
Amyloid-β Effects on Peripheral Nerve: A New Model System


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
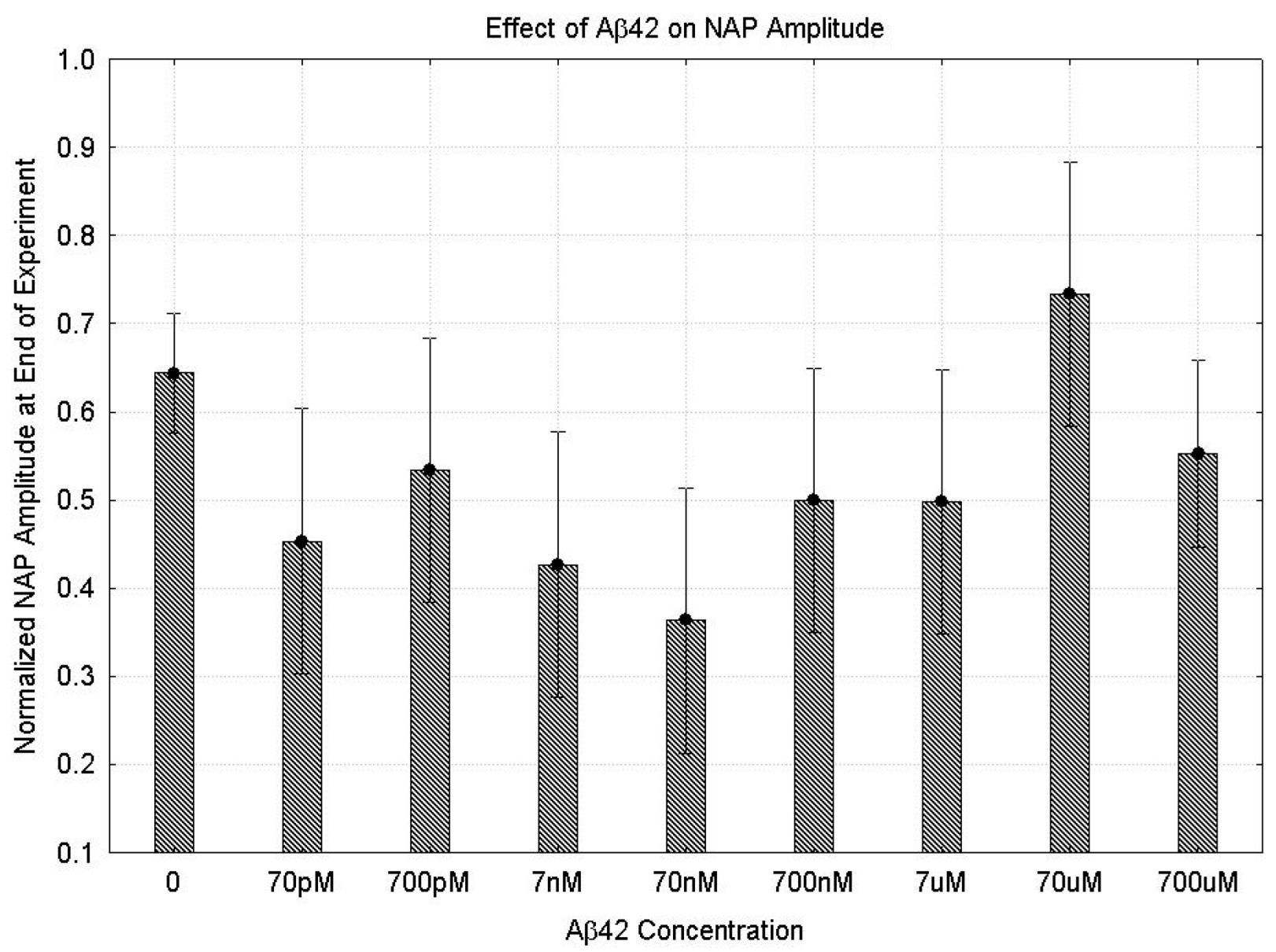
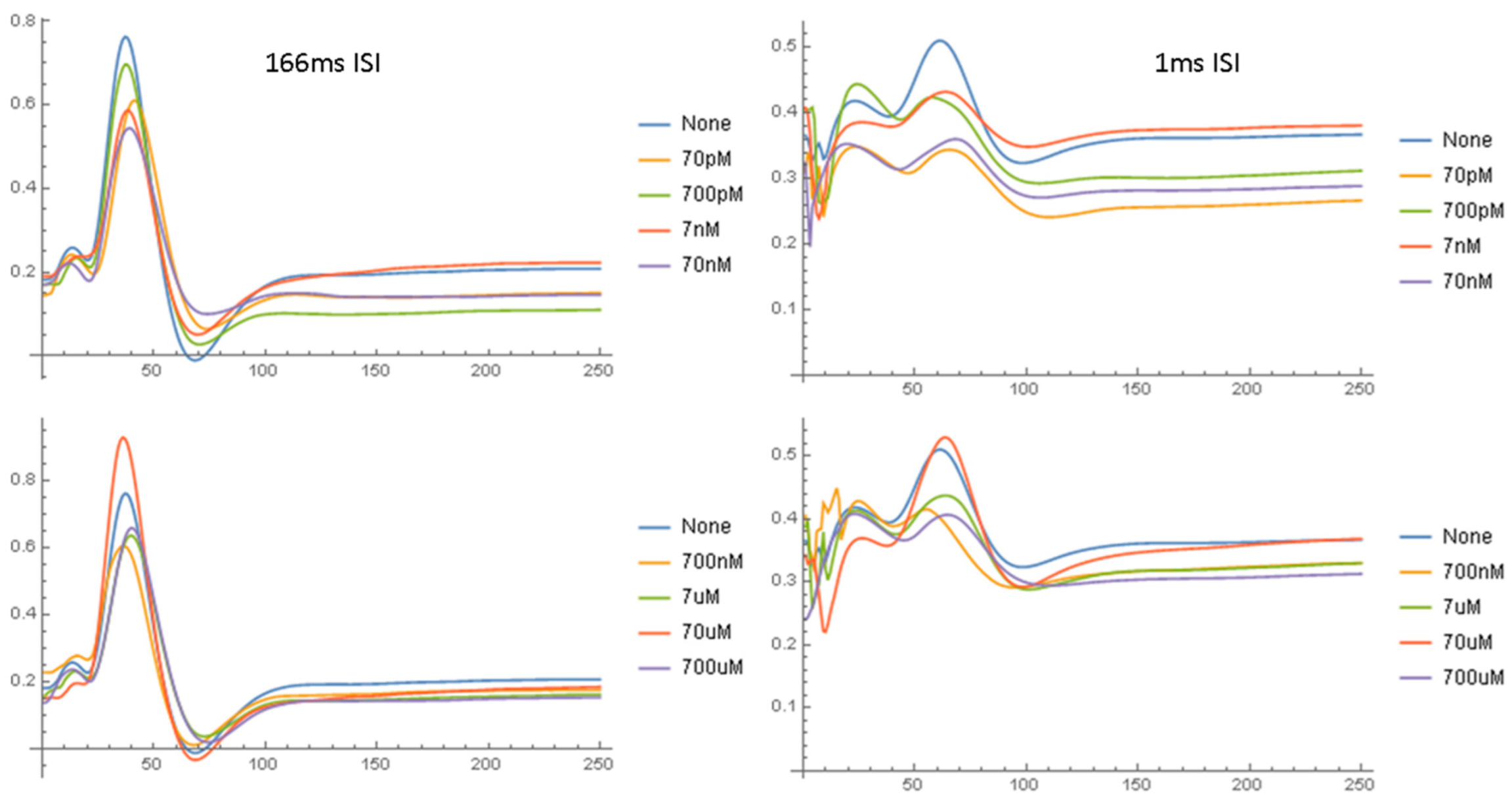
2.1. Effects of Aβ42 on the NAP
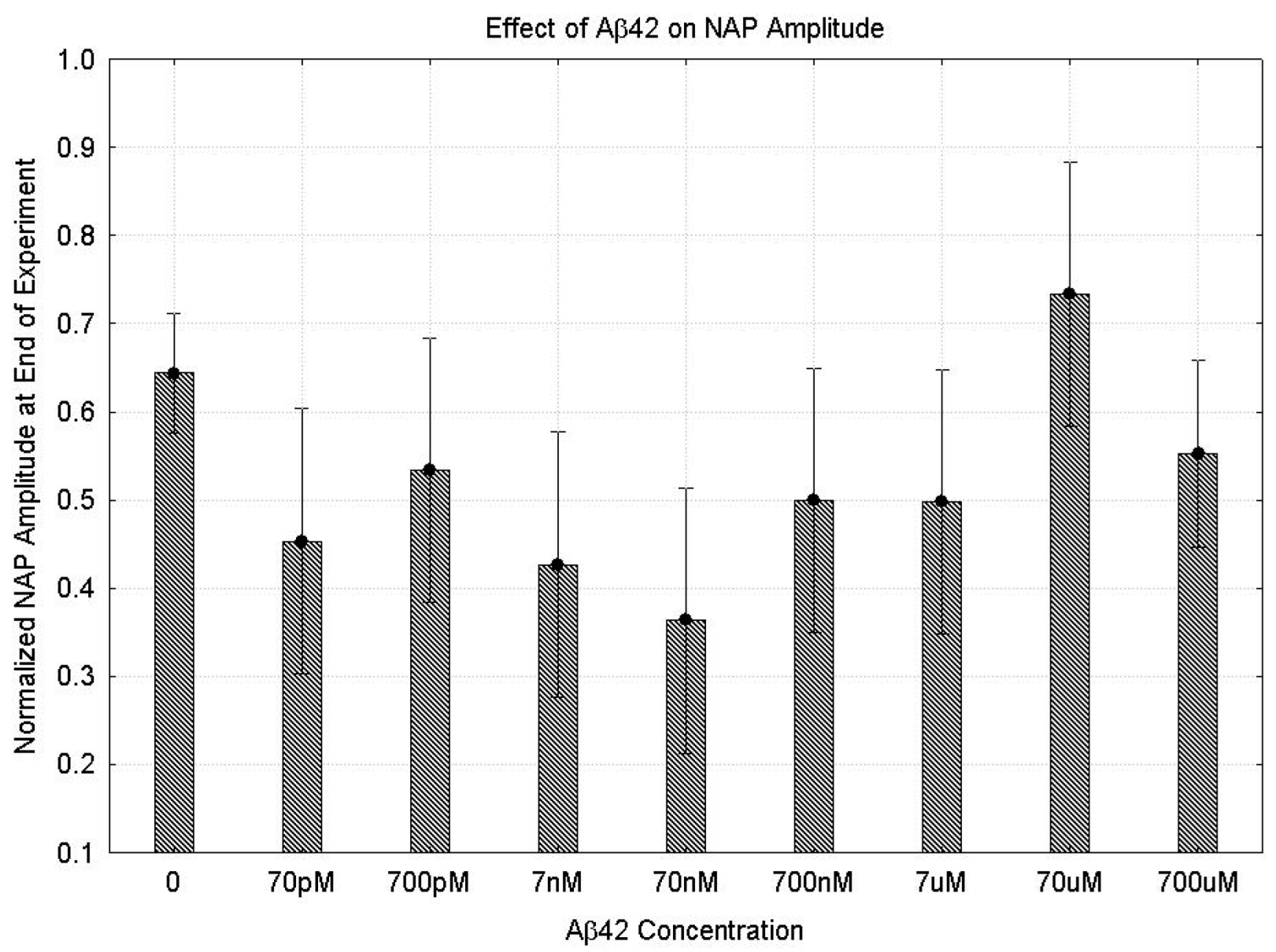
2.1.1. Parametric Data
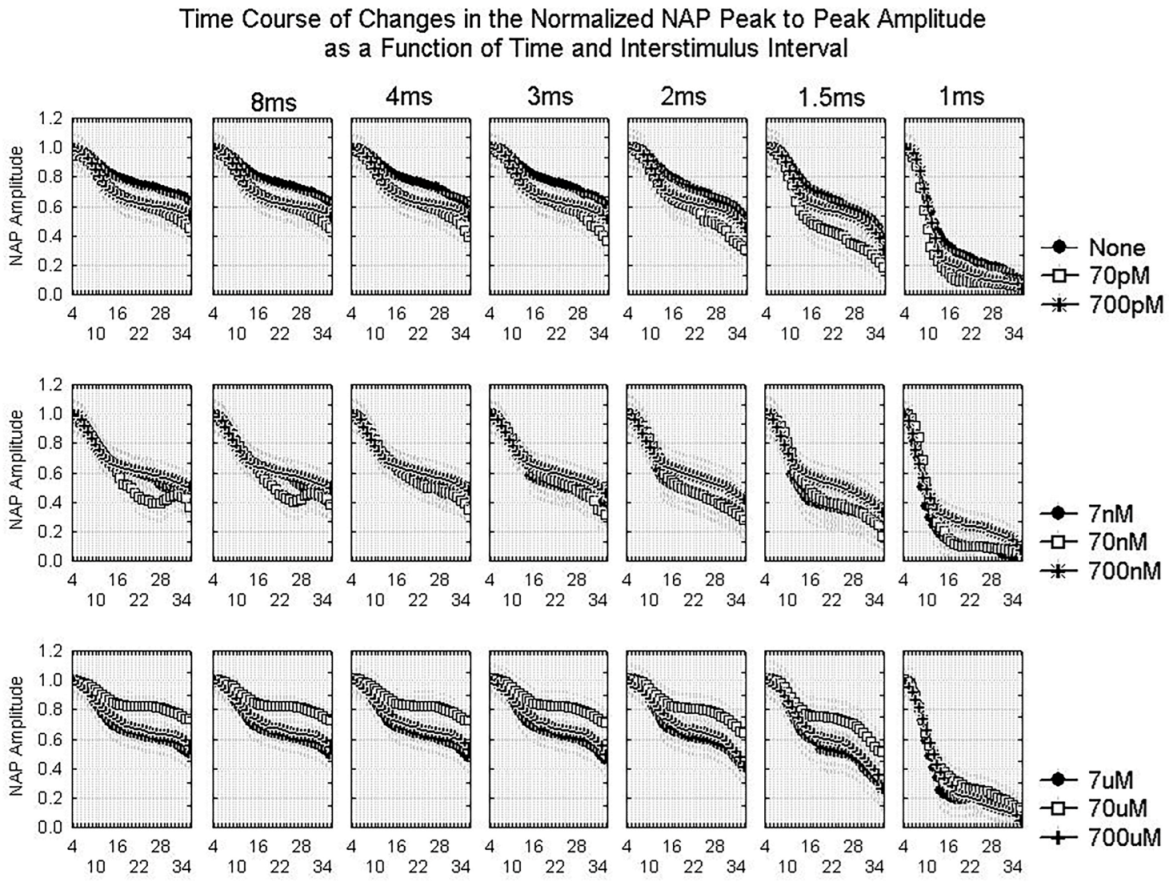
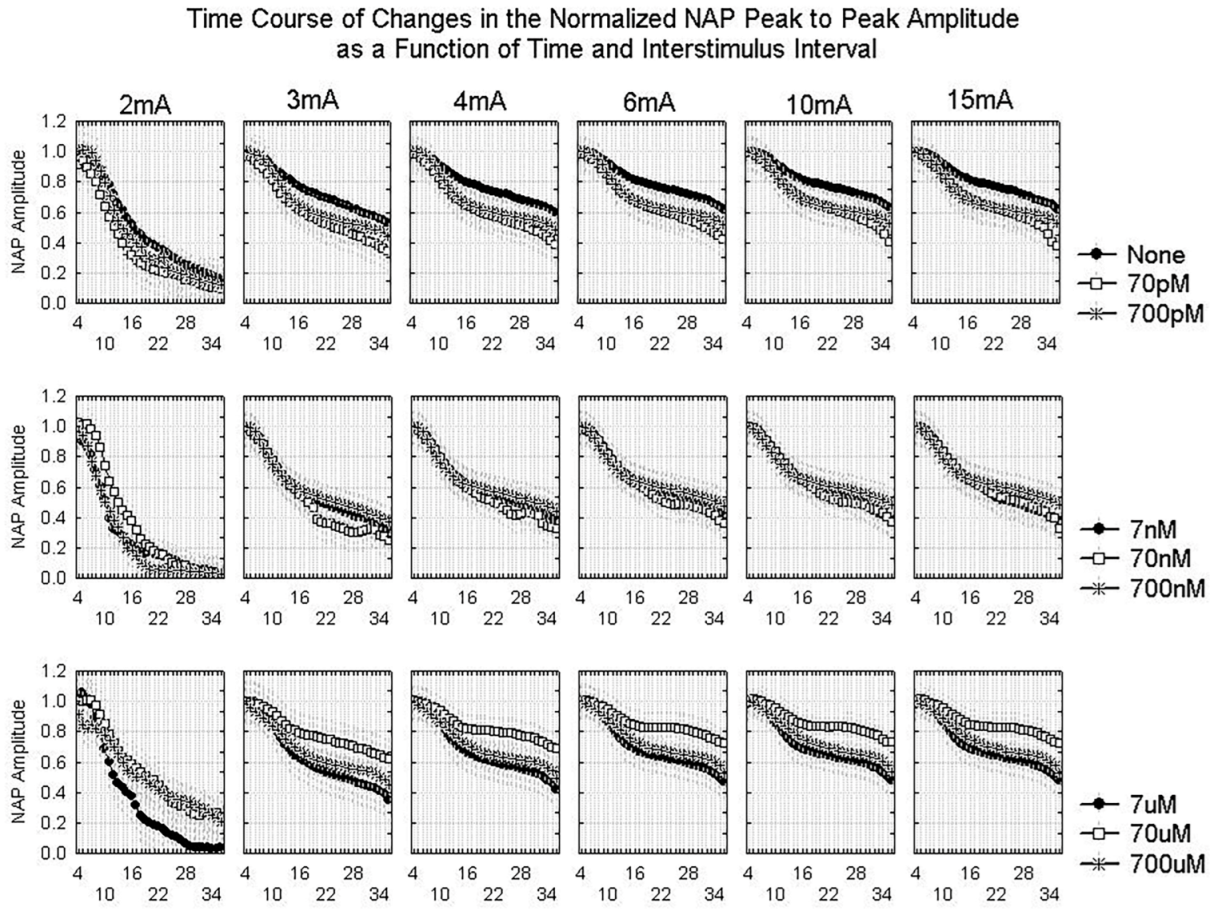
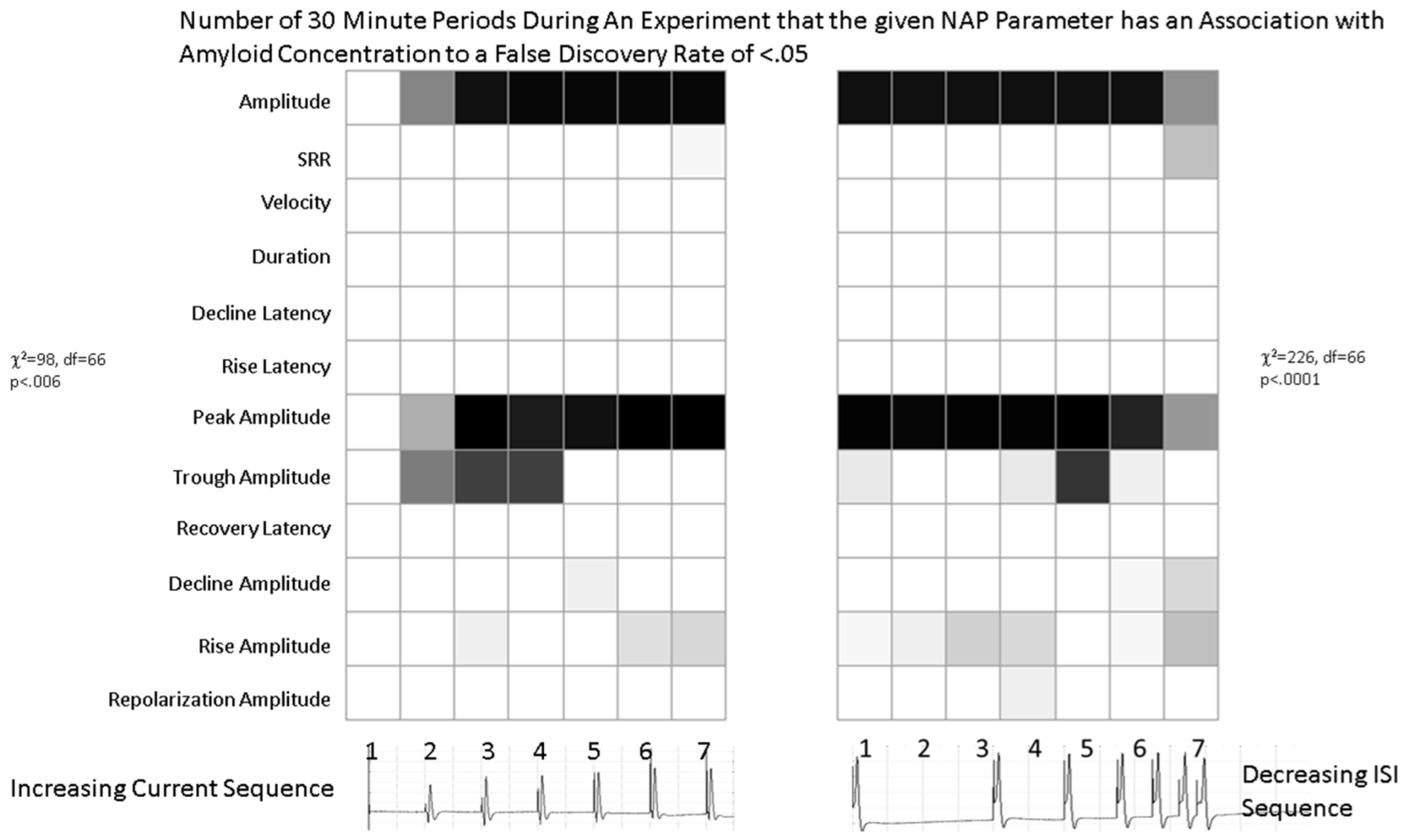
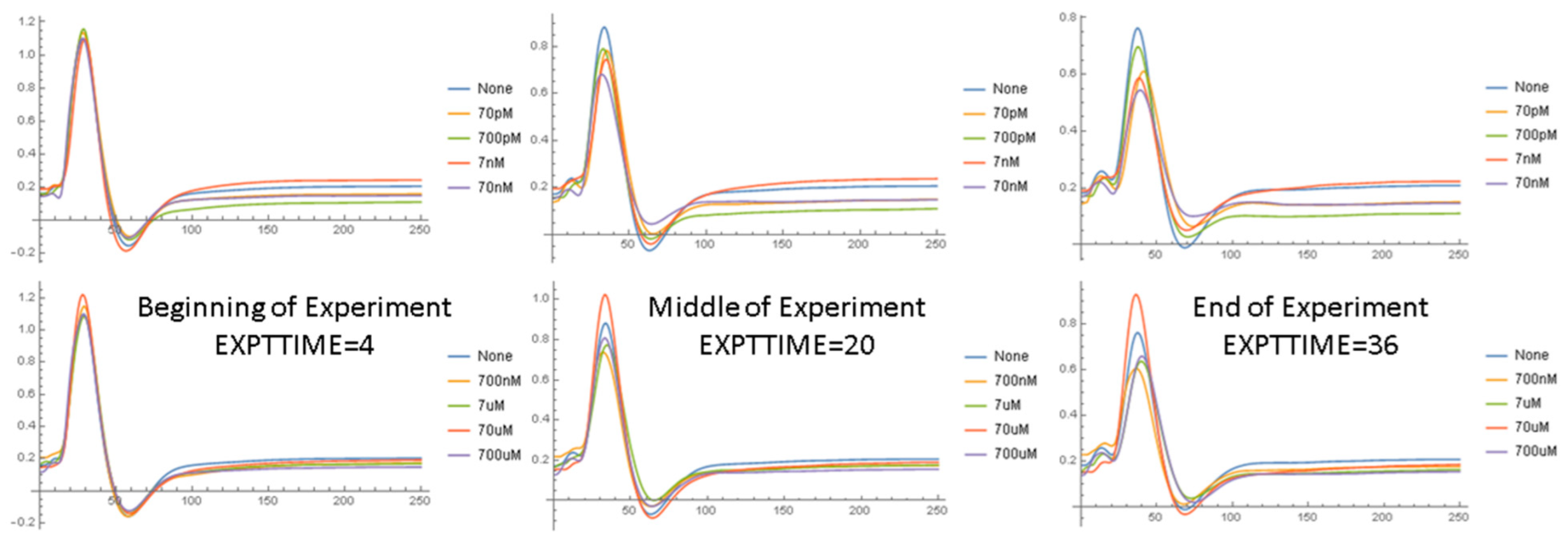
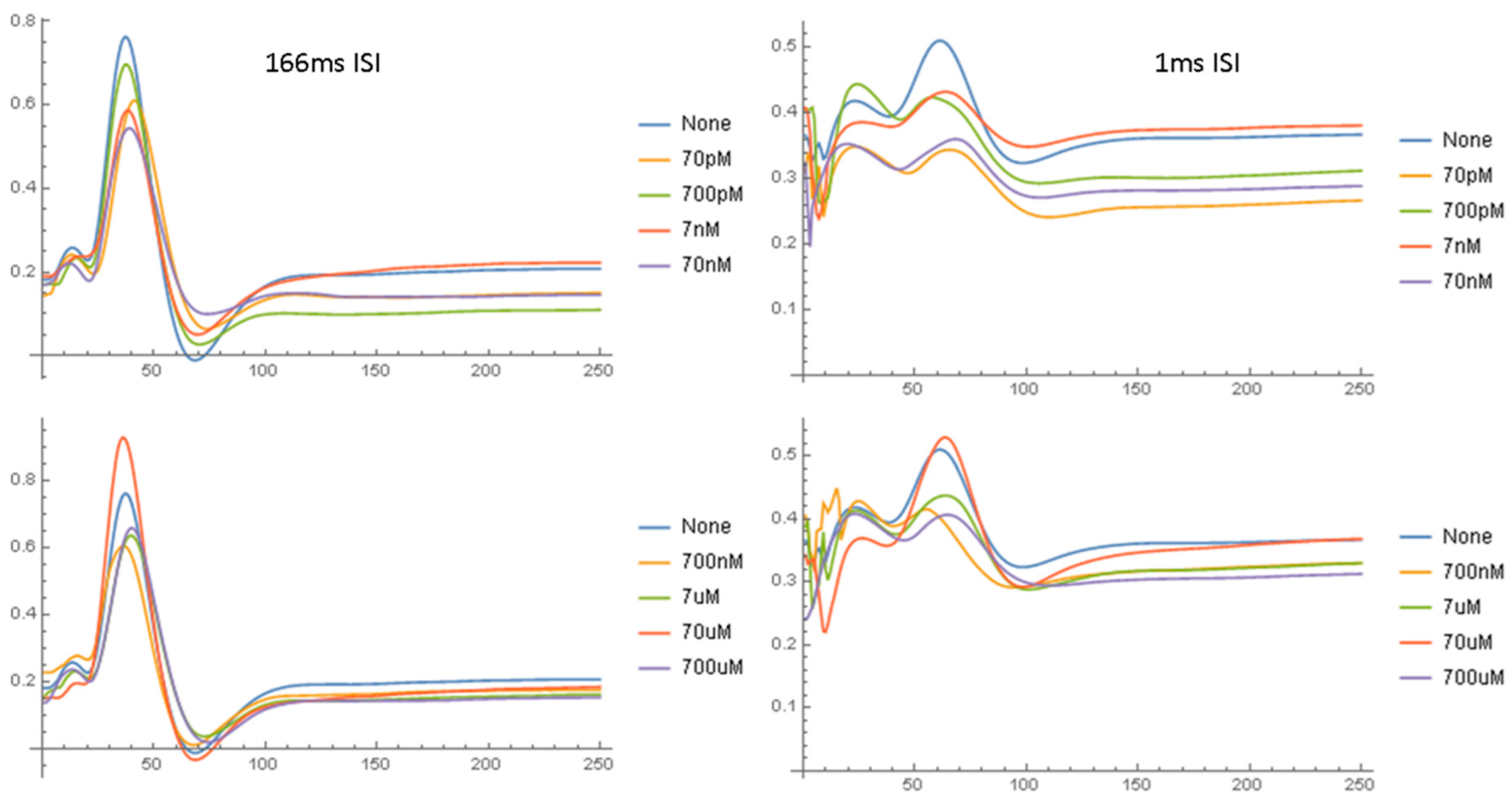
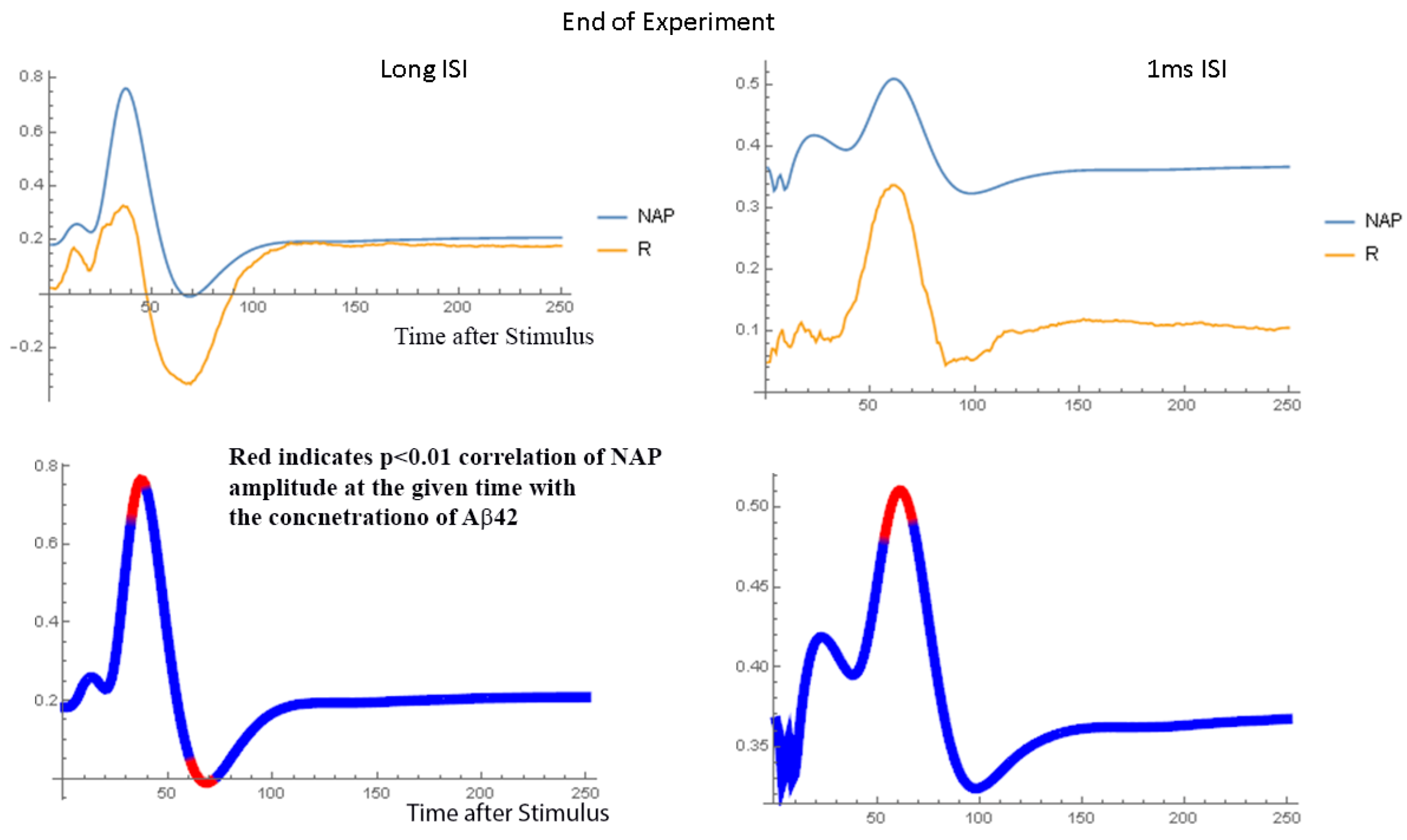
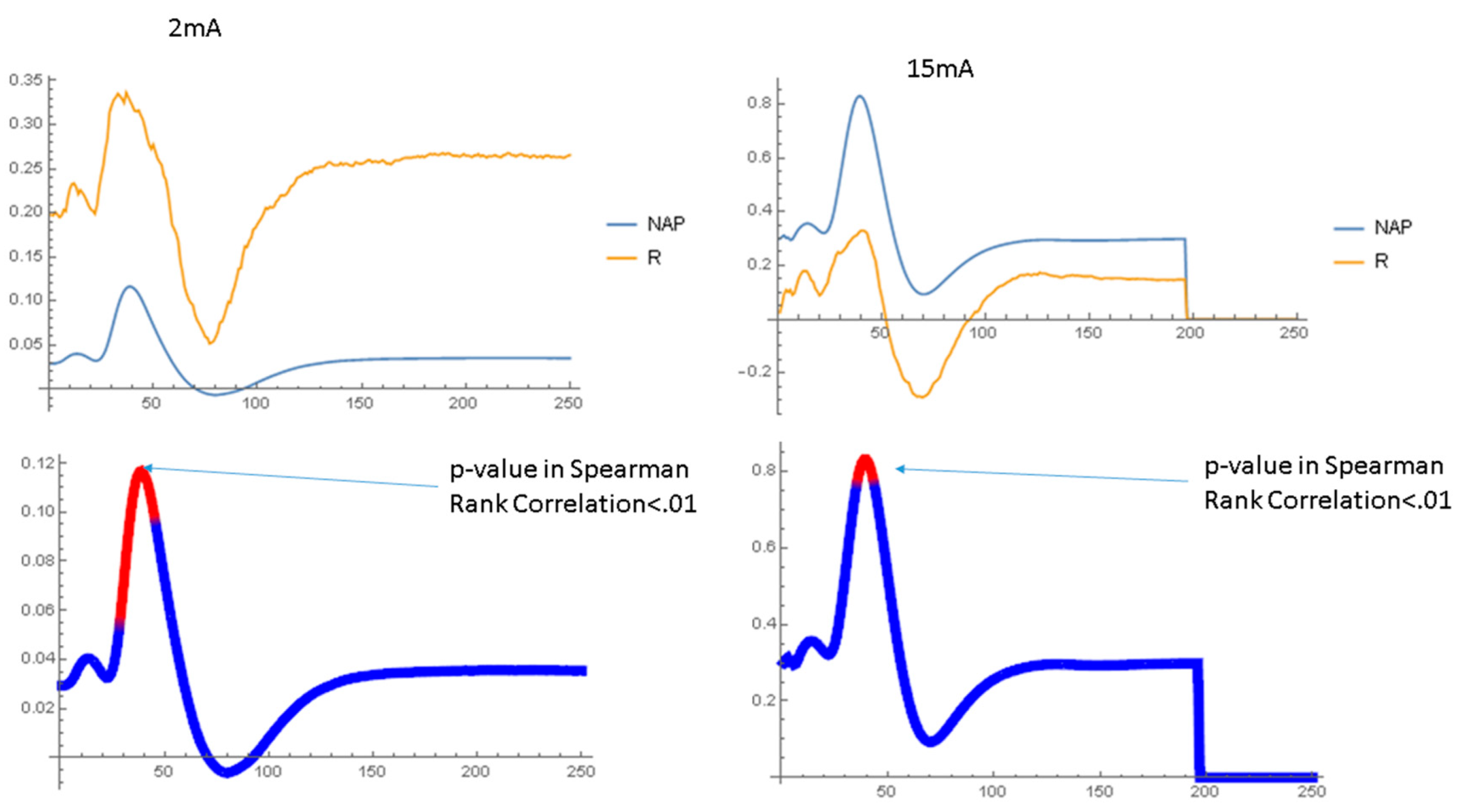
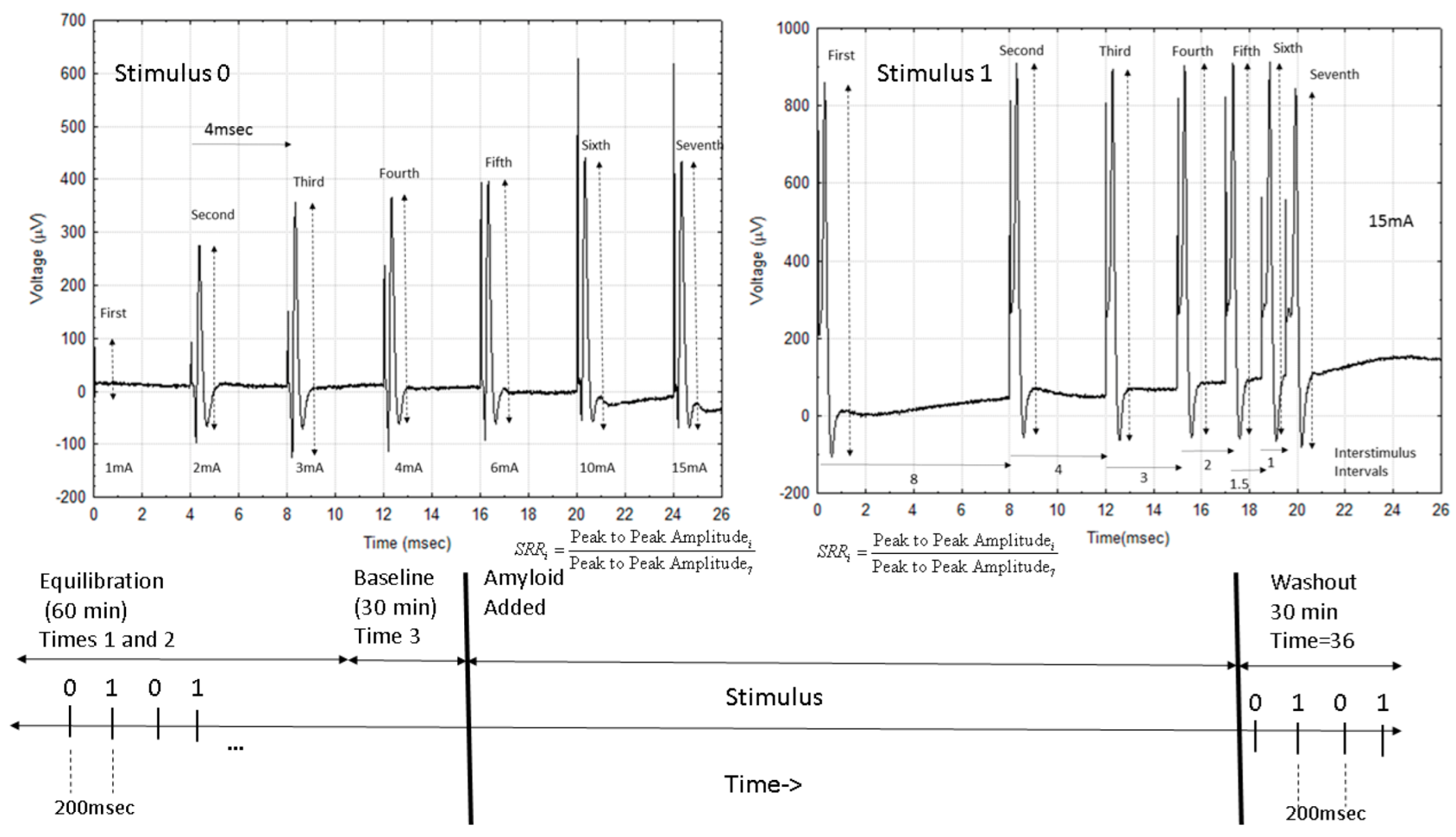
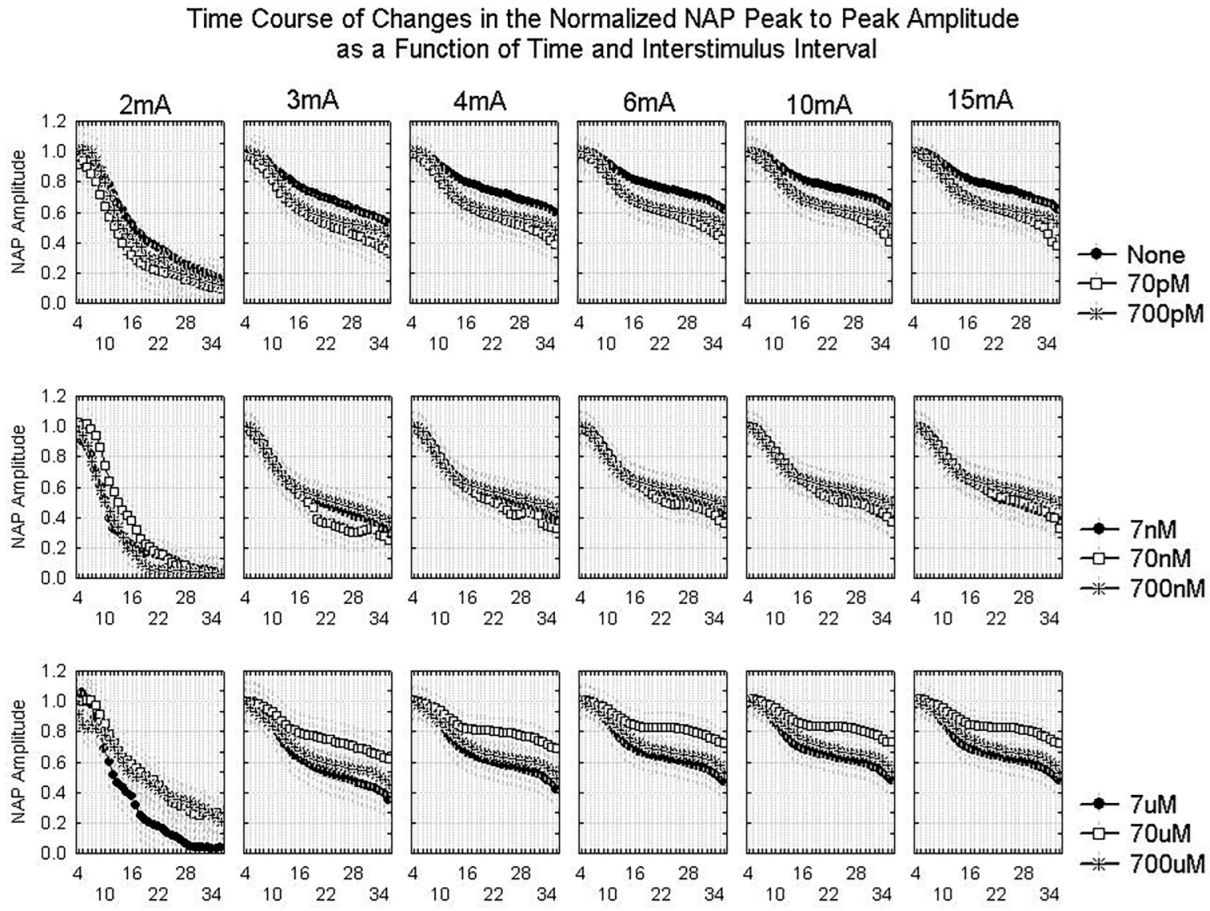
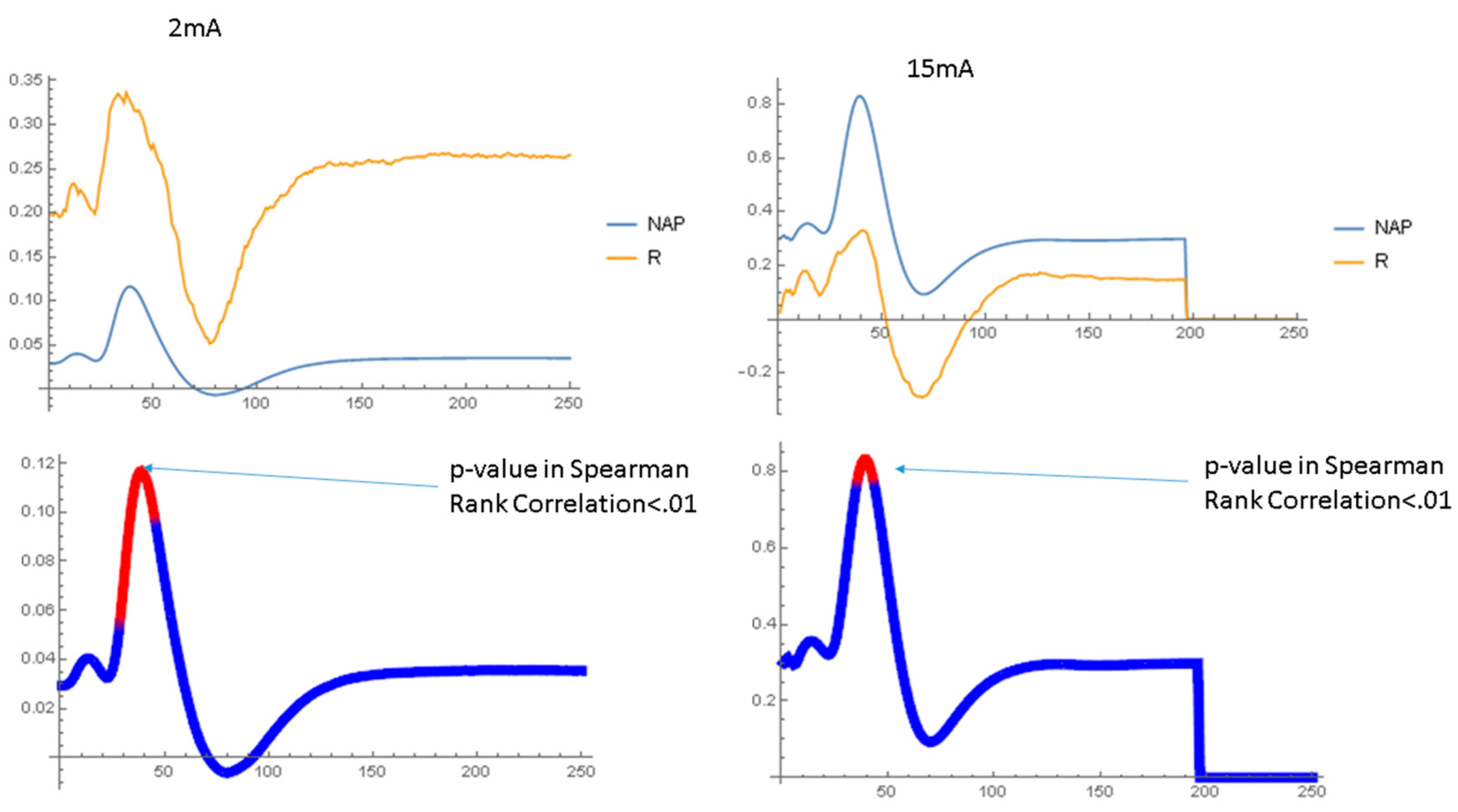
2.1.2. Waveform Data
3. Discussion
3.1. Physiologic Effects of Aβ42 on Peripheral Nerve
3.2. Concentration Effect of Aβ42
3.3. Dynamics of Aβ42 Effect
4. Materials and Methods
4.1. Experimental Setup
4.2. Preparation of Aβ42
4.3. Electrophysiology
4.4. Statistics
4.4.1. Parametric Analyses
4.4.2. Statistical Testing
4.4.3. Corrections for Multiple Testing
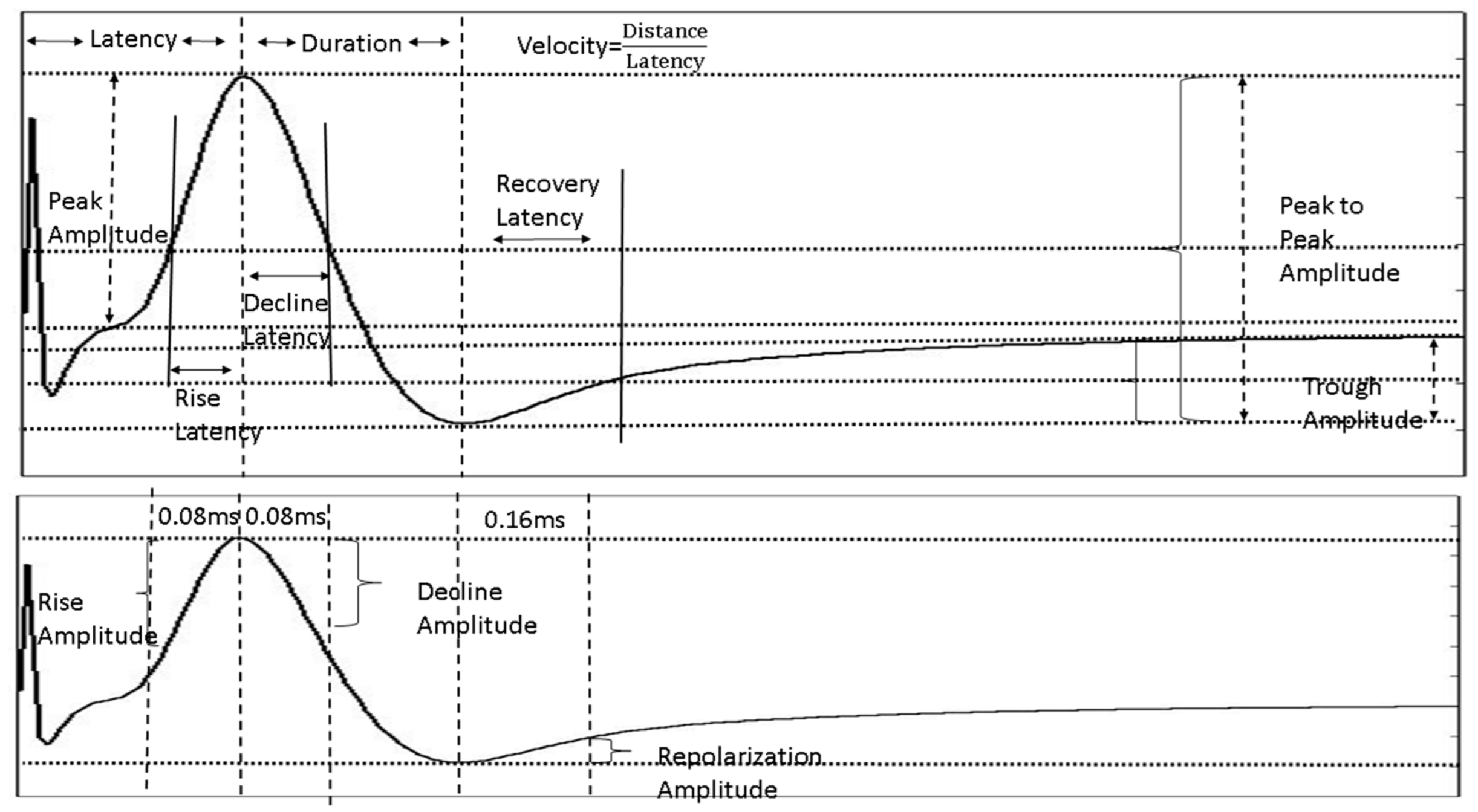
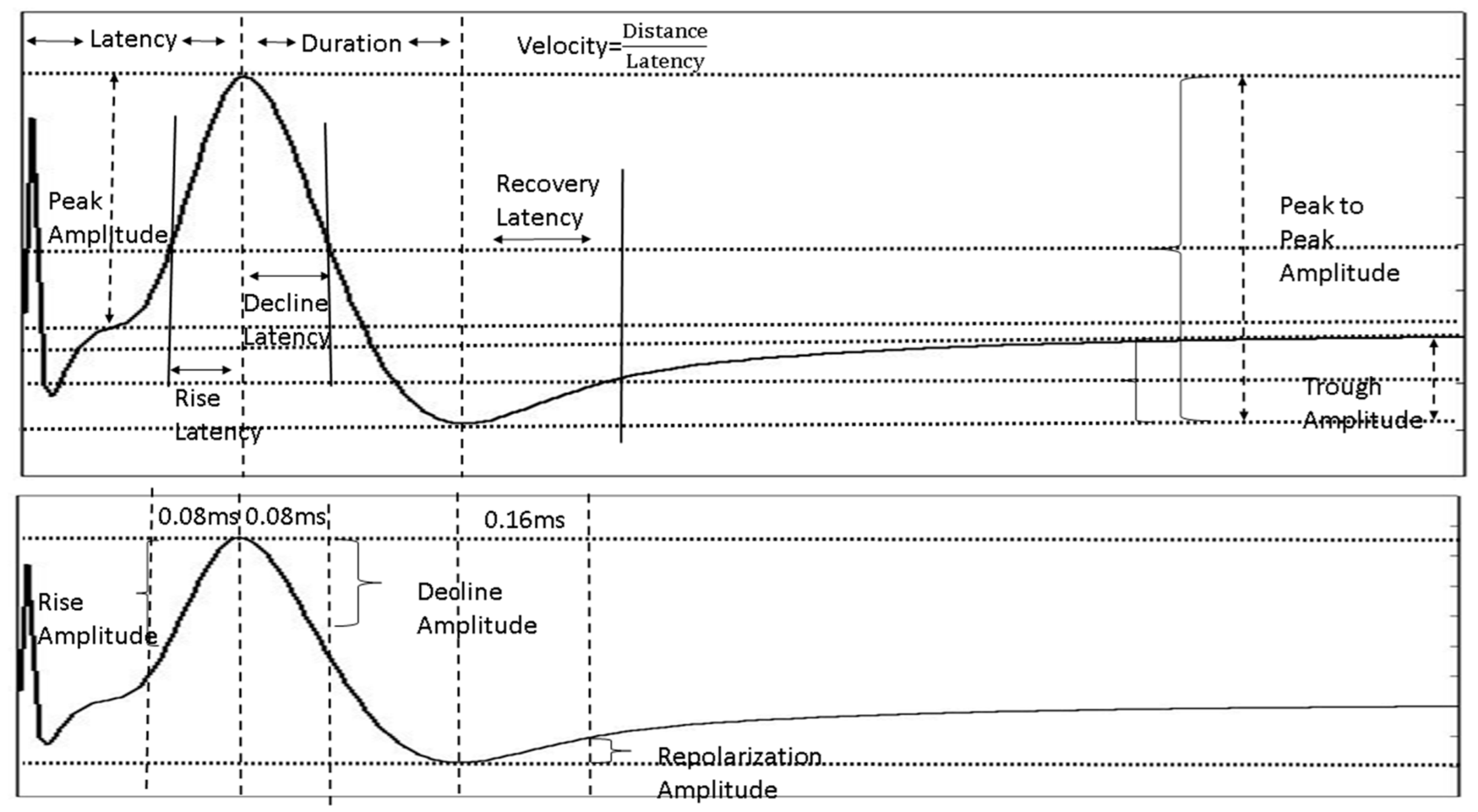
4.4.4. NAP Waveform Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-β peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef]
- Sadigh-Eteghad, S.; Sabermarouf, B.; Majdi, A.; Talebi, M.; Farhoudi, M.; Mahmoudi, J. Amyloid-β: A crucial factor in Alzheimer’s disease. Med. Princ. Pract. 2015, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lesne, S.E.; Sherman, M.A.; Grant, M.; Kuskowski, M.; Schneider, J.A.; Bennett, D.A.; Ashe, K.H. Brain amyloid-β oligomers in ageing and Alzheimer’s disease. Brain 2013, 136, 1383–1398. [Google Scholar] [PubMed]
- Reiss, A.B.; Glass, A.D.; Wisniewski, T.; Wolozin, B.; Gomolin, I.H.; Pinkhasov, A.; De Leon, J.; Stecker, M.M. Alzheimer’s disease: Many failed trials, so where do we go from here? J. Investig. Med. 2020, 68, 1135–1140. [Google Scholar] [PubMed]
- Faull, M.; Ching, S.Y.; Jarmolowicz, A.I.; Beilby, J.; Panegyres, P.K. Comparison of two methods for the analysis of CSF Aβ and tau in the diagnosis of Alzheimer’s disease. Am. J. Neurodegener. Dis. 2014, 3, 143–151. [Google Scholar]
- Kang, J.H.; Korecka, M.; Toledo, J.B.; Trojanowski, J.Q.; Shaw, L.M. Clinical utility and analytical challenges in measurement of cerebrospinal fluid amyloid-β(1–42) and tau proteins as Alzheimer disease biomarkers. Clin. Chem. 2013, 59, 903–916. [Google Scholar]
- Sojkova, J.; Resnick, S.M. In vivo human amyloid imaging. Curr. Alzheimer Res. 2011, 8, 366–372. [Google Scholar] [CrossRef]
- Ossenkoppele, R.; Jansen, W.J.; Rabinovici, G.D.; Knol, D.L.; van der Flier, W.M.; van Berckel, B.N.; Scheltens, P.; Visser, P.J.; Amyloid PET Study Group; Verfaillie, S.C.; et al. Prevalence of amyloid PET positivity in dementia syndromes: A meta-analysis. JAMA 2015, 313, 1939–1949. [Google Scholar] [CrossRef] [PubMed]
- Ruan, D.; Sun, L. Amyloid-β PET in Alzheimer’s disease: A systematic review and Bayesian meta-analysis. Brain Behav. 2023, 13, e2850. [Google Scholar] [CrossRef]
- Nikiforova, A.; Sedov, I. Molecular Design of Magnetic Resonance Imaging Agents Binding to Amyloid Deposits. Int. J. Mol. Sci. 2023, 24, 11152. [Google Scholar] [CrossRef]
- Yang, J.; Zhu, B.; Yin, W.; Han, Z.; Zheng, C.; Wang, P.; Ran, C. Differentiating Aβ40 and Aβ42 in amyloid plaques with a small molecule fluorescence probe. Chem. Sci. 2020, 11, 5238–5245. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Wirthensohn, D.C.; Flagmeier, P.; Hughes, C.; Aprile, F.A.; Ruggeri, F.S.; Whiten, D.R.; Emin, D.; Xia, Z.; Varela, J.A.; et al. Different soluble aggregates of Aβ42 can give rise to cellular toxicity through different mechanisms. Nat. Commun. 2019, 10, 1541–1543. [Google Scholar] [CrossRef]
- Miller, A.; Wei, J.; Meehan, S.; Dobson, C.M.; Welland, M.E.; Klenerman, D.; Vendruscolo, M.; Ruggeri, F.S.; Knowles, T.P.J. Formation of amyloid loops in brain tissues is controlled by the flexibility of protofibril chains. Proc. Natl. Acad. Sci. USA 2023, 120, e2216234120. [Google Scholar] [CrossRef]
- Sano, M. Neuropsychological testing in the diagnosis of dementia. J. Geriatr. Psychiatry Neurol. 2006, 19, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.J.; Bachstetter, A.D.; Nelson, P.T.; Schmitt, F.A.; Van Eldik, L.J. Using mice to model Alzheimer’s dementia: An overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 2014, 5, 88. [Google Scholar] [CrossRef]
- Gamaldo, A.A.; An, Y.; Allaire, J.C.; Kitner-Triolo, M.H.; Zonderman, A.B. Variability in performance: Identifying early signs of future cognitive impairment. Neuropsychology 2012, 26, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, X.G.; Zhou, T.T.; Li, N.; Jang, C.Y.; Xiao, Z.C.; Ma, Q.H.; Li, S. Elevated Neuronal Excitability Due to Modulation of the Voltage-Gated Sodium Channel Nav1.6 by Aβ1–42. Front. Neurosci. 2016, 10, 94. [Google Scholar] [CrossRef] [PubMed]
- Tamano, H.; Ide, K.; Adlard, P.A.; Bush, A.I.; Takeda, A. Involvement of hippocampal excitability in amyloid β-induced behavioral and psychological symptoms of dementia. J. Toxicol. Sci. 2016, 41, 449–457. [Google Scholar] [PubMed]
- Scala, F.; Fusco, S.; Ripoli, C.; Piacentini, R.; Li Puma, D.D.; Spinelli, M.; Laezza, F.; Grassi, C.; D’Ascenzo, M. Intraneuronal Aβ accumulation induces hippocampal neuron hyperexcitability through A-type K+ current inhibition mediated by activation of caspases and GSK-3. Neurobiol. Aging 2015, 36, 886–900. [Google Scholar] [CrossRef] [PubMed]
- Cummings, D.M.; Liu, W.; Portelius, E.; Bayram, S.; Yasvoina, M.; Ho, S.H.; Smits, H.; Ali, S.S.; Steinberg, R.; Pegasiou, C.M.; et al. First effects of rising amyloid-β in transgenic mouse brain: Synaptic transmission and gene expression. Brain 2015, 138, 1992–2004. [Google Scholar] [CrossRef]
- Price, K.A.; Varghese, M.; Sowa, A.; Yuk, F.; Brautigam, H.; Ehrlich, M.E.; Dickstein, D.L. Altered synaptic structure in the hippocampus in a mouse model of Alzheimer’s disease with soluble amyloid-β oligomers and no plaque pathology. Mol. Neurodegener 2014, 9, 41. [Google Scholar] [PubMed]
- Hoxha, E.; Boda, E.; Montarolo, F.; Parolisi, R.; Tempia, F. Excitability and synaptic alterations in the cerebellum of APP/PS1 mice. PLoS ONE 2012, 7, e34726. [Google Scholar]
- Lenz, M.; Eichler, A.; Kruse, P.; Galanis, C.; Kleidonas, D.; Andrieux, G.; Boerries, M.; Jedlicka, P.; Müller, U.; Deller, T.; et al. The Amyloid Precursor Protein Regulates Synaptic Transmission at Medial Perforant Path Synapses. J. Neurosci. 2023, 43, 5290–5304. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Guo, S.; Guo, F.; Liu, E.; Yang, X.; Feng, B.; Wang, J.; Vreugdenhil, M.; Lu, C. Oligomeric β-Amyloid Suppresses Hippocampal γ-Oscillations through Activation of the mTOR/S6K1 Pathway. Aging Dis. 2023, 14, 1390–1406. [Google Scholar] [CrossRef]
- Kagiava, A.; Kosmidis, E.K.; Theophilidis, G. Oxaliplatin-induced hyperexcitation of rat sciatic nerve fibers: An intra-axonal study. Anti-Cancer Agents Med. Chem. 2013, 13, 373–379. [Google Scholar] [CrossRef]
- Stecker, M.; Segelnick, J.; Wilkenfeld, M. Analysis of short-term effects of world trade center dust on rat sciatic nerve. J. Occup. Environ. Med. 2014, 56, 1024–1028. [Google Scholar] [CrossRef] [PubMed]
- Fern, R.; Davis, P.; Waxman, S.G.; Ransom, B.R. Axon conduction and survival in CNS white matter during energy deprivation: A developmental study. J. Neurophysiol. 1998, 79, 95–105. [Google Scholar]
- Stecker, M.M.; Stevenson, M.R. Anoxia-induced changes in optimal substrate for peripheral nerve. Neuroscience 2015, 284, 653–667. [Google Scholar] [PubMed]
- Brown, A.M.; Evans, R.D.; Black, J.; Ransom, B.R. Schwann cell glycogen selectively supports myelinated axon function. Ann. Neurol. 2012, 72, 406–418. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, J.M.; Straub, R.W. Oxygen consumption and phosphate efflux in mammalian non-myelinated nerve fibres [proceedings]. J. Physiol. 1979, 295, 53P–54P. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Ohkoshi, N.; Shoji, S.; Yamamoto, N.; Nagata, H. Hyperglycemia exacerbates the effect of ischemia-reperfusion on peripheral nerve in rat. Int. J. Neurosci. 2000, 105, 53–62. [Google Scholar] [CrossRef]
- Stecker, M.M.; Stevenson, M. The effect of glucose concentration on peripheral nerve and its response to anoxia. Muscle Nerve 2013, 49, 370–377. [Google Scholar]
- Matsuda, M.; Gono, T.; Morita, H.; Katoh, N.; Kodaira, M.; Ikeda, S. Peripheral nerve involvement in primary systemic AL amyloidosis: A clinical and electrophysiological study. Eur. J. Neurol. 2011, 18, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Willem, M.; Garratt, A.N.; Novak, B.; Citron, M.; Kaufmann, S.; Rittger, A.; DeStrooper, B.; Saftig, P.; Birchmeier, C.; Haass, C. Control of peripheral nerve myelination by the β-secretase BACE1. Science 2006, 314, 664–666. [Google Scholar] [PubMed]
- Sachse, C.C.; Kim, Y.H.; Agsten, M.; Huth, T.; Alzheimer, C.; Kovacs, D.M.; Kim, D.Y. BACE1 and presenilin/gamma-secretase regulate proteolytic processing of KCNE1 and 2, auxiliary subunits of voltage-gated potassium channels. FASEB J. 2013, 27, 2458–2467. [Google Scholar] [CrossRef] [PubMed]
- Huth, T.; Rittger, A.; Saftig, P.; Alzheimer, C. β-Site APP-cleaving enzyme 1 (BACE1) cleaves cerebellar Na+ channel β4-subunit and promotes Purkinje cell firing by slowing the decay of resurgent Na+ current. Pflügers Arch. 2011, 461, 355–371. [Google Scholar] [PubMed]
- Huth, T.; Schmidt-Neuenfeldt, K.; Rittger, A.; Saftig, P.; Reiss, K.; Alzheimer, C. Non-proteolytic effect of β-site APP-cleaving enzyme 1 (BACE1) on sodium channel function. Neurobiol. Dis. 2009, 33, 282–289. [Google Scholar] [CrossRef]
- Kim, D.Y.; Carey, B.W.; Wang, H.; Ingano, L.A.; Binshtok, A.M.; Wertz, M.H.; Pettingell, W.H.; He, P.; Lee, V.M.; Woolf, C.J.; et al. BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat. Cell Biol. 2007, 9, 755–764. [Google Scholar] [PubMed]
- Jolivalt, C.G.; Calcutt, N.A.; Masliah, E. Similar pattern of peripheral neuropathy in mouse models of type 1 diabetes and Alzheimer’s disease. Neuroscience 2012, 202, 405–412. [Google Scholar] [CrossRef]
- Jafari, Z.; Afrashteh, N.; Kolb, B.E.; Mohajerani, M.H. Hearing loss and impaired short-term memory in an Alzheimer’s disease mouse model of amyloid-β pathology. Exp. Neurol. 2023, 365, 114413. [Google Scholar] [CrossRef]
- O’Leary, T.P.; Shin, S.; Fertan, E.; Dingle, R.N.; Almuklass, A.; Gunn, R.K.; Yu, Z.; Wang, J.; Brown, R.E. Reduced acoustic startle response and peripheral hearing loss in the 5xFAD mouse model of Alzheimer’s disease. Genes Brain Behav. 2017, 16, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Valincius, G.; Heinrich, F.; Budvytyte, R.; Vanderah, D.J.; McGillivray, D.J.; Sokolov, Y.; Hall, J.E.; Losche, M. Soluble amyloid β-oligomers affect dielectric membrane properties by bilayer insertion and domain formation: Implications for cell toxicity. Biophys. J. 2008, 95, 4845–4861. [Google Scholar] [PubMed]
- Frankel, D.; Davies, M.; Bhushan, B.; Kulaberoglu, Y.; Urriola-Munoz, P.; Bertrand-Michel, J.; Pergande, M.R.; Smith, A.A.; Preet, S.; Park, T.J.; et al. Cholesterol-rich naked mole-rat brain lipid membranes are susceptible to amyloid β-induced damage in vitro. Aging 2020, 12, 22266–22290. [Google Scholar] [CrossRef]
- Sasahara, K.; Morigaki, K.; Shinya, K. Effects of membrane interaction and aggregation of amyloid β-peptide on lipid mobility and membrane domain structure. Phys. Chem. Chem Phys. 2013, 15, 8929–8939. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.; Lai, W.; Jin, L.; Kuo, K.; Lee, M. Electrophysiological parameters that contribute to the pathogenesis of familial amyloid polyneuropathy caused by transthyretin mutations. J. Neurol. Sci. 2020, 413, 116810. [Google Scholar] [CrossRef]
- Ohashi, N.; Kodaira, M.; Morita, H.; Sekijima, Y. Electrophysiological demyelinating features in hereditary ATTR amyloidosis. Amyloid 2019, 26, 15–23. [Google Scholar] [CrossRef]
- Davion, J.; Bocquillon, P.; Cassim, F.; Frezel, N.; Lacour, A.; Dhaenens, C.; Maurage, C.; Gibier, J.; Hachulla, E.; Nguyen The Tich, S.; et al. Electro-clinical presentation of hereditary transthyretin related amyloidosis when presenting as a polyneuropathy of unknown origin in northern France. Rev. Neurol. 2021, 177, 1160–1167. [Google Scholar] [CrossRef]
- Tozza, S.; Severi, D.; Spina, E.; Iovino, A.; Aruta, F.; Ruggiero, L.; Dubbioso, R.; Iodice, R.; Nolano, M.; Manganelli, F. The neuropathy in hereditary transthyretin amyloidosis: A narrative review. J. Peripher. Nerv. Syst. 2021, 26, 155–159. [Google Scholar] [CrossRef]
- Carrillo-Mora, P.; Luna, R.; Colin-Barenque, L. Amyloid β: Multiple mechanisms of toxicity and only some protective effects? Oxid. Med. Cell Longev. 2014, 2014, 795375. [Google Scholar] [CrossRef]
- Bukanova, J.V.; Sharonova, I.N.; Skrebitsky, V.G. Amyloid β peptide (25–35) in picomolar concentrations modulates the function of glycine receptors in rat hippocampal pyramidal neurons through interaction with extracellular site(s). Brain Res. 2014, 1558, 1–10. [Google Scholar]
- Kontush, A.; Berndt, C.; Weber, W.; Akopyan, V.; Arlt, S.; Schippling, S.; Beisiegel, U. Amyloid-β is an antioxidant for lipoproteins in cerebrospinal fluid and plasma. Free Radic. Biol. Med. 2001, 30, 119–128. [Google Scholar] [CrossRef]
- Sinha, M.; Bhowmick, P.; Banerjee, A.; Chakrabarti, S. Antioxidant role of amyloid β protein in cell-free and biological systems: Implication for the pathogenesis of Alzheimer disease. Free Radic. Biol. Med. 2013, 56, 184–192. [Google Scholar] [CrossRef]
- Giuffrida, M.L.; Caraci, F.; Pignataro, B.; Cataldo, S.; De Bona, P.; Bruno, V.; Molinaro, G.; Pappalardo, G.; Messina, A.; Palmigiano, A.; et al. β-amyloid monomers are neuroprotective. J. Neurosci. 2009, 29, 10582–10587. [Google Scholar] [CrossRef] [PubMed]
- Niidome, T.; Goto, Y.; Kato, M.; Wang, P.L.; Goh, S.; Tanaka, N.; Akaike, A.; Kihara, T.; Sugimoto, H. Non-fibrillar amyloid-β peptide reduces NMDA-induced neurotoxicity, but not AMPA-induced neurotoxicity. Biochem. Biophys. Res. Commun. 2009, 386, 734–738. [Google Scholar] [CrossRef]
- Ghosh, P.; Kumar, A.; Datta, B.; Rangachari, V. Dynamics of protofibril elongation and association involved in Aβ42 peptide aggregation in Alzheimer’s disease. BMC Bioinform. 2010, 11 (Suppl. 6), S24. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Knowles, T.P.; Linse, S. On the lag phase in amyloid fibril formation. Phys. Chem. Chem. Phys. 2015, 17, 7606–7618. [Google Scholar]
- Walsh, D.M.; Hartley, D.M.; Kusumoto, Y.; Fezoui, Y.; Condron, M.M.; Lomakin, A.; Benedek, G.B.; Selkoe, D.J.; Teplow, D.B. Amyloid β-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J. Biol. Chem. 1999, 274, 25945–25952. [Google Scholar] [CrossRef] [PubMed]
- Lomakin, A.; Teplow, D.B.; Kirschner, D.A.; Benedek, G.B. Kinetic theory of fibrillogenesis of amyloid β-protein. Proc. Natl. Acad. Sci. USA 1997, 94, 7942–7947. [Google Scholar] [CrossRef]
- Bergeron, D.; Ossenkoppele, R.; Jr Laforce, R. Evidence-based Interpretation of Amyloid-β PET Results: A Clinician’s Tool. Alzheimer Dis. Assoc. Disord. 2018, 32, 28–34. [Google Scholar] [CrossRef]
- Volloch, V.; Olsen, B.R.; Rits, S. Precursor-Independent Overproduction of β-Amyloid in AD: Mitochondrial Dysfunction as Possible Initiator of Asymmetric RNA-Dependent βAPP mRNA Amplification. An Engine that Drives Alzheimer’s Disease. Ann. Integr. Mol. Med. 2019, 1, 61–74. [Google Scholar] [CrossRef]
- Puzzo, D.; Gulisano, W.; Arancio, O.; Palmeri, A. The keystone of Alzheimer pathogenesis might be sought in Aβ physiology. Neuroscience 2015, 307, 26–36. [Google Scholar] [CrossRef]
- Burlando, B.; Losacco, S.; Villa, V.; Fedele, E.; Ricciarelli, R. A New Bistable Switch Model of Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2022, 23, 7061. [Google Scholar] [CrossRef] [PubMed]
- Stecker, M.; Wolfe, J.; Stevenson, M. Neurophysiologic responses of peripheral nerve to repeated episodes of anoxia. Clin. Neurophysiol. 2013, 124, 792–800. [Google Scholar] [CrossRef]
- Baylor, K.; Stecker, M.M. Peripheral nerve at extreme low temperatures 2: Pharmacologic modulation of temperature effects. Cryobiology 2009, 59, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Stecker, M.M.; Baylor, K. Peripheral nerve at extreme low temperatures 1: Effects of temperature on the action potential. Cryobiology 2009, 59, 1–11. [Google Scholar] [CrossRef]
- Hansson, O.; Lehmann, S.; Otto, M.; Zetterberg, H.; Lewczuk, P. Advantages and disadvantages of the use of the CSF Amyloid β (Aβ) 42/40 ratio in the diagnosis of Alzheimer’s Disease. Alzheimers Res. Ther. 2019, 11, 34. [Google Scholar] [CrossRef]
- Lazarevic, V.; Fieńko, S.; Andres-Alonso, M.; Anni, D.; Ivanova, D.; Montenegro-Venegas, C.; Gundelfinger, E.D.; Cousin, M.A.; Fejtova, A. Physiological Concentrations of Amyloid β Regulate Recycling of Synaptic Vesicles via Alpha7 Acetylcholine Receptor and CDK5/Calcineurin Signaling. Front. Mol. Neurosci. 2017, 10, 221. [Google Scholar] [CrossRef]
- Bjerke, M.; Portelius, E.; Minthon, L.; Wallin, A.; Anckarsater, H.; Anckarsater, R.; Andreasen, N.; Zetterberg, H.; Andreasson, U.; Blennow, K. Confounding factors influencing amyloid β concentration in cerebrospinal fluid. Int. J. Alzheimers Dis. 2010, 2010, 986310. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.D.; Pirttila, T.; Mehta, S.P.; Sersen, E.A.; Aisen, P.S.; Wisniewski, H.M. Plasma and cerebrospinal fluid levels of amyloid β proteins 1-40 and 1-42 in Alzheimer disease. Arch. Neurol. 2000, 57, 100–105. [Google Scholar] [CrossRef]
- Puzzo, D.; Privitera, L.; Leznik, E.; Fa, M.; Staniszewski, A.; Palmeri, A.; Arancio, O. Picomolar amyloid-β positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 2008, 28, 14537–14545. [Google Scholar] [PubMed]
- Fagan, A.M.; Mintun, M.A.; Mach, R.H.; Lee, S.Y.; Dence, C.S.; Shah, A.R.; LaRossa, G.N.; Spinner, M.L.; Klunk, W.E.; Mathis, C.A.; et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Aβ42 in humans. Ann. Neurol. 2006, 59, 512–519. [Google Scholar] [CrossRef]
- Weigand, S.D.; Vemuri, P.; Wiste, H.J.; Senjem, M.L.; Pankratz, V.S.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Shaw, L.M.; Trojanowski, J.Q.; et al. Alzheimer’s Disease Neuroimaging Initiative Transforming cerebrospinal fluid Aβ42 measures into calculated Pittsburgh Compound B units of brain Aβ amyloid. Alzheimers Dement. 2011, 7, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Raskatov, J.A. What Is the “Relevant” Amyloid β42 Concentration? Chembiochem 2019, 20, 1725–1726. [Google Scholar] [CrossRef] [PubMed]
- Pachahara, S.K.; Chaudhary, N.; Subbalakshmi, C.; Nagaraj, R. Hexafluoroisopropanol induces self-assembly of β-amyloid peptides into highly ordered nanostructures. J. Pept. Sci. 2012, 18, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Pachahara, S.K.; Adicherla, H.; Nagaraj, R. Self-Assembly of Aβ40, Aβ42 and Aβ43 Peptides in Aqueous Mixtures of Fluorinated Alcohols. PLoS ONE 2015, 10, e0136567. [Google Scholar] [CrossRef]
- Benseny-Cases, N.; Klementieva, O.; Cladera, J. In vitro oligomerization and fibrillogenesis of amyloid-β peptides. Subcell. Biochem. 2012, 65, 53–74. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stecker, M.M.; Srivastava, A.; Reiss, A.B. Amyloid-β Effects on Peripheral Nerve: A New Model System. Int. J. Mol. Sci. 2023, 24, 14488. https://doi.org/10.3390/ijms241914488
Stecker MM, Srivastava A, Reiss AB. Amyloid-β Effects on Peripheral Nerve: A New Model System. International Journal of Molecular Sciences. 2023; 24(19):14488. https://doi.org/10.3390/ijms241914488
Chicago/Turabian StyleStecker, Mark M., Ankita Srivastava, and Allison B. Reiss. 2023. "Amyloid-β Effects on Peripheral Nerve: A New Model System" International Journal of Molecular Sciences 24, no. 19: 14488. https://doi.org/10.3390/ijms241914488
APA StyleStecker, M. M., Srivastava, A., & Reiss, A. B. (2023). Amyloid-β Effects on Peripheral Nerve: A New Model System. International Journal of Molecular Sciences, 24(19), 14488. https://doi.org/10.3390/ijms241914488