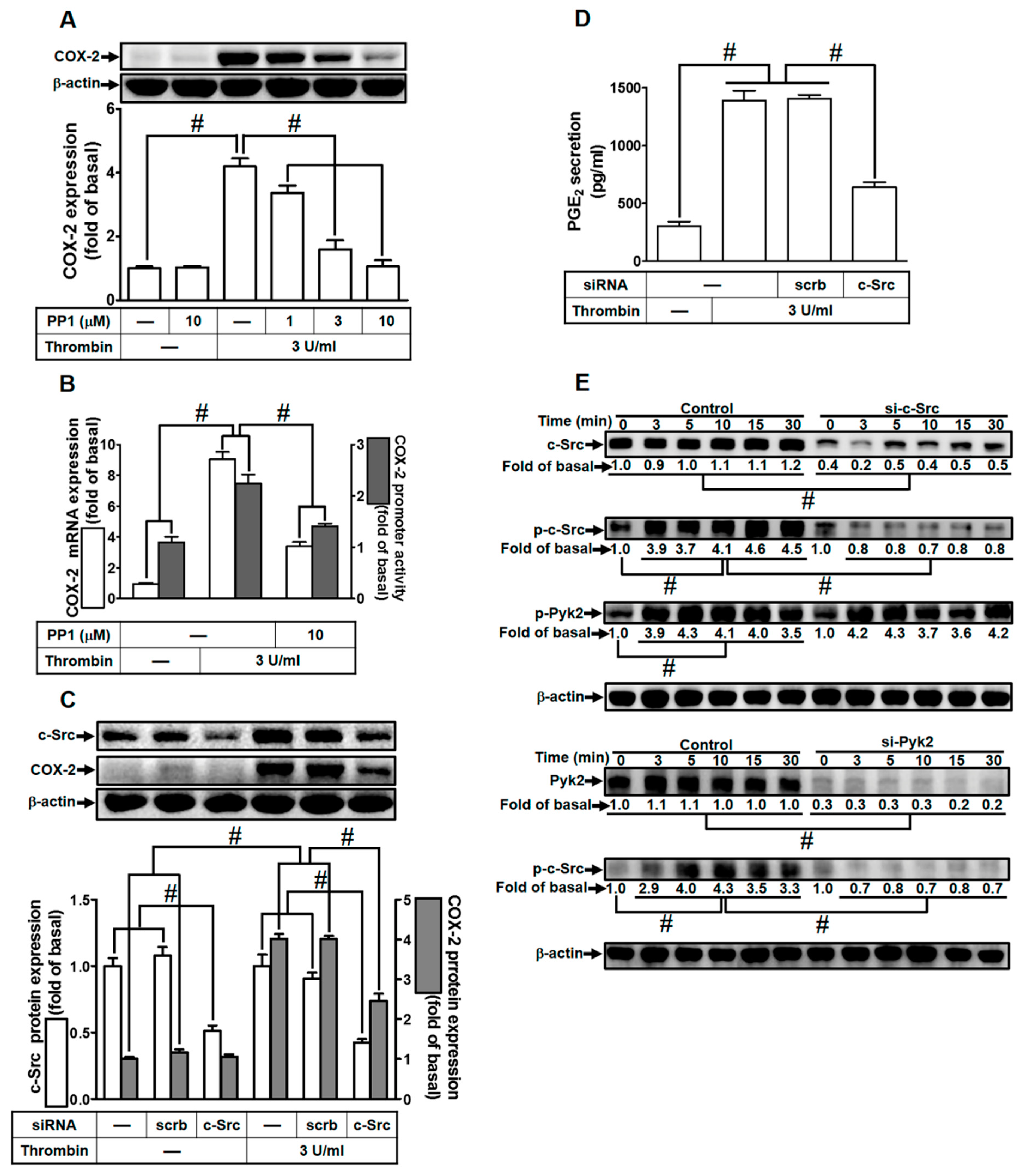
2.3. Thrombin Up-Regulates COX-2 Expression through c-Src Activation
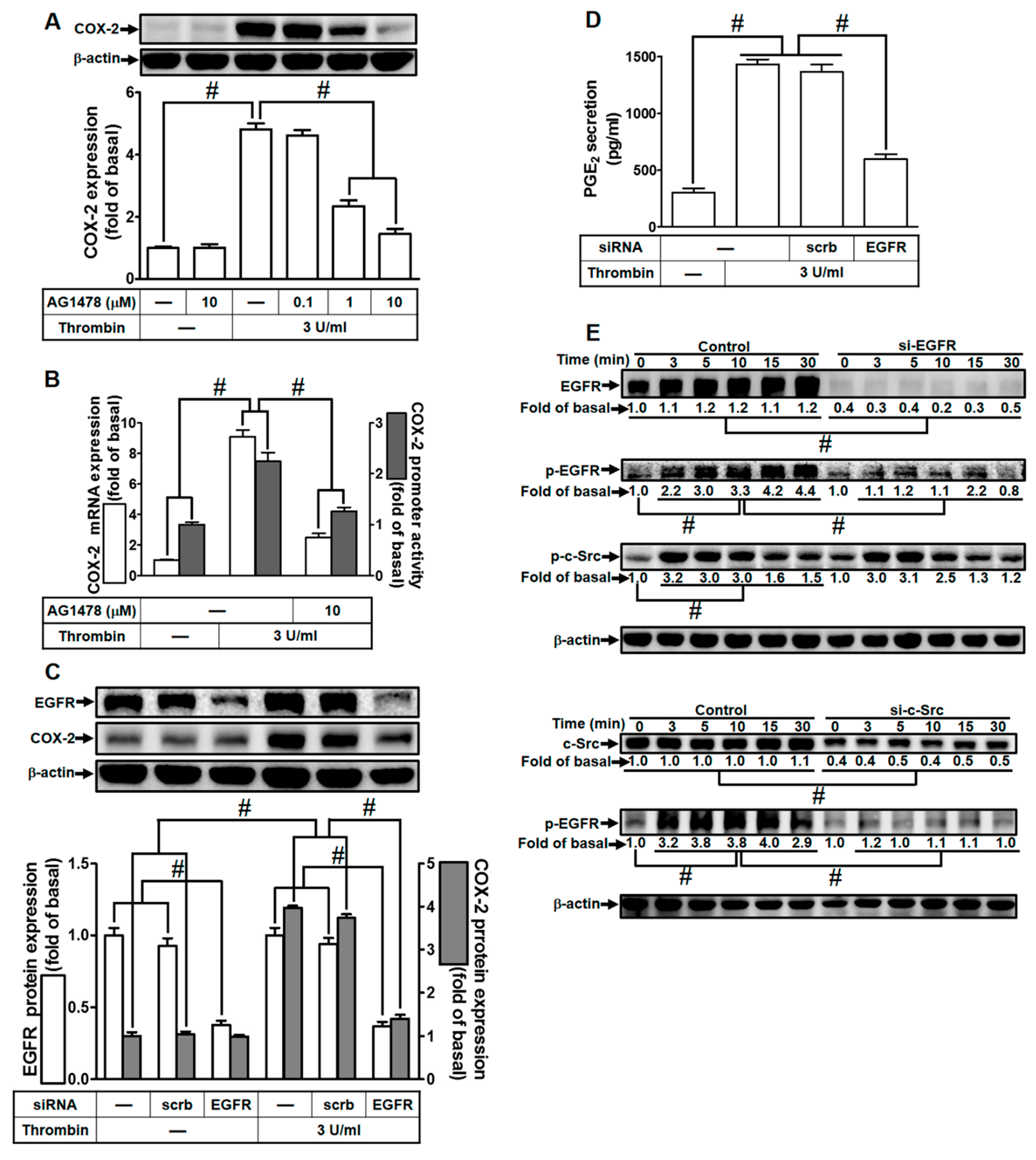
2.4. Thrombin Induces COX-2 Expression through EGFR Activation
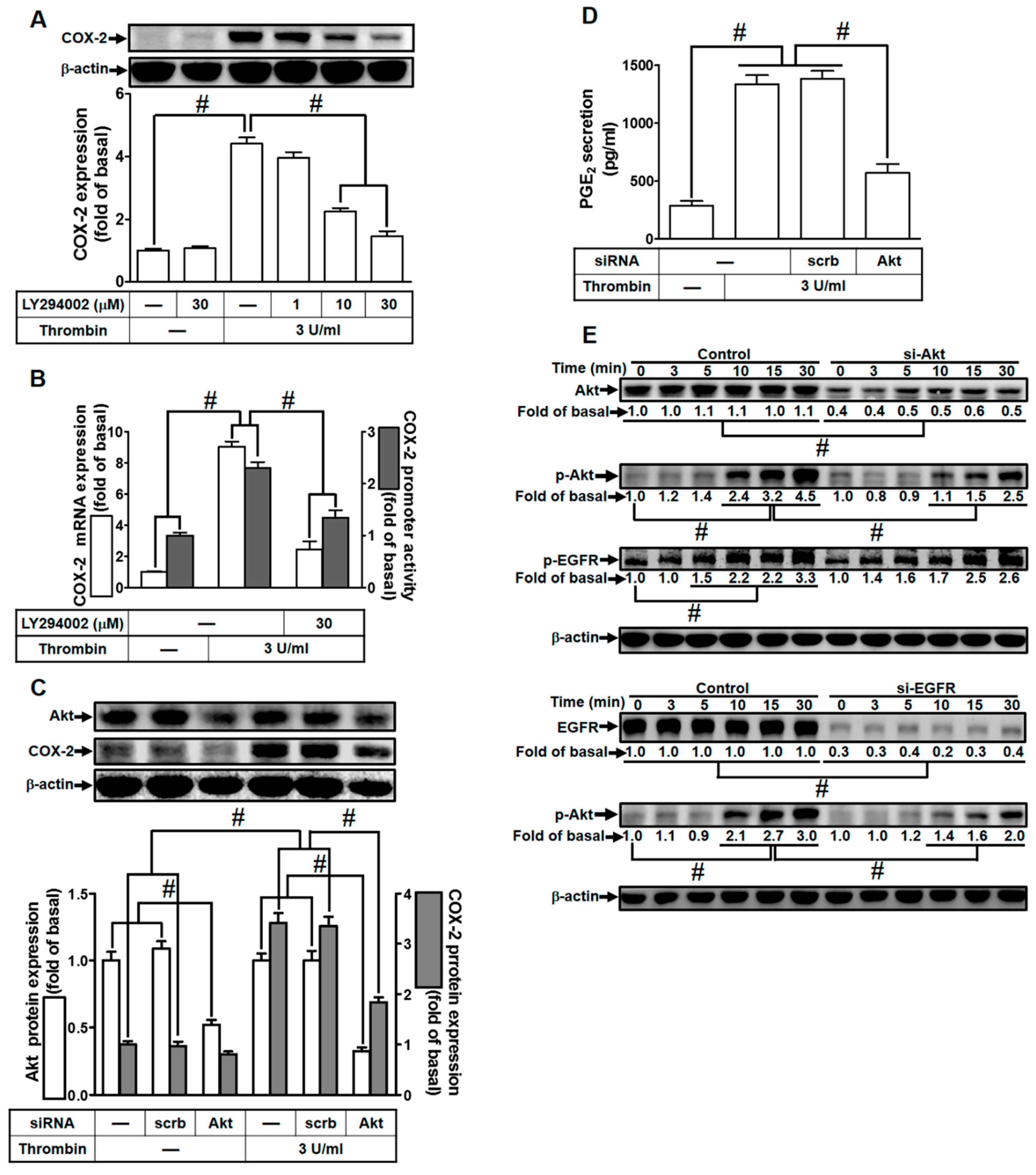
2.5. Thrombin Induces COX-2 Expression via PI3K/Akt
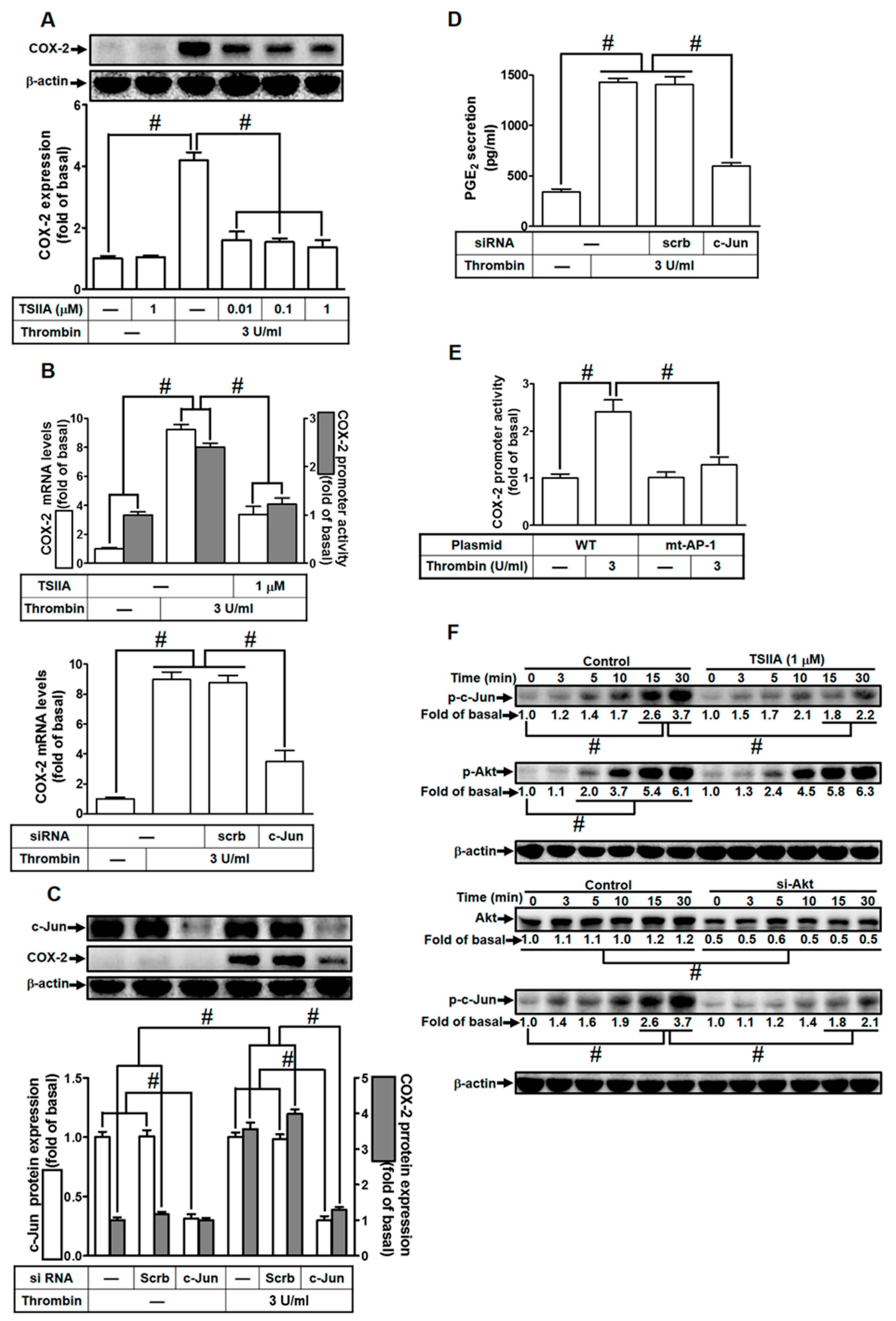
2.6. Thrombin Induces COX-2 Expression via AP-1
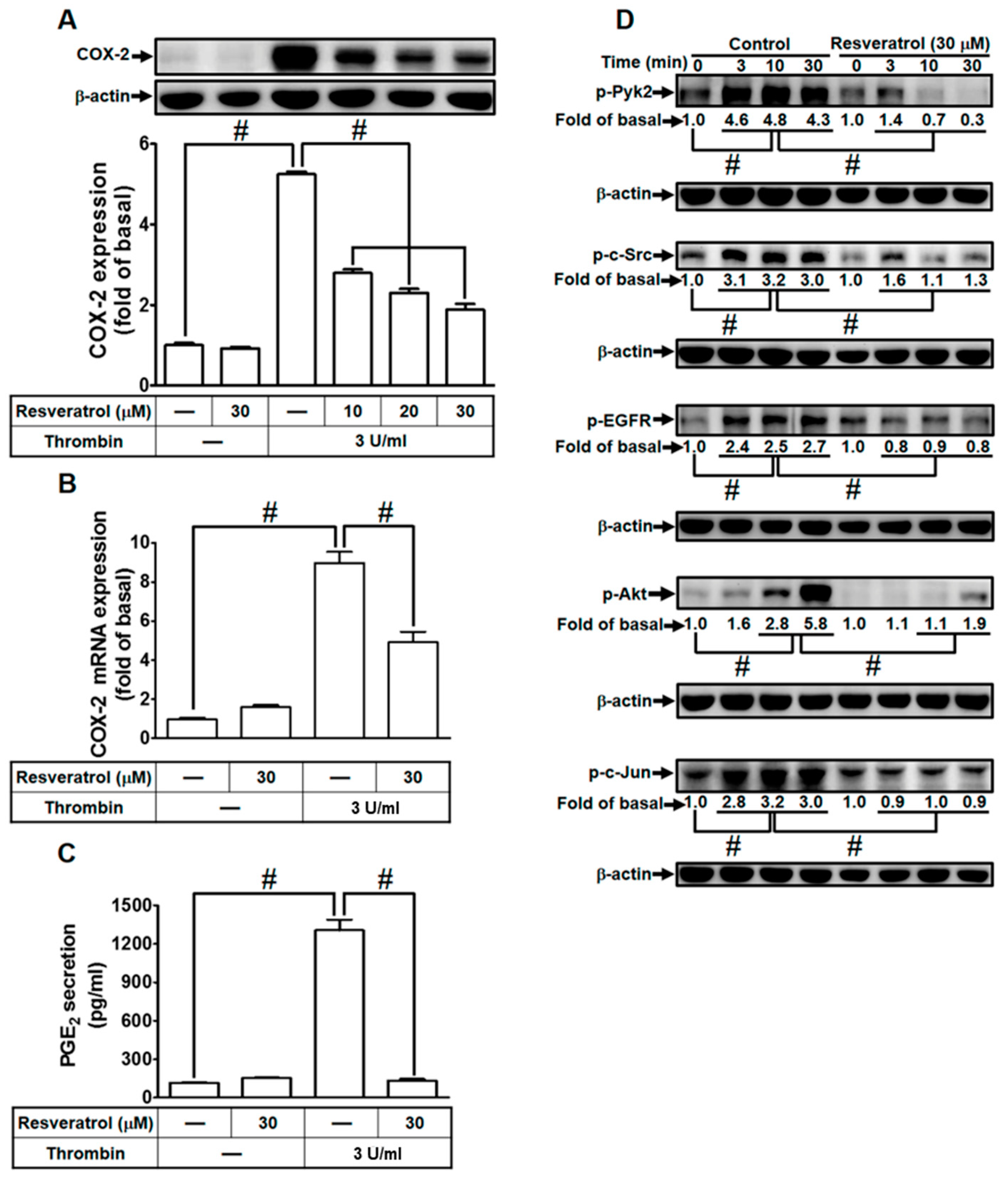
2.7. Resveratrol Blocks Protein Kinase Activation to Attenuate Thrombin-Induced COX-2 Expression and PGE2 Secretion
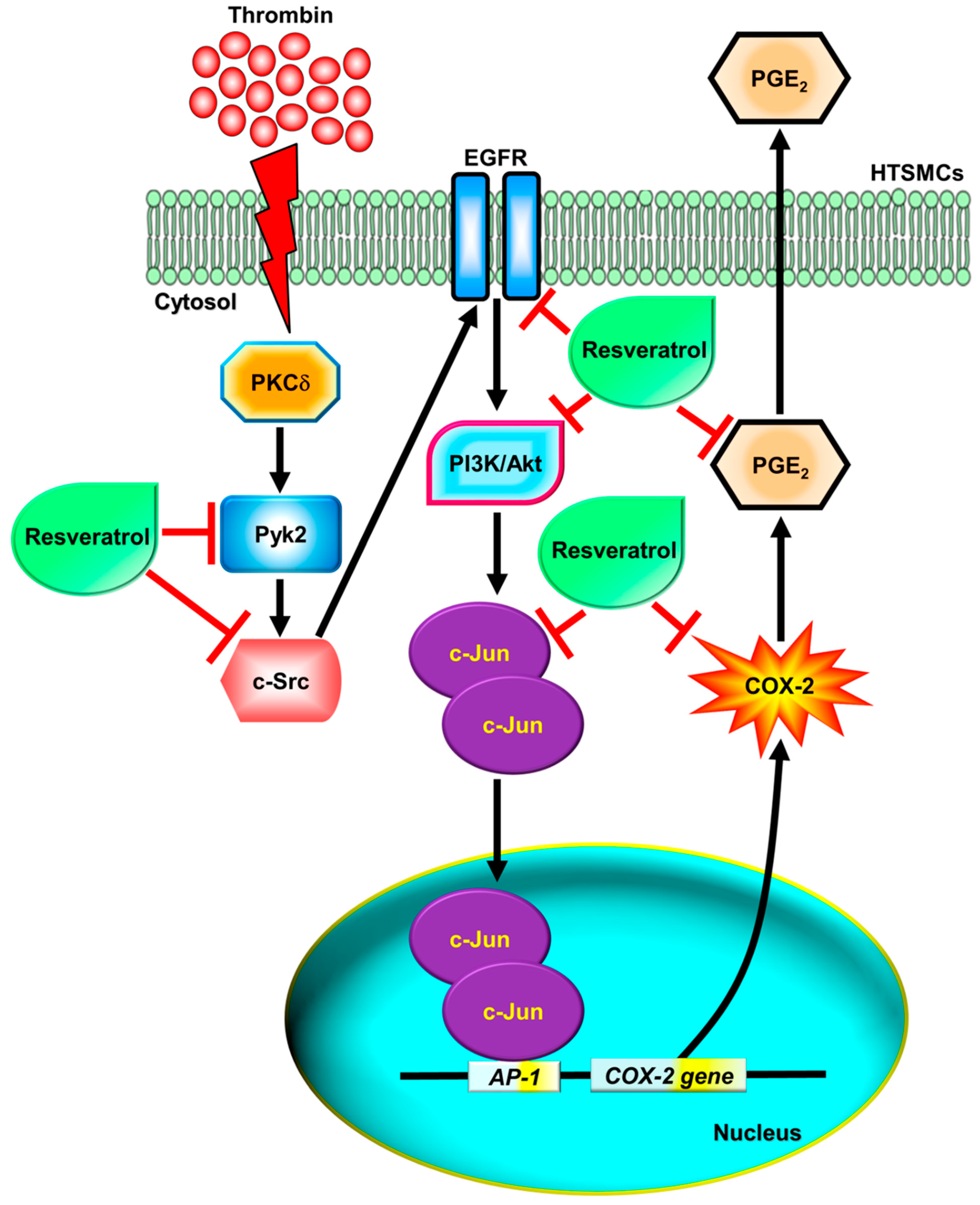
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Western Blot
4.4. Real-Time PCR
4.5. Measurement of PGE2 Generation
4.6. Measurement of COX-2 Luciferase Promoter Activity
4.7. Transient Transfection with Small Interfering RNAs (siRNAs)
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lam, M.; Lamanna, E.; Bourke, J.E. Regulation of airway smooth muscle contraction in health and disease. Adv. Exp. Med. Biol. 2019, 1124, 381–422. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.T.; Yang, C.M. Inflammatory signalings involved in airway and pulmonary diseases. Mediat. Inflamm. 2013, 2013, 791231. [Google Scholar] [CrossRef] [PubMed]
- Kolmert, J.; Gómez, C.; Balgoma, D.; Sjödin, M.; Bood, J.; Konradsen, J.R.; Ericsson, M.; Thörngren, J.O.; James, A.; Mikus, M.; et al. Urinary Leukotriene E4 and Prostaglandin D2 metabolites increase in adult and childhood severe asthma characterized by type 2 inflammation. A clinical observational study. Am. J. Respir. Crit. Care Med. 2021, 203, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Pucsok, J.M.; Györe, I.; Argay, K.; Huszár, E.; Barát, E.; Pucsok, J.; Horváth, I. Effect of exercise on levels of cyclo-oxygenase mediators in exhaled breath condensate in elite athletes. J. Sports Med. Phys. Fitness 2007, 47, 223–227. [Google Scholar] [PubMed]
- Lee, I.-T.; Lee, C.-W.; Tung, W.-H.; Wang, S.-W.; Lin, C.-C.; Shu, J.-C.; Yang, C.-M. Cooperation of TLR2 with MyD88, PI3K, and Rac1 in lipoteichoic acid–induced cPLA2/COX-2–dependent airway inflammatory responses. Am. J. Pathol. 2010, 176, 1671–1684. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, P.; Hanaoka, M.; Droma, Y.; Kubo, K. Enhanced levels of prostaglandin E2 and matrix metalloproteinase-2 correlate with the severity of airflow limitation in stable COPD. Respirology 2008, 13, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Säfholm, J.; Dahlén, S.E.; Delin, I.; Maxey, K.; Stark, K.; Cardell, L.O.; Adner, M. PGE2 maintains the tone of the guinea pig trachea through a balance between activation of contractile EP1 receptors and relaxant EP2 receptors. Br. J. Pharmacol. 2013, 168, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Birrell, M.A.; Maher, S.A.; Dekkak, B.; Jones, V.; Wong, S.; Brook, P.; Belvisi, M.G. Anti-inflammatory effects of PGE2 in the lung: Role of the EP4 receptor subtype. Thorax 2015, 70, 740–747. [Google Scholar] [CrossRef]
- Dagouassat, M.; Gagliolo, J.M.; Chrusciel, S.; Bourin, M.C.; Duprez, C.; Caramelle, P.; Boyer, L.; Hue, S.; Stern, J.B.; Validire, P.; et al. The cyclooxygenase-2-prostaglandin E2 pathway maintains senescence of chronic obstructive pulmonary disease fibroblasts. Am. J. Respir. Crit. Care Med. 2013, 187, 703–714. [Google Scholar] [CrossRef]
- Siller-Matula, J.M.; Schwameis, M.; Blann, A.; Mannhalter, C.; Jilma, B. Thrombin as a multi-functional enzyme. Focus on in vitro and in vivo effects. Thromb. Haemost. 2011, 106, 1020–1033. [Google Scholar] [CrossRef]
- Bazan-Socha, S.; Mastalerz, L.; Cybulska, A.; Zareba, L.; Kremers, R.; Zabczyk, M.; Pulka, G.; Iwaniec, T.; Hemker, C.; Undas, A. Asthma is associated with enhanced thrombin formation and impaired fibrinolysis. Clin. Exp. Allergy 2016, 46, 932–944. [Google Scholar] [CrossRef] [PubMed]
- Undas, A.; Jankowski, M.; Kaczmarek, P.; Sladek, K.; Brummel-Ziedins, K. Thrombin generation in chronic obstructive pulmonary disease: Dependence on plasma factor composition. Thromb. Res. 2011, 128, e24–e28. [Google Scholar] [CrossRef] [PubMed]
- Wygrecka, M.; Didiasova, M.; Berscheid, S.; Piskulak, K.; Taborski, B.; Zakrzewicz, D.; Kwapiszewska, G.; Preissner, K.T.; Markart, P. Protease-activated receptors (PAR)-1 and -3 drive epithelial-mesenchymal transition of alveolar epithelial cells—Potential role in lung fibrosis. Thromb. Haemost. 2013, 110, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wu, M.; Hu, Z.; Wang, T.; Yu, J.; Ma, Y.; Wang, Q.; Zhang, Y.; Chen, D.; Li, T.; et al. A novel oncotherapy strategy: Direct thrombin inhibitors suppress progression, dissemination and spontaneous metastasis in non-small cell lung cancer. Br. J. Pharmacol. 2021, 179, 5056–5073. [Google Scholar] [CrossRef] [PubMed]
- Asokananthan, N.; Graham, P.T.; Fink, J.; Knight, D.A.; Bakker, A.J.; McWilliam, A.S.; Thompson, P.J.; Stewart, G.A. Activation of protease-activated receptor (PAR)-1, PAR-2, and PAR-4 stimulates IL-6, IL-8, and prostaglandin E2 release from human respiratory epithelial cells. J. Immunol. 2002, 168, 3577–3585. [Google Scholar] [CrossRef] [PubMed]
- Asokananthan, N.; Lan, R.S.; Graham, P.T.; Bakker, A.J.; Tokanović, A.; Stewart, G.A. Activation of protease-activated receptors (PARs)-1 and -2 promotes alpha-smooth muscle actin expression and release of cytokines from human lung fibroblasts. Physiol. Rep. 2015, 3, e12295. [Google Scholar] [CrossRef] [PubMed]
- Moriyuki, K.; Sekiguchi, F.; Matsubara, K.; Nishikawa, H.; Kawabata, A. Curcumin Inhibits the proteinase-activated receptor-2-triggered prostaglandin E2 production by suppressing cyclooxygenase-2 upregulation and Akt-dependent activation of nuclear factor-κB in human lung epithelial cells. J. Pharmacol. Sci. 2010, 114, 225–229. [Google Scholar] [CrossRef]
- Yang, C.C.; Hsiao, L.D.; Shih, Y.F.; Hsu, C.K.; Hu, C.Y.; Yang, C.M. Thrombin induces COX-2 and PGE2 expression via PAR1/PKCα/MAPK-dependent NF-κB activation in human tracheal smooth muscle cells. Mediat. Inflamm. 2022, 2022, 4600029. [Google Scholar] [CrossRef]
- Chien, P.T.; Hsieh, H.L.; Chi, P.L.; Yang, C.M. PAR1-dependent COX-2/PGE2 production contributes to cell proliferation via EP2 receptors in primary human cardiomyocytes. Br. J. Pharmacol. 2014, 171, 4504–4519. [Google Scholar] [CrossRef]
- Shih, C.H.; Bien, M.Y.; Chiang, L.L.; Su, C.L.; Lin, C.H.; Chen, B.C. Thrombin induces cyclooxygenase-2 expression via the ERK and NF-κB pathways in human lung fibroblasts. Eur. J. Pharmacol. 2009, 618, 70–75. [Google Scholar] [CrossRef]
- Kikkawa, U.; Matsuzaki, H.; Yamamoto, T. Protein kinase Cδ (PKCδ): Activation mechanisms and functions. J. Biochem. 2002, 132, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Cui, X.Y.; Wu, W.; Yu, F.Y.; Yao, H.R.; Liu, Q.; Song, E.W.; Chen, J.Q. Pyk2 and Src mediate signaling to CCL18-induced breast cancer metastasis. J. Cell. Biochem. 2014, 115, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Hsiao, L.D.; Yang, C.M.; Lin, C.C. Thrombin enhanced matrix metalloproteinase-9 expression and migration of SK-N-SH Cells via PAR-1, c-Src, PYK2, EGFR, Erk1/2 and AP-1. Mol. Neurobiol. 2017, 54, 3476–3491. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Reiser, G. The role of the Ca2+-sensitive tyrosine kinase Pyk2 and Src in thrombin signalling in rat astrocytes. J. Neurochem. 2003, 84, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Wrenn, R.W. Carbachol stimulates TYR phosphorylation and association of PKCδ and PYK2 in pancreas. Biochem. Biophys. Res. Commun. 2001, 282, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Jang, B.C. Induction of COX-2 in human airway cells by manganese: Role of PI3K/PKB, p38 MAPK, PKCs, Src, and glutathione depletion. Toxicol. In Vitro 2009, 23, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Chen, S.Y.; Tsai, H.C.; Hsu, H.C.; Tang, C.H. Thrombin induces epidermal growth factor receptor transactivation and CCL2 expression in human osteoblasts. Arthritis Rheum. 2012, 64, 3344–3354. [Google Scholar] [CrossRef]
- Chien, P.T.; Lin, C.C.; Hsiao, L.D.; Yang, C.M. c-Src/Pyk2/EGFR/PI3K/Akt/CREB-activated pathway contributes to human cardiomyocyte hypertrophy: Role of COX-2 induction. Mol. Cell. Endocrinol. 2015, 409, 59–72. [Google Scholar] [CrossRef]
- Khanal, T.; Kim, H.G.; Do, M.T.; Choi, J.H.; Chung, Y.C.; Kim, H.S.; Park, Y.J.; Jeong, T.C.; Jeong, H.G. Genipin induces cyclooxygenase-2 expression via NADPH oxidase, MAPKs, AP-1, and NF-κB in RAW 264.7 cells. Food Chem. Toxicol. 2014, 64, 126–134. [Google Scholar] [CrossRef]
- Tiwari, R.L.; Singh, V.; Singh, A.; Barthwal, M.K. IL-1R-associated kinase-1 mediates protein kinase Cδ-induced IL-1β production in monocytes. J. Immunol. 2011, 187, 2632–2645. [Google Scholar] [CrossRef]
- Frémont, L. Biological effects of resveratrol. Life Sci. 2000, 66, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Gal, R.; Deres, L.; Toth, K.; Halmosi, R.; Habon, T. The effect of resveratrol on the cardiovascular system from molecular mechanisms to clinical results. Int. J. Mol. Sci. 2021, 22, 10152. [Google Scholar] [CrossRef] [PubMed]
- Alghetaa, H.; Mohammed, A.; Zhou, J.; Singh, N.; Nagarkatti, M.; Nagarkatti, P. Resveratrol-mediated attenuation of superantigen-driven acute respiratory distress syndrome is mediated by microbiota in the lungs and gut. Pharmacol. Res. 2021, 167, 105548. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.T.; Lin, C.C.; Yang, C.C.; Hsiao, L.D.; Wu, M.Y.; Yang, C.M. Resveratrol attenuates Staphylococcus aureus-induced monocyte adhesion through downregulating PDGFR/AP-1 activation in human lung epithelial cells. Int. J. Mol. Sci. 2018, 19, 3058. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-M.; Chen, Y.-W.; Chi, P.-L.; Lin, C.-C.; Hsiao, L.-D. Resveratrol inhibits BK-induced COX-2 transcription by suppressing acetylation of AP-1 and NF-κB in human rheumatoid arthritis synovial fibroblasts. Biochem. Pharmacol. 2017, 132, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.S.; Tsai, C.W.; Yang, J.S.; Hsu, Y.M.; Shih, L.C.; Chiu, H.Y.; Bau, D.T.; Tsai, F.J. Resveratrol inhibited the metastatic behaviors of cisplatin-resistant human oral cancer cells via phosphorylation of ERK/p-38 and suppression of MMP-2/9. J. Food Biochem. 2021, 45, e13666. [Google Scholar] [CrossRef] [PubMed]
- Culpitt, S.V.; Rogers, D.F.; Fenwick, P.S.; Shah, P.; De Matos, C.; Russell, R.E.; Barnes, P.J.; Donnelly, L.E. Inhibition by red wine extract, resveratrol, of cytokine release by alveolar macrophages in COPD. Thorax 2003, 58, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, L.E.; Newton, R.; Kennedy, G.E.; Fenwick, P.S.; Leung, R.H.; Ito, K.; Russell, R.E.; Barnes, P.J. Anti-inflammatory effects of resveratrol in lung epithelial cells: Molecular mechanisms. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L774–L783. [Google Scholar] [CrossRef]
- Hsieh, H.L.; Sun, C.C.; Wang, T.S.; Yang, C.M. PKC-δ/c-Src-mediated EGF receptor transactivation regulates thrombin-induced COX-2 expression and PGE2 production in rat vascular smooth muscle cells. Biochim. Biophys. Acta 2008, 1783, 1563–1575. [Google Scholar] [CrossRef]
- Farshori, P.Q.; Shah, B.H.; Arora, K.K.; Martinez-Fuentes, A.; Catt, K.J. Activation and nuclear translocation of PKCδ, Pyk2 and ERK1/2 by gonadotropin releasing hormone in HEK293 cells. J. Steroid Biochem. Mol. Biol. 2003, 85, 337–347. [Google Scholar] [CrossRef]
- Moffatt, J.D.; Lever, R.; Page, C.P. Effects of inhaled thrombin receptor agonists in mice. Br. J. Pharmacol. 2004, 143, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Ossovskaya, V.S.; Bunnett, N.W. Protease-activated receptors: Contribution to physiology and disease. Physiol. Rev. 2004, 84, 579–621. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.; Henry, P.J. Protease-activated receptors and prostaglandins in inflammatory lung disease. Br. J. Pharmacol. 2009, 158, 1017–1033. [Google Scholar] [CrossRef] [PubMed]
- Wu-Zhang, A.X.; Newton, A.C. Protein kinase C pharmacology: Refining the toolbox. Biochem. J. 2013, 452, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Carlin, S.; Yang, K.X.; Donnelly, R.; Black, J.L. Protein kinase C isoforms in human airway smooth muscle cells: Activation of PKC-zeta during proliferation. Am. J. Physiol. 1999, 276, L506–L512. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Ryer, E.J.; Kundi, R.; Kamiya, K.; Itoh, H.; Faries, P.L.; Sakakibara, K.; Kent, K.C. Protein kinase C-δ regulates migration and proliferation of vascular smooth muscle cells through the extracellular signal-regulated kinase 1/2. J. Vasc. Surg. 2007, 45, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Parihar, S.P.; Ozturk, M.; Marakalala, M.J.; Loots, D.T.; Hurdayal, R.; Maasdorp, D.B.; Van Reenen, M.; Zak, D.E.; Darboe, F.; Penn-Nicholson, A.; et al. Protein kinase C-δ (PKCδ), a marker of inflammation and tuberculosis disease progression in humans, is important for optimal macrophage killing effector functions and survival in mice. Mucosal Immunol. 2018, 11, 496–511. [Google Scholar] [CrossRef]
- Koyasu, S. The role of PI3K in immune cells. Nat. Immunol. 2003, 4, 313–319. [Google Scholar] [CrossRef]
- Yoon, H.S.; Park, C.M. Chrysoeriol ameliorates COX-2 expression through NF-κB, AP-1 and MAPK regulation via the TLR4/MyD88 signaling pathway in LPS-stimulated murine macrophages. Exp. Ther. Med. 2021, 22, 718. [Google Scholar] [CrossRef]
- Woo, J.H.; Lim, J.H.; Kim, Y.H.; Suh, S.I.; Min, D.S.; Chang, J.S.; Lee, Y.H.; Park, J.W.; Kwon, T.K. Resveratrol inhibits phorbol myristate acetate-induced matrix metalloproteinase-9 expression by inhibiting JNK and PKCδ signal transduction. Oncogene 2004, 23, 1845–1853. [Google Scholar] [CrossRef]
- Atten, M.J.; Godoy-Romero, E.; Attar, B.M.; Milson, T.; Zopel, M.; Holian, O. Resveratrol regulates cellular PKCα and δ to inhibit growth and induce apoptosis in gastric cancer cells. Investig. New Drugs 2005, 23, 111–119. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.-C.; Lee, I.-T.; Lin, Y.-J.; Wu, W.-B.; Hsiao, L.-D.; Yang, C.-M. Thrombin-Induced COX-2 Expression and PGE2 Synthesis in Human Tracheal Smooth Muscle Cells: Role of PKCδ/Pyk2-Dependent AP-1 Pathway Modulation. Int. J. Mol. Sci. 2023, 24, 15130. https://doi.org/10.3390/ijms242015130
Yang C-C, Lee I-T, Lin Y-J, Wu W-B, Hsiao L-D, Yang C-M. Thrombin-Induced COX-2 Expression and PGE2 Synthesis in Human Tracheal Smooth Muscle Cells: Role of PKCδ/Pyk2-Dependent AP-1 Pathway Modulation. International Journal of Molecular Sciences. 2023; 24(20):15130. https://doi.org/10.3390/ijms242015130
Chicago/Turabian StyleYang, Chien-Chung, I-Ta Lee, Yan-Jyun Lin, Wen-Bin Wu, Li-Der Hsiao, and Chuen-Mao Yang. 2023. "Thrombin-Induced COX-2 Expression and PGE2 Synthesis in Human Tracheal Smooth Muscle Cells: Role of PKCδ/Pyk2-Dependent AP-1 Pathway Modulation" International Journal of Molecular Sciences 24, no. 20: 15130. https://doi.org/10.3390/ijms242015130
APA StyleYang, C.-C., Lee, I.-T., Lin, Y.-J., Wu, W.-B., Hsiao, L.-D., & Yang, C.-M. (2023). Thrombin-Induced COX-2 Expression and PGE2 Synthesis in Human Tracheal Smooth Muscle Cells: Role of PKCδ/Pyk2-Dependent AP-1 Pathway Modulation. International Journal of Molecular Sciences, 24(20), 15130. https://doi.org/10.3390/ijms242015130