

Prioritizing Endangered Species in Genome Sequencing: Conservation Genomics in Action with the First Platinum-Standard Reference-Quality Genome of the Critically Endangered European Mink Mustela lutreola L., 1761

 ,
,  , , , ,
, , , ,
Abstract
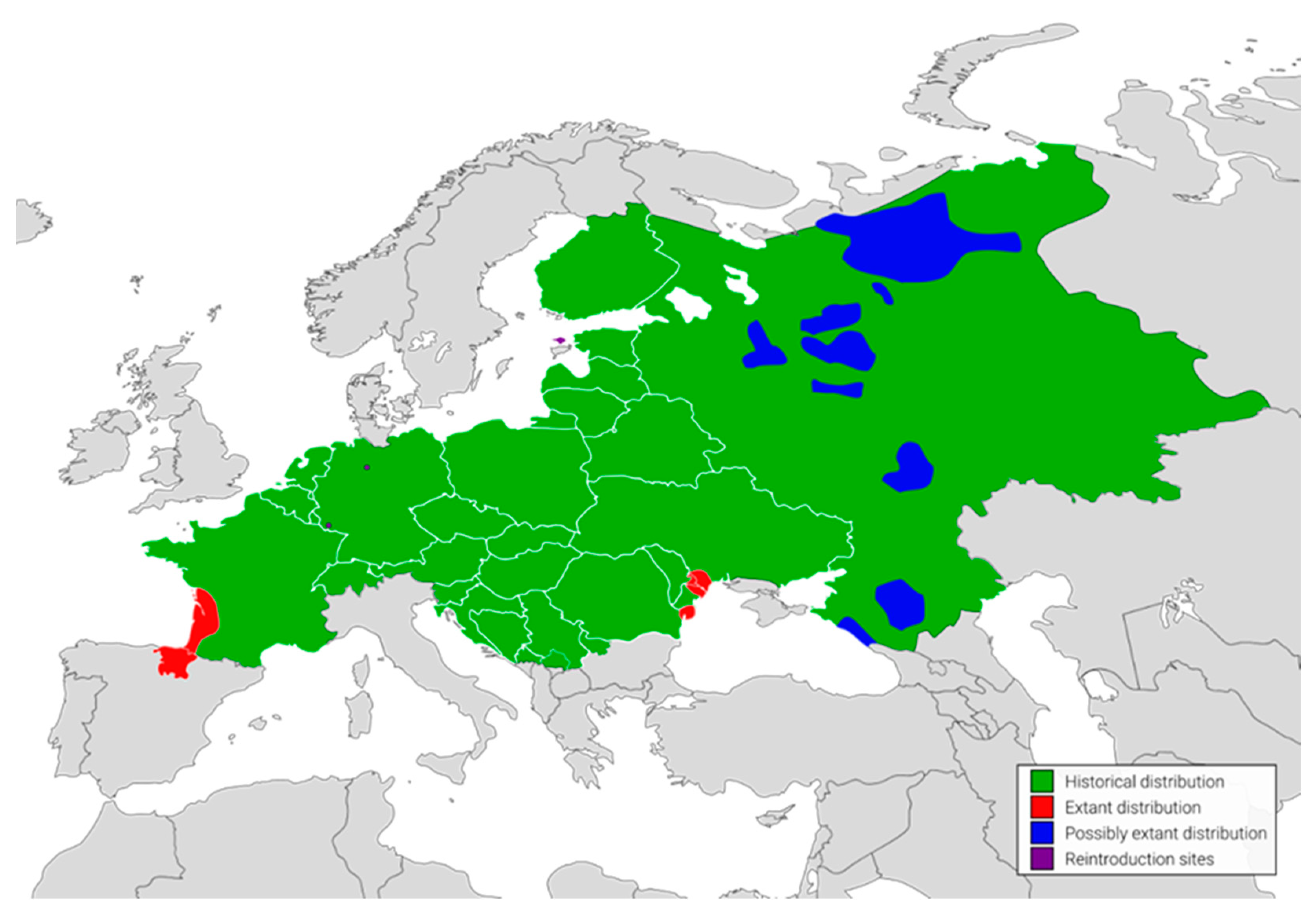
:1. Introduction
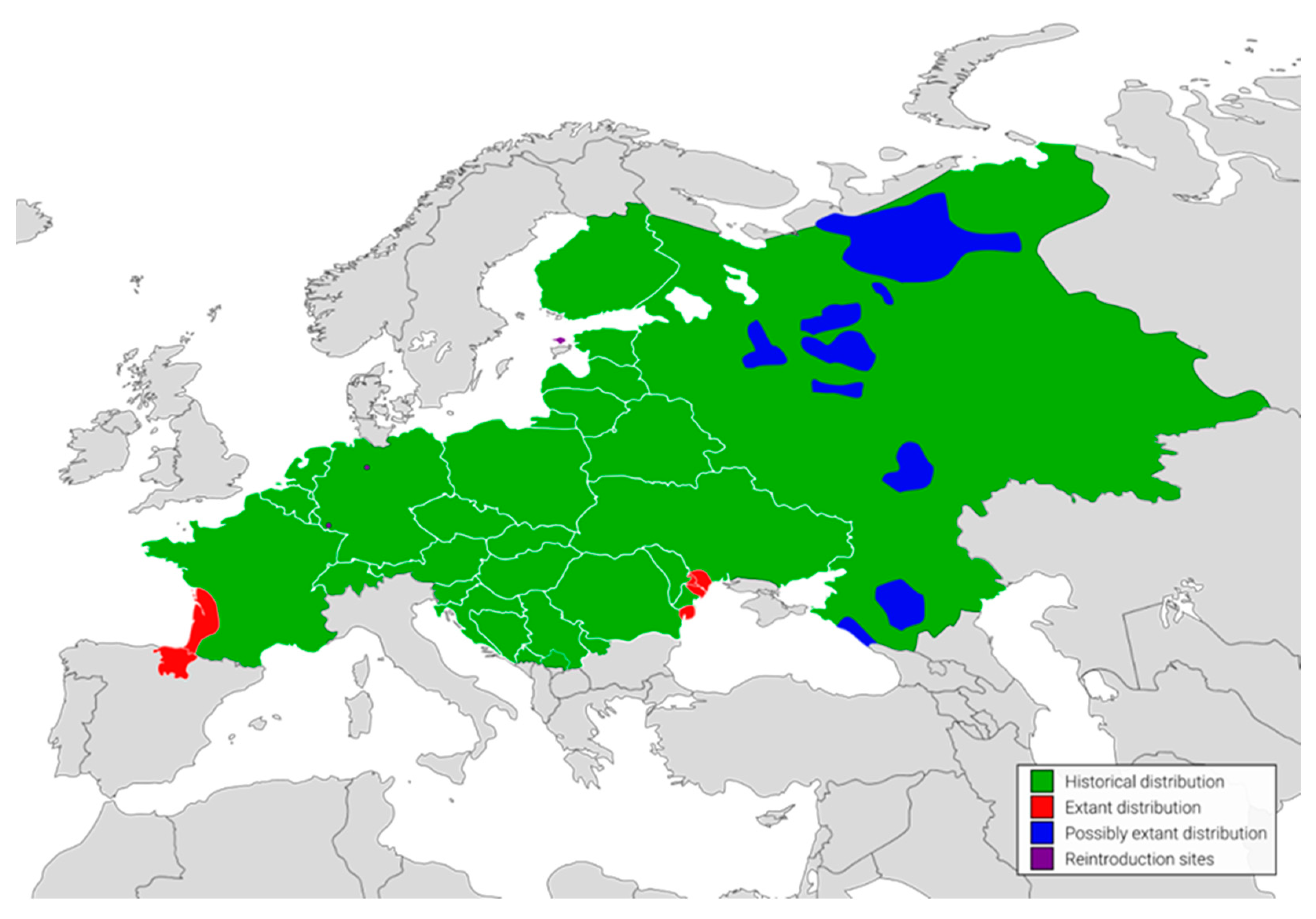
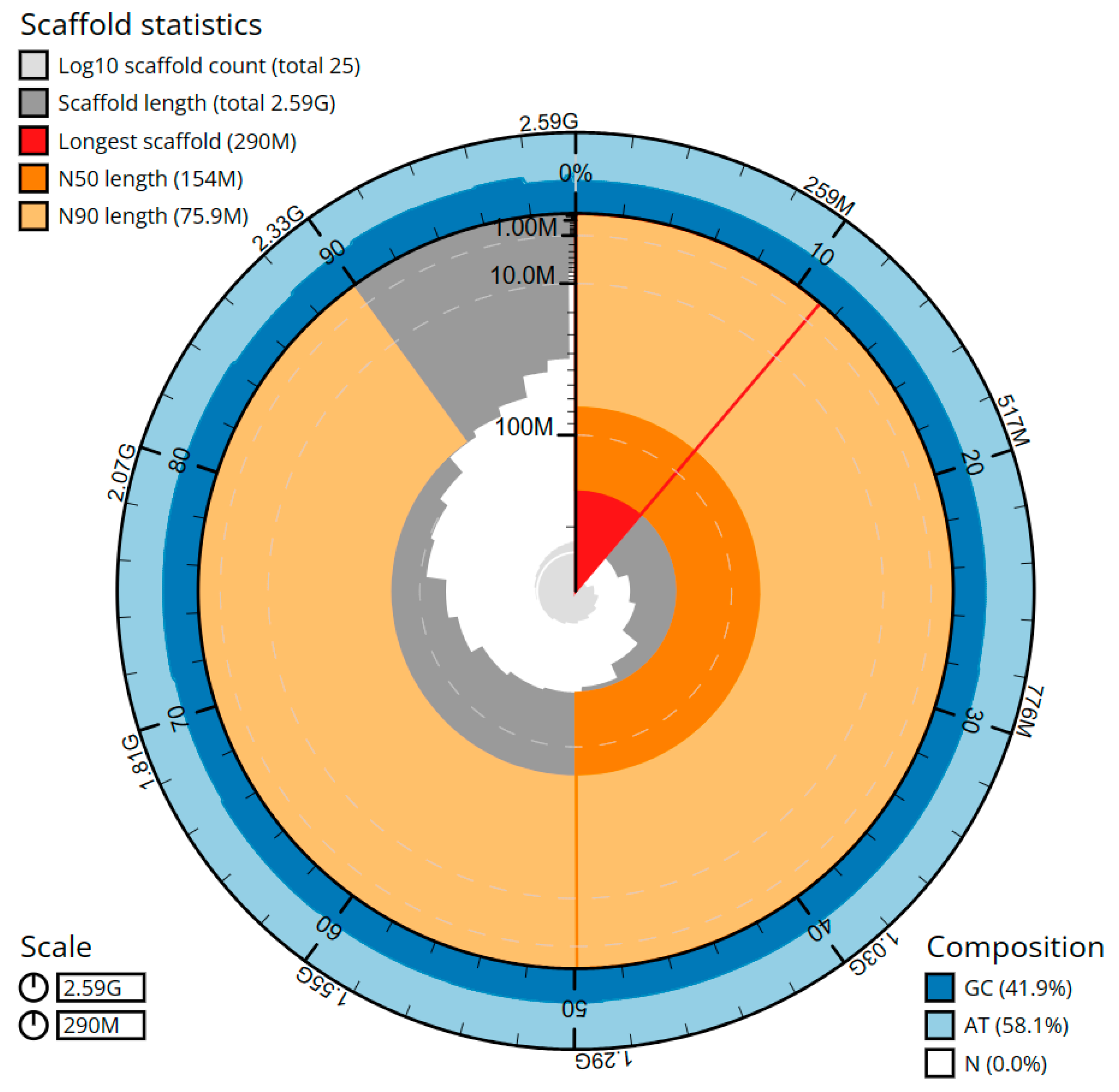
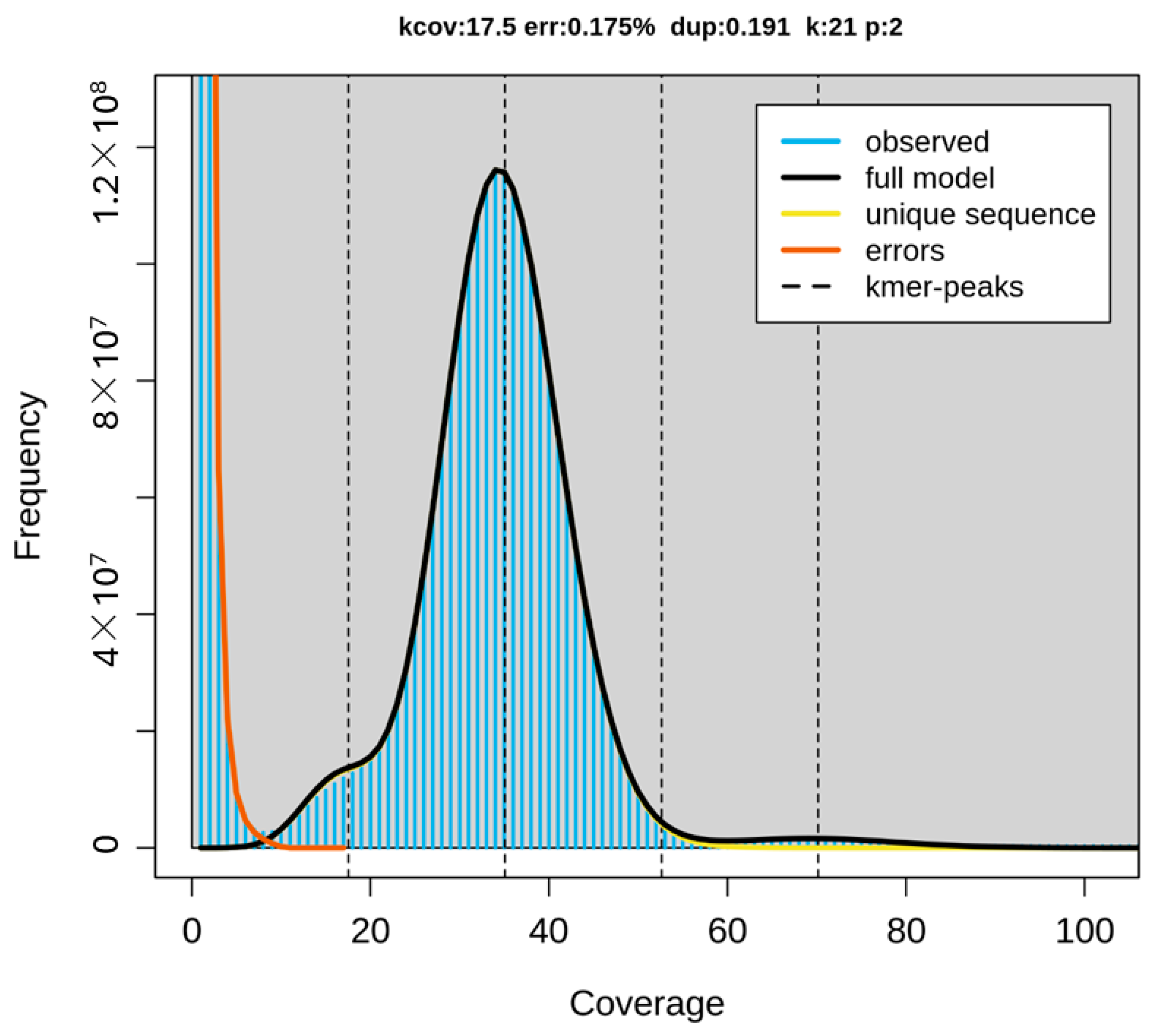
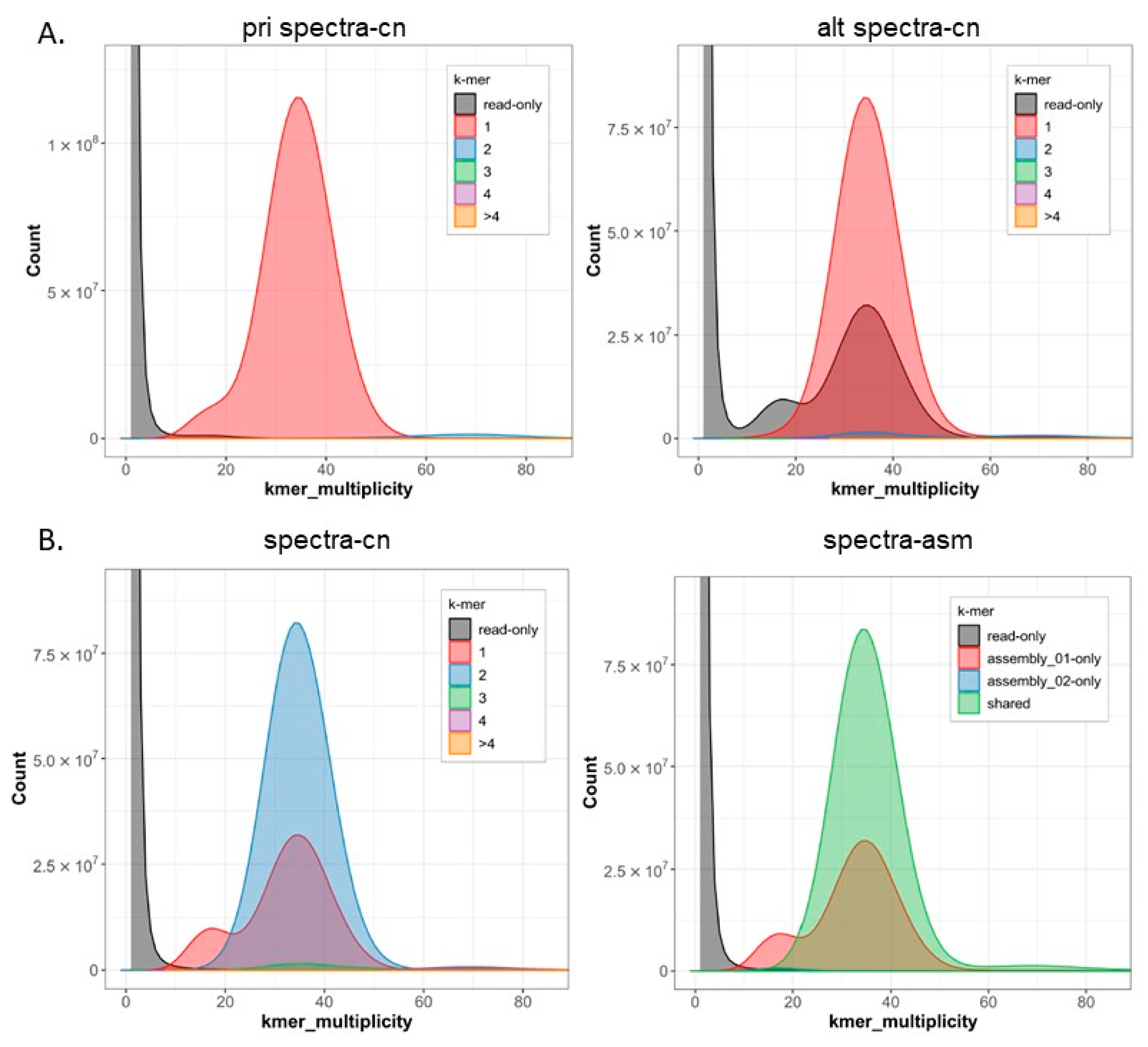
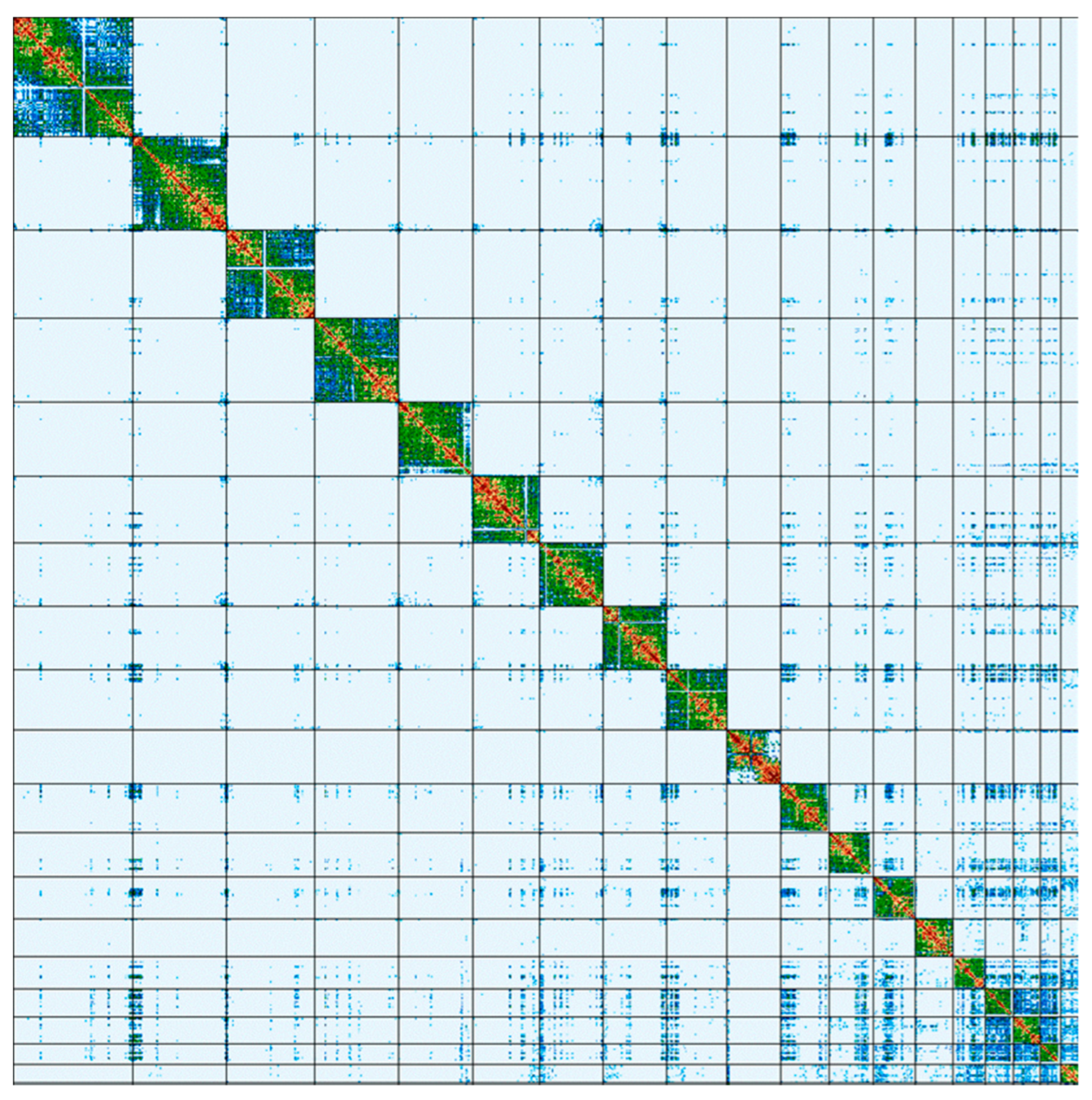
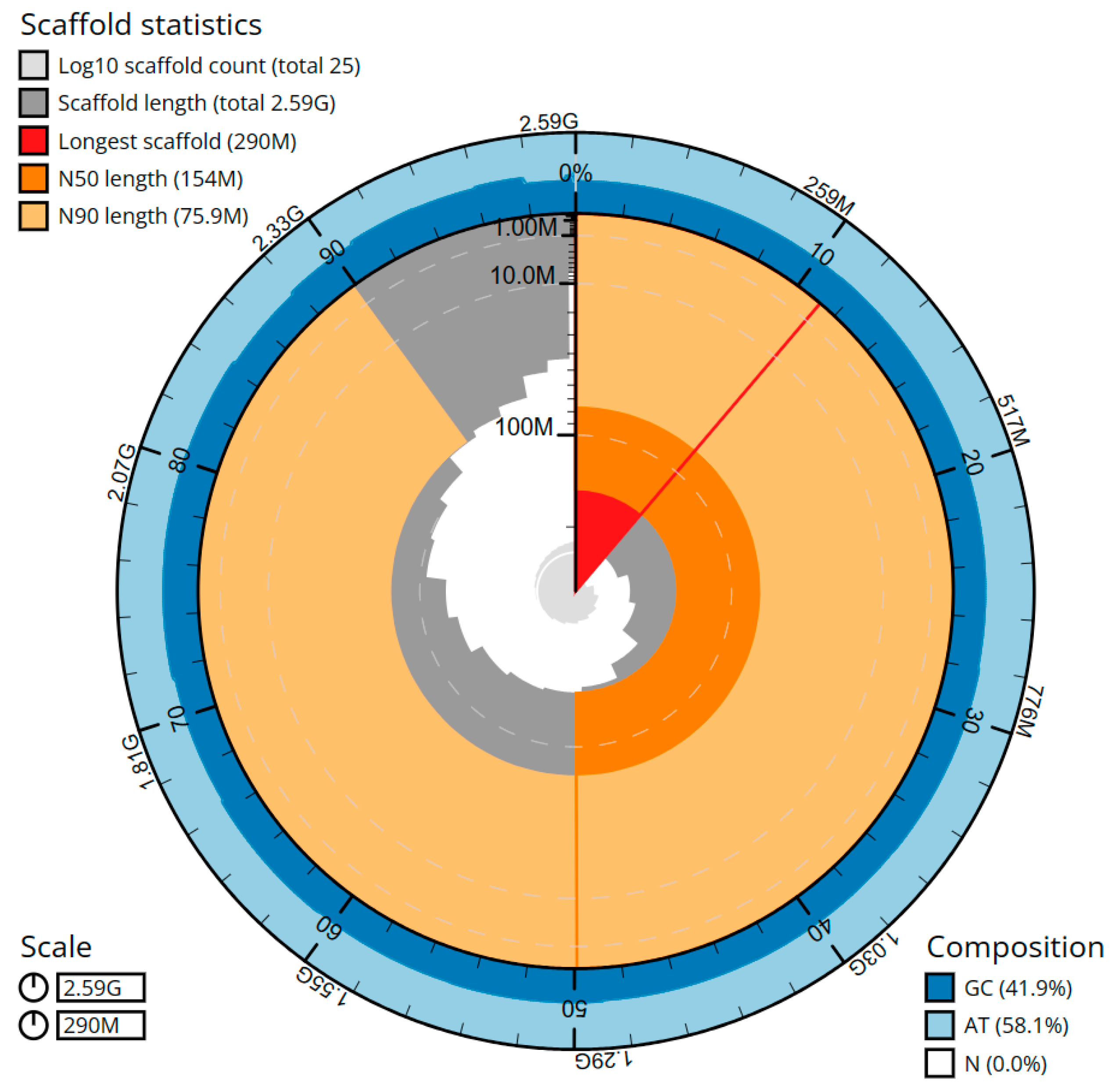
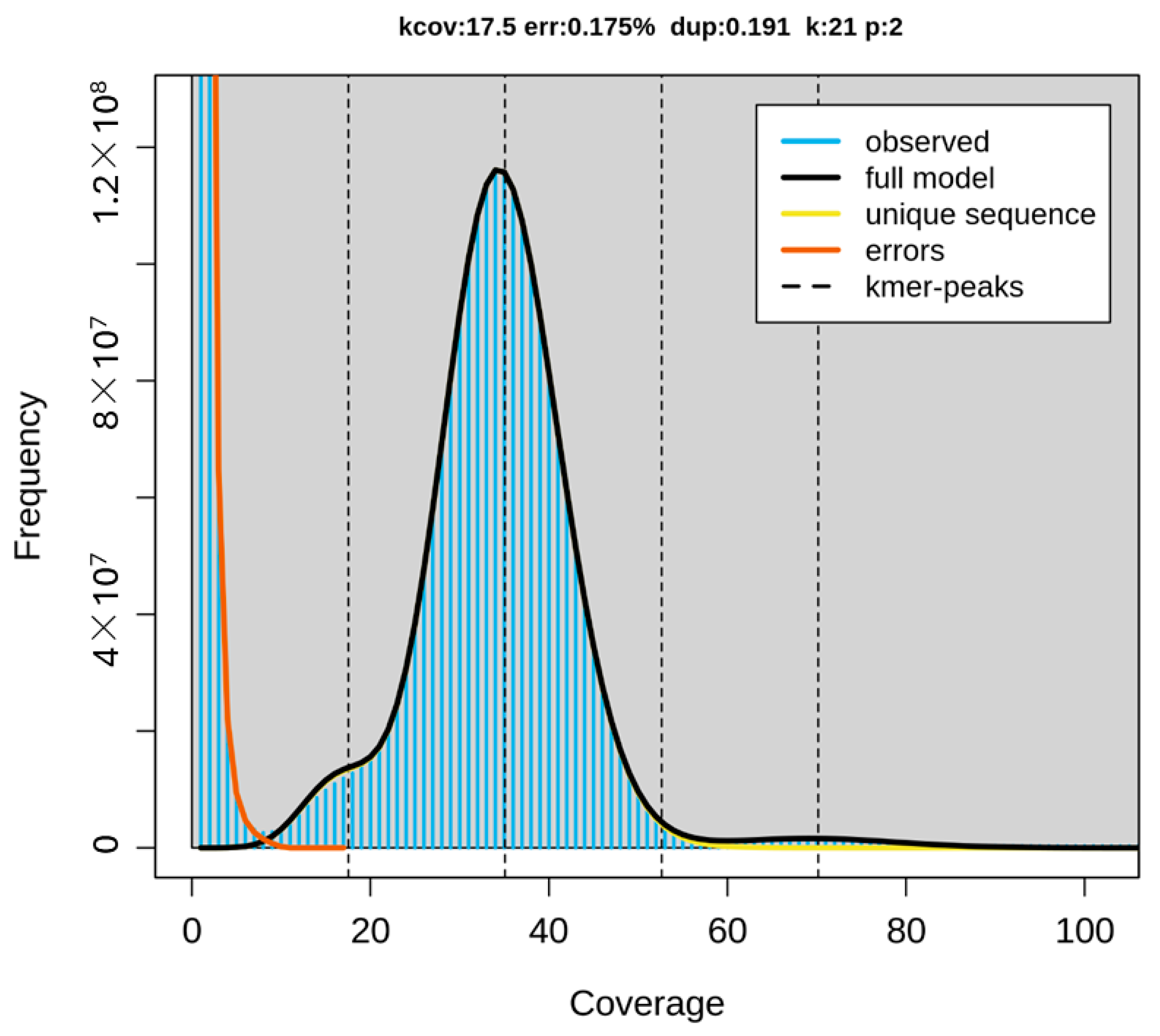
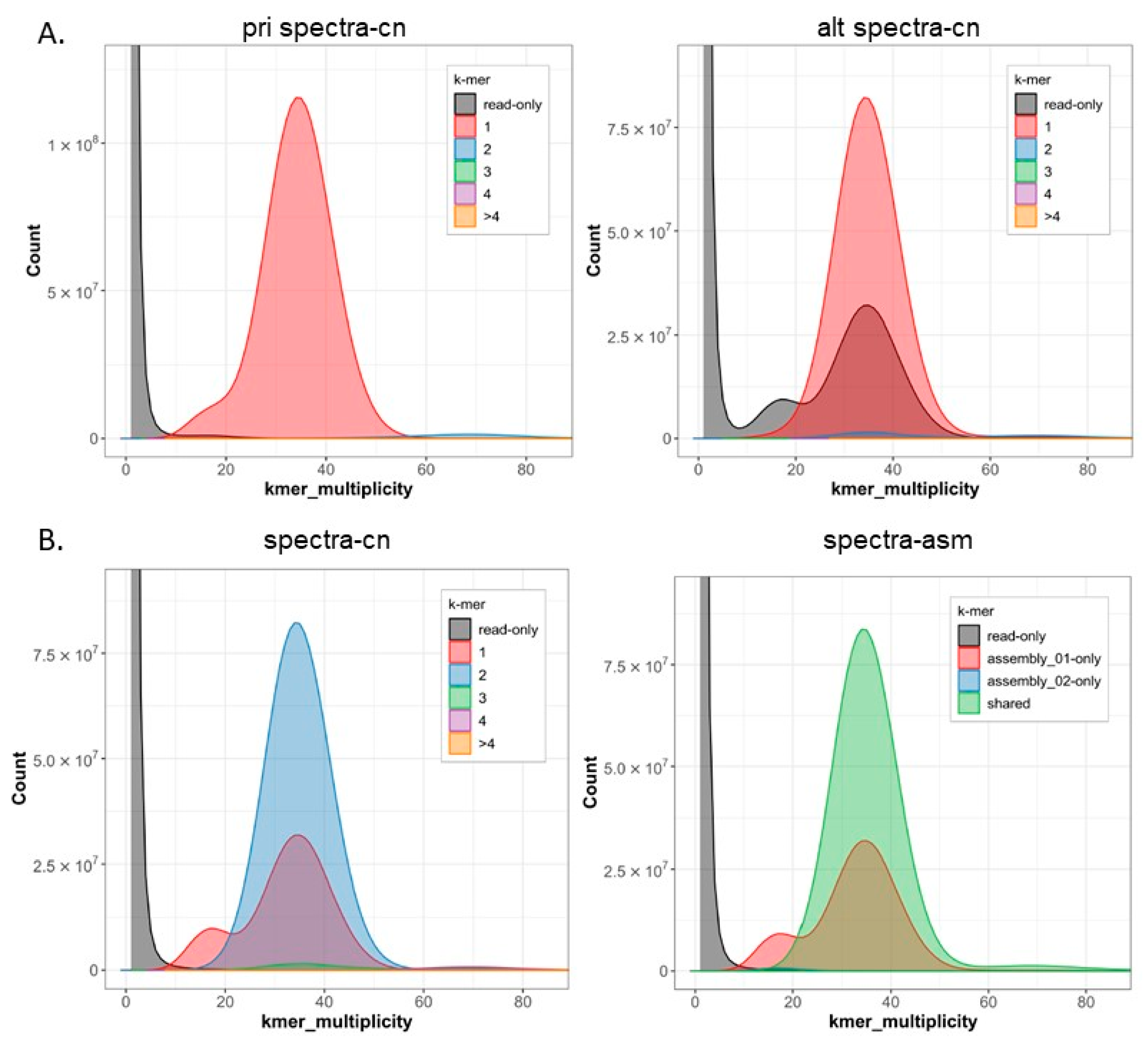
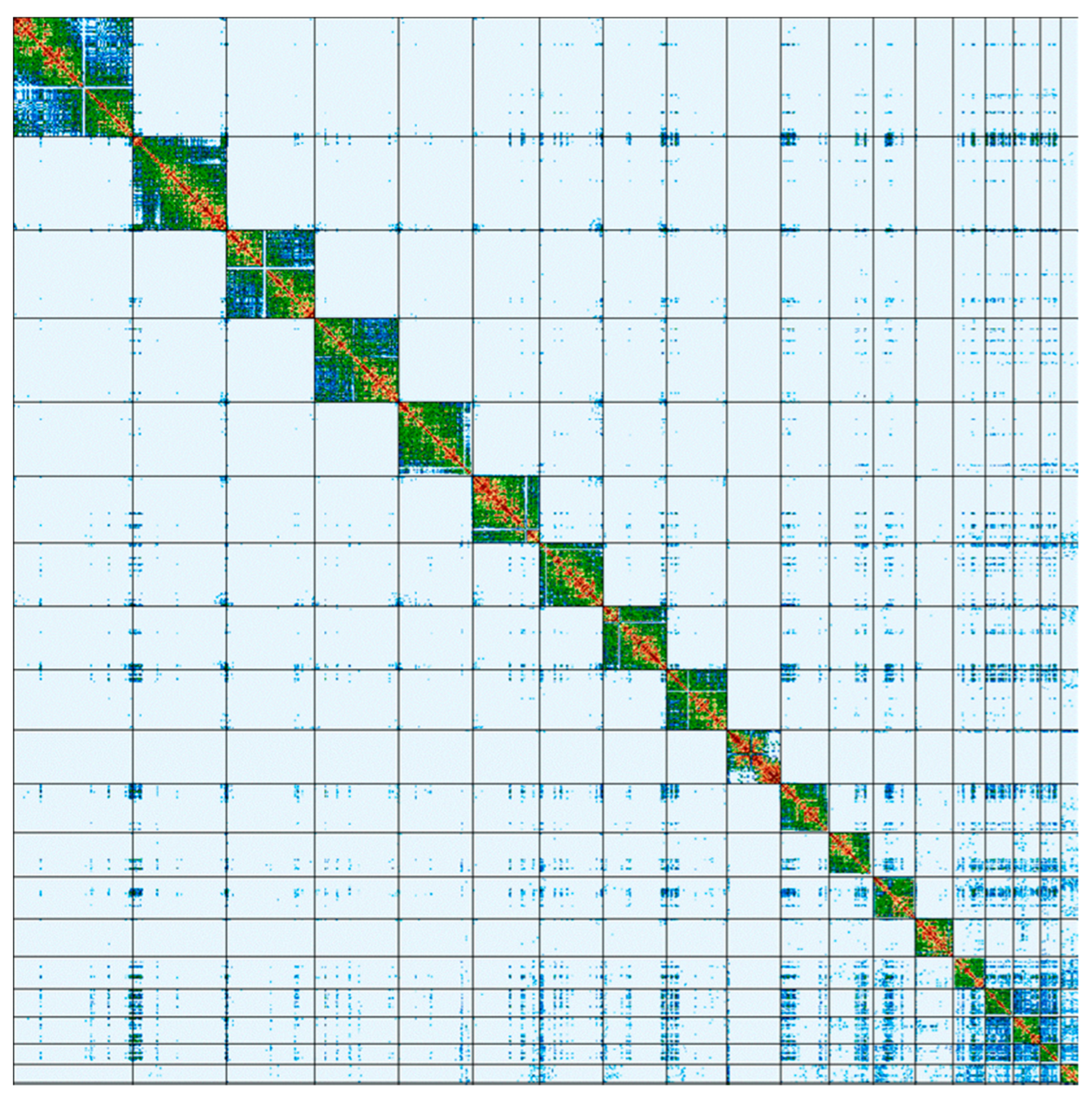
2. Results
3. Discussion
4. Materials and Methods
4.1. Sample Collection and DNA Extraction
4.2. Sequencing
4.3. De Novo Genome Assembly, Curation, and Quality Control
4.4. Genome Annotation and Data Availability
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- IUCN. The IUCN Red List of Threatened Species—Summary Statistics. 2023. Available online: https://www.iucnredlist.org/resources/summary-statistics (accessed on 14 July 2023).
- Youngman, P.M. Mustela lutreola. Mamm. Species 1990, 362, 1–3. [Google Scholar]
- Heptner, V.G.; Naumov, N.P.; Yurgenson, P.B.; Sludskii, A.A.; Chirkova, A.F.; Bannikov, A.G. Part 1b: Carnivora. Mammals of the Soviet Union, 1st ed.; Smithsonian Institution Libraries: Washington, DC, USA; The National Science Foundation Press: New Delhi, India, 2001; Volume 2, pp. 1078–1106.
- Witkowski, Z.; Król, W.; Solarz, W. Carpathian List of Endangered Species; WWF: Vienna, Austria; Institute of Nature Conservation, Polish Academy of Sciences: Kraków, Poland, 2003; 64p. [Google Scholar]
- Maran, T.; Skumatov, D.; Gomez, A.; Põdra, M.; Abramov, A.V.; Dinets, V. Mustela lutreola. IUCN Red List Threat. Species 2016, 13, e.T14018A45199861. [Google Scholar] [CrossRef]
- Maran, T. European mink: Setting of goal for conservation and Estonian case study. Galemys 2003, 15, 1–11. [Google Scholar]
- Harrington, L.A.; Põdra, M.; Gómez, A.; Maran, T. Raising awareness of the plight of the critically endangered European mink in Spain is not miscommunication: A response to Melero. Biodivers. Conserv. 2018, 27, 269–271. [Google Scholar] [CrossRef]
- Skorupski, J. Fifty Years of Research on European Mink Mustela lutreola L., 1761 Genetics: Where Are We Now in Studies on One of the Most Endangered Mammals? Genes 2020, 11, 1332. [Google Scholar] [CrossRef]
- Fuentes-Pardo, A.P.; Ruzzante, D.E. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Mol. Ecol. 2017, 26, 5369–5406. [Google Scholar] [CrossRef]
- Brandies, P.; Peel, E.; Hogg, C.J.; Belov, K. The Value of Reference Genomes in the Conservation of Threatened Species. Genes 2019, 10, 846. [Google Scholar] [CrossRef]
- Steiner, C.C.; Putnam, A.S.; Hoeck, P.E.; Ryder, O.A. Conservation Genomics of Threatened Animal Species. Annu. Rev. Anim. Biosci. 2013, 1, 261–281. [Google Scholar] [CrossRef]
- Khan, S.; Nabi, G.; Ullah, M.W.; Yousaf, M.; Manan, S.; Siddique, R.; Hou, H. Overview on the Role of Advance Genomics in Conservation Biology of Endangered Species. Int. J. Genom. 2016, 2016, 1–8. [Google Scholar] [CrossRef]
- Supple, M.A.; Shapiro, B. Conservation of biodiversity in the genomics era. Genome Biol. 2018, 19, 131. [Google Scholar] [CrossRef]
- Stronen, A.V.; Iacolina, L.; Ruiz-Gonzalez, A. Rewilding and conservation genomics: How developments in (re)colonization ecology and genomics can offer mutual benefits for understanding contemporary evolution. Glob. Ecol. Conserv. 2019, 17, e00502. [Google Scholar] [CrossRef]
- Wright, B.; Farquharson, K.A.; McLennan, E.A.; Belov, K.; Hogg, C.J.; Grueber, C.E. From reference genomes to population genomics: Comparing three reference-aligned reduced-representation sequencing pipelines in two wildlife species. BMC Genom. 2019, 20, 453. [Google Scholar] [CrossRef] [PubMed]
- Skorupski, J. Characterisation of the Complete Mitochondrial Genome of Critically Endangered Mustela lutreola (Carnivora: Mustelidae) and Its Phylogenetic and Conservation Implications. Genes 2022, 13, 125. [Google Scholar] [CrossRef] [PubMed]
- Lewin, H.A.; Graves, J.A.M.; Ryder, O.A.; Graphodatsky, A.S.; O’Brien, S.J. Precision nomenclature for the new genomics. Gigascience 2019, 8, giz086. [Google Scholar] [CrossRef] [PubMed]
- Kollias, G.V.; Fernandez-Moran, J. Mustelidae. In Fowler’s Zoo and Wild Animal Medicine; Miller, R.E., Fowler, M.E., Eds.; Elsevier Inc.: St. Louis, MI, USA, 2015; Volume 8, pp. 476–491. [Google Scholar] [CrossRef]
- NCBI. Genome List. 2023. Available online: https://www.ncbi.nlm.nih.gov/datasets/genome/?taxon=9655 (accessed on 14 July 2023).
- Morin, P.A.; Alexander, A.; Blaxter, M.; Caballero, S.; Fedrigo, O.; Fontaine, M.C.; Foote, A.D.; Kuraku, S.; Maloney, B.; McCarthy, M.L.; et al. Building genomic infrastructure: Sequencing platinum-standard reference-quality genomes of all cetacean species. Mar. Mammal Sci. 2020, 36, 1356–1366. [Google Scholar] [CrossRef]
- Rhie, A.; McCarthy, S.A.; Fedrigo, O.; Damas, J.; Formenti, G.; Koren, S.; Uliano-Silva, M.; Chow, W.; Fungtammasan, A.; Kim, J.; et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature 2021, 592, 737–746. [Google Scholar] [CrossRef]
- Koepfli, K.-P.; Paten, B.; the Genome 10K Community of Scientists; O’brien, S.J. The Genome 10K Project: A Way Forward. Annu. Rev. Anim. Biosci. 2015, 3, 57–111. [Google Scholar] [CrossRef]
- Anonymous. A reference standard for genome biology. Nat. Biotechnol. 2018, 36, 1121. [Google Scholar] [CrossRef]
- Davison, A.; Griffiths, H.I.; Brookes, R.C.; Maran, T.; Macdonald, D.W.; Sidorovich, V.E.; Kitchener, A.C.; Irizar, I.; Villate, I.; Gonzalez-Esteban, J.; et al. Mitochondrial DNA and palaeontological evidence for the origins of endangered European mink, Mustela lutreola. Anim. Conserv. 2000, 3, 345–355. [Google Scholar] [CrossRef]
- Lodé, T.; Guiral, G.; Peltier, D. European Mink–Polecat Hybridization Events: Hazards From Natural Process? J. Hered. 2005, 96, 89–96. [Google Scholar] [CrossRef]
- Graphodatsky, A.S. Mustela lutreola (European Mink). In Atlas of Mammalian Chromosomes; O’Brien, S.J., Menninger, J.C., Nash, W.G., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2006; p. 487. [Google Scholar]
- Franco-De-Sá, J.F.O.; Rosas, F.C.W.; Feldberg, E. Cytogenetic study of the giant otter Pteronura brasiliensis Zimmermann 1780 (Carnivora, Mustelidae, Lutrinae). Genet. Mol. Biol. 2007, 30, 1093–1096. [Google Scholar] [CrossRef]
- Maran, T.; Fienieg, E.; Schad, K. Long-Term Management Plan for European Mink (Mustela lutreola) European Endangered Species Programme (EEP); Tallinn Zoo: Tallinn, Estonia; European Association of Zoos: Amsterdam, The Netherlands, 2017; pp. 1–46. [Google Scholar]
- Formenti, G.; Theissinger, K.; Fernandes, C.; Bista, I.; Bombarely, A.; Bleidorn, C.; Ciofi, C.; Crottini, A.; Godoy, J.A.; Malukiewicz, J.; et al. The era of reference genomes in conservation genomics. Trends Ecol. Evol. 2022, 37, 197–202. [Google Scholar] [CrossRef]
- Campana, M.G.; Parker, L.D.; Hawkins, M.T.; Young, H.S.; Helgen, K.M.; Gunther, M.S.; Woodroffe, R.; Maldonado, J.E.; Fleischer, R.C. Genome sequence, population history, and pelage genetics of the endangered African wild dog (Lycaon pictus). BMC Genom. 2016, 17, 1013. [Google Scholar] [CrossRef]
- Ekblom, R.; Brechlin, B.; Persson, J.; Smeds, L.; Johansson, M.; Magnusson, J.; Flagstad, Ø.; Ellegren, H. Genome sequencing and conservation genomics in the Scandinavian wolverine population. Conserv. Biol. 2018, 32, 1301–1312. [Google Scholar] [CrossRef] [PubMed]
- Saremi, N.F.; Supple, M.A.; Byrne, A.; Cahill, J.A.; Coutinho, L.L.; Dalén, L.; Figueiró, H.V.; Johnson, W.E.; Milne, H.J.; O’brien, S.J.; et al. Puma genomes from North and South America provide insights into the genomic consequences of inbreeding. Nat. Commun. 2019, 10, 227–245. [Google Scholar] [CrossRef]
- Hohenlohe, P.A.; Funk, W.C.; Rajora, O.P. Population genomics for wildlife conservation and management. Mol. Ecol. 2020, 30, 62–82. [Google Scholar] [CrossRef]
- Michaux, J.; Libois, R.; Davison, A.; Chevret, P.; Rosoux, R. Is the western population of the European mink, (Mustela lutreola), a distinct Management Unit for conservation? Biol. Conserv. 2004, 115, 357–367. [Google Scholar] [CrossRef]
- Arthur, C.; Aulagnier, S.; des Neiges de Bellefroid, M.; Delas, G.; Fournier, P.; Gourreau, J.-M.; Lodé, T.; Michaux, J.; Rosoux, R.; Ruette, S. Deuxième Plan National de Restauration du Vison d’Europe (Mustela lutreola) 2007–2011; Ministère de l’Ecologie, du Développement et de l’Aménagement Durables: Paris, France, 2007; pp. 1–102.
- Cabria, M.T.; González, E.G.; Gómez-Moliner, B.J.; Zardoya, R. Microsatellite markers for the endangered European mink (Mustela lutreola) and closely related mustelids. Mol. Ecol. Notes 2007, 7, 1185–1188. [Google Scholar] [CrossRef]
- Zuberogoitia, I.; Põdra, M.; Palazón, S.; Gómez, A.; Zabala, N.; Zabala, J. Misleading interpretation of shifting baseline syndrome in the conservation of European mink. Biodivers. Conserv. 2016, 25, 1795–1800. [Google Scholar] [CrossRef]
- Emerson, K.J.; Merz, C.R.; Catchen, J.M.; Hohenlohe, P.A.; Cresko, W.A.; Bradshaw, W.E.; Holzapfel, C.M. Resolving postglacial phylogeography using high-throughput sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 16196–16200. [Google Scholar] [CrossRef]
- McGaughran, A.; Liggins, L.; Marske, K.A.; Dawson, M.N.; Schiebelhut, L.M.; Lavery, S.D.; Knowles, L.L.; Moritz, C.; Riginos, C. Comparative phylogeography in the genomic age: Opportunities and challenges. J. Biogeogr. 2022, 49, 2130–2144. [Google Scholar] [CrossRef]
- Lynch, M. The genetic interpretation of inbreeding depression and outbreeding depression. Evolution 1991, 45, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D.J.; Bernatchez, L. Adaptive evolutionary conservation: Towards a unified concept for defining conservation units. Mol. Ecol. 2001, 10, 2741–2752. [Google Scholar] [CrossRef]
- Carbonell, R. Managing Spanish European mink populations: Moving from a precautionary approach towards knowledge-based management. J. Nat. Conserv. 2015, 25, 58–61. [Google Scholar] [CrossRef]
- Cabria, M.T.; Gonzalez, E.G.; Gomez-Moliner, B.J.; Michaux, J.R.; Skumatov, D.; Kranz, A.; Fournier, P.; Palazon, S.; Zardoya, R. Patterns of genetic variation in the endangered European mink (Mustela lutreola L., 1761). BMC Evol. Biol. 2015, 15, 141. [Google Scholar] [CrossRef]
- Lodé, T. The European Mink’s Paradox: Near Extinction but Colonizing New Habitats. JSM Biol. 2017, 2, 1011. [Google Scholar] [CrossRef]
- Farquharson, K.A.; Hogg, C.J.; Grueber, C.E. Offspring survival changes over generations of captive breeding. Nat. Commun. 2021, 12, 1–9. [Google Scholar] [CrossRef]
- Haage, M.; Maran, T.; Bergvall, U.A.; Elmhagen, B.; Angerbjörn, A. The influence of spatiotemporal conditions and personality on survival in reintroductions–evolutionary implications. Oecologia 2016, 183, 45–56. [Google Scholar] [CrossRef]
- Kneidinger, N.; Nagl, A.; Kiik, K.; Schwarzenberger, F.; Maran, T. The individual courtship behaviour of male European mink (Mustela lutreola) is a good indicator for their breeding success. Appl. Anim. Behav. Sci. 2018, 205, 98–106. [Google Scholar] [CrossRef]
- van Oers, K.; de Jong, G.; van Noordwijk, A.J.; Drent, P.J.; Kempenaers, B. Contribution of genetics to the study of animal personalities: A review of case studies. Behaviour 2005, 142, 1185–1206. [Google Scholar] [CrossRef]
- Whiteley, A.R.; Fitzpatrick, S.W.; Funk, W.C.; Tallmon, D.A. Genetic rescue to the rescue. Trends Ecol. Evol. 2015, 30, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Serrouya, R.; Seip, D.R.; Hervieux, D.; McLellan, B.N.; McNay, R.S.; Steenweg, R.; Heard, D.C.; Hebblewhite, M.; Gillingham, M.; Boutin, S. Saving endangered species using adaptive management. Proc. Natl. Acad. Sci. USA 2019, 116, 6181–6186. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Prado-Martinez, J.; Sudmant, P.H.; Narasimhan, V.; Ayub, Q.; Szpak, M.; Frandsen, P.; Chen, Y.; Yngvadottir, B.; Cooper, D.N.; et al. Mountain gorilla genomes reveal the impact of long-term population decline and inbreeding. Science 2015, 348, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Batut, B.; Hiltemann, S.; Bagnacani, A.; Baker, D.; Bhardwaj, V.; Blank, C.; Bretaudeau, A.; Brillet-Guéguen, L.; Čech, M.; Chilton, J.; et al. Community-Driven Data Analysis Training for Biology. Cell Syst. 2018, 6, 752–758.e1. [Google Scholar] [CrossRef] [PubMed]
- Hiltemann, S.; Rasche, H.; Gladman, S.; Hotz, H.-R.; Larivière, D.; Blankenberg, D.; Jagtap, P.D.; Wollmann, T.; Bretaudeau, A.; Goué, N.; et al. Galaxy Training: A powerful framework for teaching! PLoS Comput. Biol. 2023, 19, e1010752. [Google Scholar] [CrossRef]
- Lariviere, D.; Ostrovsky, A.; Gallardo, C.; Syme, A.; Abueg, L.; Pickett, B.; Formenti, G.; Sozzoni, M. VGP Assembly Pipeline (Galaxy Training Materials). 2023. Available online: https://training.galaxyproject.org/training-material/topics/assembly/tutorials/vgp_genome_assembly/tutorial.html (accessed on 14 July 2023).
- Bixler, H.; Ellis, C. Ferret care and husbandry. Veter Clin. N. Am. Exot. Anim. Pract. 2004, 7, 227–255. [Google Scholar] [CrossRef]
- Wolf, T.M. Ferrets. In Manual of Exotic Pet Practice; Mitchell, M.A., Tully, T.N., Jr., Eds.; Saunders: St. Louis, MI, USA, 2009; pp. 345–374. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Levene, M.J.; Korlach, J.; Turner, S.W.; Foquet, M.; Craighead, H.G.; Webb, W.W. Zero-Mode Waveguides for Single-Molecule Analysis at High Concentrations. Science 2003, 299, 682–686. [Google Scholar] [CrossRef]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef]
- Zhou, C.; McCarthy, S.A.; Durbin, R. YaHS: Yet another Hi-C scaffolding tool. Bioinformatics 2023, 39, btac808. [Google Scholar] [CrossRef]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Rhie, A.; Walenz, B.P.; Koren, S.; Phillippy, A.M. Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 2020, 21, 245. [Google Scholar] [CrossRef] [PubMed]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Uliano-Silva, M.; Ferreira, J.G.R.N.; Krasheninnikova, K.; Blaxter, M.; Mieszkowska, N.; Hall, N.; Holland, P.; Durbin, R.; Richards, T.; Kersey, P.; et al. MitoHiFi: A python pipeline for mitochondrial genome assembly from PacBio high fidelity reads. bioRxiv 2023, 24, 521667. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W.; Karami, A.; Movaghar, A.F.; Mercier, S.; Ferre, L.; Seligmann, H.; Proença, D.; et al. A Greedy Algorithm for Aligning DNA Sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Formenti, G.; Abueg, L.; Brajuka, A.; Brajuka, N.; Gallardo-Alba, C.; Giani, A.; Fedrigo, O.; Jarvis, E.D. Gfastats: Conversion, evaluation and manipulation of genome sequences using assembly graphs. Bioinformatics 2022, 38, 4214–4216. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol. Biol. 2019, 1962, 227–245. [Google Scholar] [CrossRef]
- Guan, D.; McCarthy, S.A.; Wood, J.; Howe, K.; Wang, Y.; Durbin, R. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 2020, 36, 2896–2898. [Google Scholar] [CrossRef]
- Kerpedjiev, P.; Abdennur, N.; Lekschas, F.; McCallum, C.; Dinkla, K.; Strobelt, H.; Luber, J.M.; Ouellette, S.B.; Azhir, A.; Kumar, N.; et al. HiGlass: Web-based visual exploration and analysis of genome interaction maps. Genome Biol. 2018, 19, 125. [Google Scholar] [CrossRef]
- Harry, E. PretextView (Paired REad TEXTure Viewer): A Desktop Application for Viewing Pretext Contact Maps. 2022. Available online: https://github.com/wtsi-hpag/PretextView (accessed on 15 July 2020).
- NCBI. The NCBI Eukaryotic Genome Annotation Pipeline. 2023. Available online: https://www.ncbi.nlm.nih.gov/genome/annotation_euk/process/ (accessed on 15 July 2023).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NG | Primary Assembly | Alternate Assembly | ||||||
|---|---|---|---|---|---|---|---|---|
| Contigs | Scaffolds | Contigs | Scaffolds | |||||
| LG | Length | LG | Length | LG | Length | LG | Length | |
| 10 | 2 | 161.66 Mbp | 1 | 290.10 Mbp | 137 | 0.79 Mbp | 137 | 0.79 Mbp |
| 20 | 4 | 146.10 Mbp | 3 | 211.29 Mbp | 417 | 0.53 Mbp | 417 | 0.53 Mbp |
| 30 | 6 | 117.74 Mbp | 4 | 202.57 Mbp | 811 | 398.20 Kbp | 811 | 398.20 Kbp |
| 40 | 8 | 94.97 Mbp | 5 | 177.47 Mbp | 1321 | 311.74 Kbp | 1321 | 311.74 Kbp |
| 50 | 11 | 83.36 Mbp | 7 | 154.08 Mbp | 1968 | 245.42 Kbp | 1968 | 245.42 Kbp |
| 60 | 14 | 72.93 Mbp | 8 | 151.51 Mbp | 2788 | 193.83 Kbp | 2788 | 193.83 Kbp |
| 70 | 18 | 63.47 Mbp | 10 | 133.08 Mbp | 3841 | 148.62 Kbp | 3841 | 148.62 Kbp |
| 80 | 23 | 42.37 Mbp | 12 | 104.55 Mbp | 5262 | 105.12 Kbp | 5262 | 105.12 Kbp |
| 90 | 31 | 25.02 Mbp | 15 | 75.87 Mbp | 7424 | 64.08 Kbp | 7424 | 64.08 Kbp |
| 100 | 64 | 38.84 Kbp | 25 | 38.84 Kbp | 12,043 | 10.04 Kbp | 12,043 | 10.04 Kbp |
| 1.000x | 64 | 2.59 Gbp | 25 | 2.59 Gbp | 137 | 0.79 Mbp | 137 | 0.79 Mbp |
| Feature | Pre-Curation Assembly | Curated Assembly |
|---|---|---|
| Expected genome size | 2,572,597,696 | 2,586,268,927 |
| Number of scaffolds | 34 | 25 |
| Total scaffolds length | 2,586,267,127 | 2,586,268,927 |
| Average scaffold length | 76,066,680.21 | 103,450,757.08 |
| Scaffold N50 | 154,078,643 | 154,078,643 |
| Scaffold auN | 163,788,738.52 | 165,291,595.19 |
| Scaffold L50 | 7 | 7 |
| Largest Scaffold | 289,168,877 | 290,104,894 |
| Smallest scaffold | 38,844 | 38,844 |
| Number of contigs | 64 | 64 |
| Total contig length | 2,586,261,127 | 2,586,261,127 |
| Average contig length | 40,410,330.11 | 40,410,330.11 |
| Contig N50 | 83,356,672 | 83,356,672 |
| Contig auN | 90,760,552.39 | 90,760,552.39 |
| Contig L50 | 11 | 11 |
| Largest contig | 171,611,272 | 171,611,272 |
| Smallest contig | 38,844 | 38,844 |
| Number of gaps in scaffolds | 30 | 39 |
| Total gap length in scaffolds | 6000 | 7800 |
| Average gap length in scaffolds | 200 | 200 |
| Gap N50 in scaffolds | 200 | 200 |
| Gap auN in scaffolds | 200 | 200 |
| Gap L50 in scaffolds | 15 | 20 |
| Largest gap in scaffolds | 200 | 200 |
| Smallest gap in scaffolds | 200 | 200 |
| Base composition (A:C:G:T) | 752,087,122:541,204,141:541,131,424:751,838,440 | 752,018,420:541,573,618:540,761,947:751,907,142 |
| GC content (%) | 41.85 | 41.85 |
| Molecule Name | GenBank Sequence | Size (bp) | GC-Content (%) | Unlocalized Count | Merqury’s Quality Value | Merqury’s Error Rate |
|---|---|---|---|---|---|---|
| Chromosome 1 | CM059626.1 | 290,104,894 | 40.0 | 0 | 65.9388 | 2.5475 × 10−7 |
| Chromosome 2 | CM059627.1 | 225,180,311 | 40.5 | 0 | 64.2117 | 3.7917 × 10−7 |
| Chromosome 3 | CM059628.1 | 211,291,007 | 39.5 | 0 | 64.7304 | 3.3648 × 10−7 |
| Chromosome 4 | CM059629.1 | 202,574,368 | 41.5 | 1 | 64.7407 | 3.3568 × 10−7 |
| Chromosome 5 | CM059630.1 | 177,472,721 | 41.0 | 1 | 63.785 | 4.1831 × 10−7 |
| Chromosome 6 | CM059631.1 | 160,010,566 | 40.5 | 0 | 65.6828 | 2.7022 × 10−7 |
| Chromosome 7 | CM059632.1 | 154,078,643 | 42.0 | 0 | 63.1434 | 4.8491 × 10−7 |
| Chromosome 8 | CM059633.1 | 151,511,187 | 41.5 | 0 | 63.7556 | 4.2116 × 10−7 |
| Chromosome 9 | CM059634.1 | 146,271,530 | 42.5 | 0 | 65.0556 | 3.1221 × 10−7 |
| Chromosome 10 | CM059635.1 | 123,813,674 | 43.0 | 0 | 64.5547 | 3.5038 × 10−7 |
| Chromosome 11 | CM059636.1 | 104,548,976 | 43.5 | 0 | 63.8729 | 4.0993 × 10−7 |
| Chromosome 12 | CM059637.1 | 100,611,554 | 42.5 | 0 | 64.2944 | 3.7201 × 10−7 |
| Chromosome 13 | CM059638.1 | 92,103,190 | 39.5 | 0 | 63.4847 | 4.4826 × 10−7 |
| Chromosome 14 | CM059639.1 | 75,872,633 | 43.0 | 0 | 62.7699 | 5.2846 × 10−7 |
| Chromosome 15 | CM059640.1 | 70,922,371 | 47.0 | 0 | 63.3164 | 4.6598 × 10−7 |
| Chromosome 16 | CM059641.1 | 64,033,846 | 46.0 | 1 | 63.6445 | 4.3207 × 10−7 |
| Chromosome 17 | CM059642.1 | 49,398,020 | 48.5 | 0 | 65.3592 | 2.9113 × 10−7 |
| Chromosome 18 | CM059643.1 | 42,373,272 | 40.5 | 0 | 64.4825 | 3.5625 × 10−7 |
| Chromosome X | CM059644.1 | 133,082,756 | 40.0 | 0 | 66.1191 | 2.4439 × 10−7 |
| Chromosome Y | CM059645.1 | 6,758,561 | 47.5 | 1 | 65.6101 | 2.7479 × 10−7 |
| Mitochondrion MT | CM059646.1 | 16,552 | 39.5 | 0 | - | - |
| Unplaced | 38,844 | - | - | - | - | |
| Species | Assembly Size (Gbp) 1 | Assembly Level 2 | Number of Assembled Chromosomes | Number of Assemblies | Assembly Release Date 3 | Reference Genome |
|---|---|---|---|---|---|---|
| Eira barbara | 2.4676–2.4697 | S | - | 2 | 05.10.2021 | ✓ |
| Enhydra lutris kenyoni | 2.4553 | S | - | 1 | 11.09.2017 | ✓ |
| Enhydra lutris nereid | 2.4256 | S | - | 1 | 24.06.2019 | - |
| Gulo gulo | 2.4232 | S | - | 1 | 06.12.2018 | - |
| Gulo gulo luscus | 2.2495–2.3882 | S | - | 3 | 04.08.2022 | ✓ |
| Lontra canadensis | 2.4057 | S | - | 1 | 27.01.2020 | ✓ |
| Lutra lutra | 1.0815–2.4384 | C, Ch | 20 | 2 | 09.12.2019 | ✓ |
| Martes flavigula | 2.4487 | Ch | 21 | 1 | 27.03.2023 | ✓ |
| Martes zibellina | 2.4207 | S | - | 1 | 20.04.2020 | ✓ |
| Meles meles | 2.6927–2.7387 | S, Ch | 23 | 2 | 18.12.2021 | ✓ |
| Mellivora capensis | 3.0912 | S | - | 1 | 15.01.2019 | ✓ |
| Mustela erminea | 1.6402–2.4452 | C, Ch | 23 | 2 | 03.01.2020 | ✓ |
| Mustela lutreola | 1.7870–2.5862 | Ch | 21 | 2 | 11.07.2023 | ✓ |
| Mustela nigripes | 2.4985 | Ch | 19 | 1 | 24.02.2022 | ✓ |
| Mustela nivalis | 2.5010–2.5012 | S | - | 2 | 06.07.2021 | ✓ |
| Mustela putorius | 2.4566–2.9416 | S | - | 10 | 07.08.2019 | - |
| Mustela putorius furo | 1.0954–2.5771 | C, S | - | 15 | 02.06.2011 | ✓ |
| Neogale vison | 2.4472–2.6812 | S, Ch | 15 | 2 | 02.01.2018 | ✓ |
| Pteronura brasiliensis | 2.6023 | S | - | 1 | 15.01.2019 | ✓ |
| Taxidea taxus jeffersonii | 2.4160 | S | - | 1 | 30.10.2018 | ✓ |
| Feature | Individual | |
|---|---|---|
| Assembly identifier | mMusLut2 | mMustLut3 |
| Ex-situ Programme (EEP) studbook number | 3587 | 3708 |
| Specimen | M1207 | M1287 |
| Sex | ♂ | ♂ |
| Age at sampling (months) | 22 | 9 |
| Site of birth | the Zoological Garden in Osnabrück (Lower Saxony, Germany) | the Zoological Garden in Mönchengladbach (North Rhine-Westphalia, Germany) |
| Degree of kinship | father (P) | son (F1) |
| Research use of sample | HiFi (high-fidelity) reads, genome assembly | Hi-C (all-versus-all chromatin conformation capture) data |
| Information | Individual | ||
|---|---|---|---|
| General identifiers | |||
| Isolate (assembly identifier) | mMusLut2 | mMustLut3 | |
| Assembly type | principal pseudohaplotype | alternate pseudohaplotype | - |
| Whole genome sequencing project accession data | |||
| BioProject | PRJNA984926 | PRJNA984927 | - |
| BioSample ID | SAMN35784236 | - | |
| WGS project | JAUCGO01 | JAUCGP01 | - |
| Raw data accessions | |||
| PacBio Sequel IIe HiFi data | https://genomeark.s3.amazonaws.com/index.html?prefix=species/Mustela_lutreola/mMusLut2/ (accesed on 1 July 2023) | - | |
| Hi-C Dovetail Genomics data | - | - | https://genomeark.s3.amazonaws.com/index.html?prefix=species/Mustela_lutreola/mMusLut3/ (accesed on 1 July 2023) |
| Genome assembly | |||
| GenBank Accession | GCA_030435805.1 | GCA_030435785.1 | - |
| Mitochondrial Assembly (GenBank) | CM059646.1 | - | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skorupski, J.; Brandes, F.; Seebass, C.; Festl, W.; Śmietana, P.; Balacco, J.; Jain, N.; Tilley, T.; Abueg, L.; Wood, J.; et al. Prioritizing Endangered Species in Genome Sequencing: Conservation Genomics in Action with the First Platinum-Standard Reference-Quality Genome of the Critically Endangered European Mink Mustela lutreola L., 1761. Int. J. Mol. Sci. 2023, 24, 14816. https://doi.org/10.3390/ijms241914816
Skorupski J, Brandes F, Seebass C, Festl W, Śmietana P, Balacco J, Jain N, Tilley T, Abueg L, Wood J, et al. Prioritizing Endangered Species in Genome Sequencing: Conservation Genomics in Action with the First Platinum-Standard Reference-Quality Genome of the Critically Endangered European Mink Mustela lutreola L., 1761. International Journal of Molecular Sciences. 2023; 24(19):14816. https://doi.org/10.3390/ijms241914816
Chicago/Turabian StyleSkorupski, Jakub, Florian Brandes, Christian Seebass, Wolfgang Festl, Przemysław Śmietana, Jennifer Balacco, Nivesh Jain, Tatiana Tilley, Linelle Abueg, Jonathan Wood, and et al. 2023. "Prioritizing Endangered Species in Genome Sequencing: Conservation Genomics in Action with the First Platinum-Standard Reference-Quality Genome of the Critically Endangered European Mink Mustela lutreola L., 1761" International Journal of Molecular Sciences 24, no. 19: 14816. https://doi.org/10.3390/ijms241914816
APA StyleSkorupski, J., Brandes, F., Seebass, C., Festl, W., Śmietana, P., Balacco, J., Jain, N., Tilley, T., Abueg, L., Wood, J., Sims, Y., Formenti, G., Fedrigo, O., & Jarvis, E. D. (2023). Prioritizing Endangered Species in Genome Sequencing: Conservation Genomics in Action with the First Platinum-Standard Reference-Quality Genome of the Critically Endangered European Mink Mustela lutreola L., 1761. International Journal of Molecular Sciences, 24(19), 14816. https://doi.org/10.3390/ijms241914816