
(+)-Lipoic Acid Reduces Lipotoxicity and Regulates Mitochondrial Homeostasis and Energy Balance in an In Vitro Model of Liver Steatosis


 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results
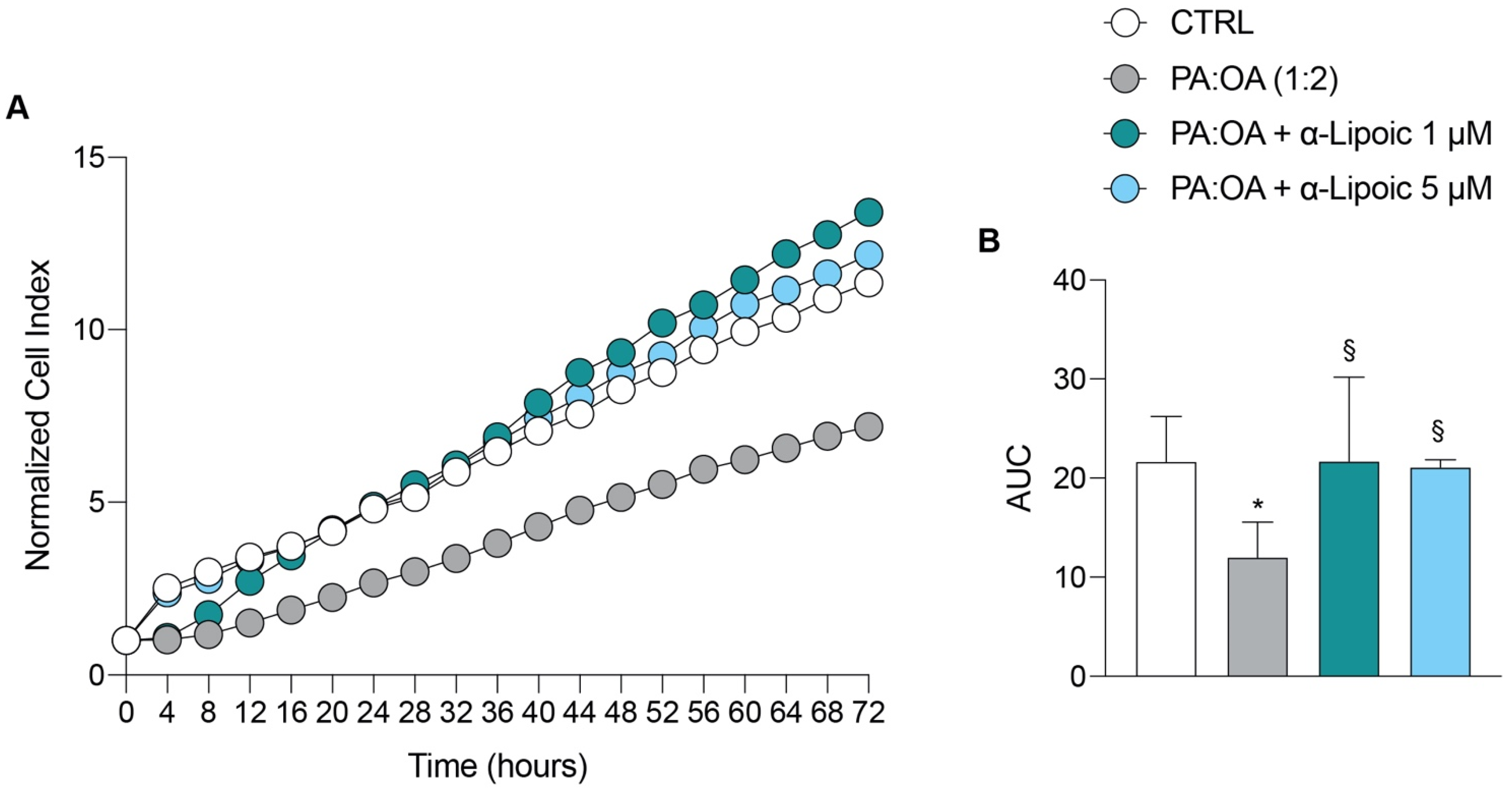
2.1. Lipoic Acid Restores the Effect of Palmitic Acid/Oleic Acid on HepG2 Cell Viability
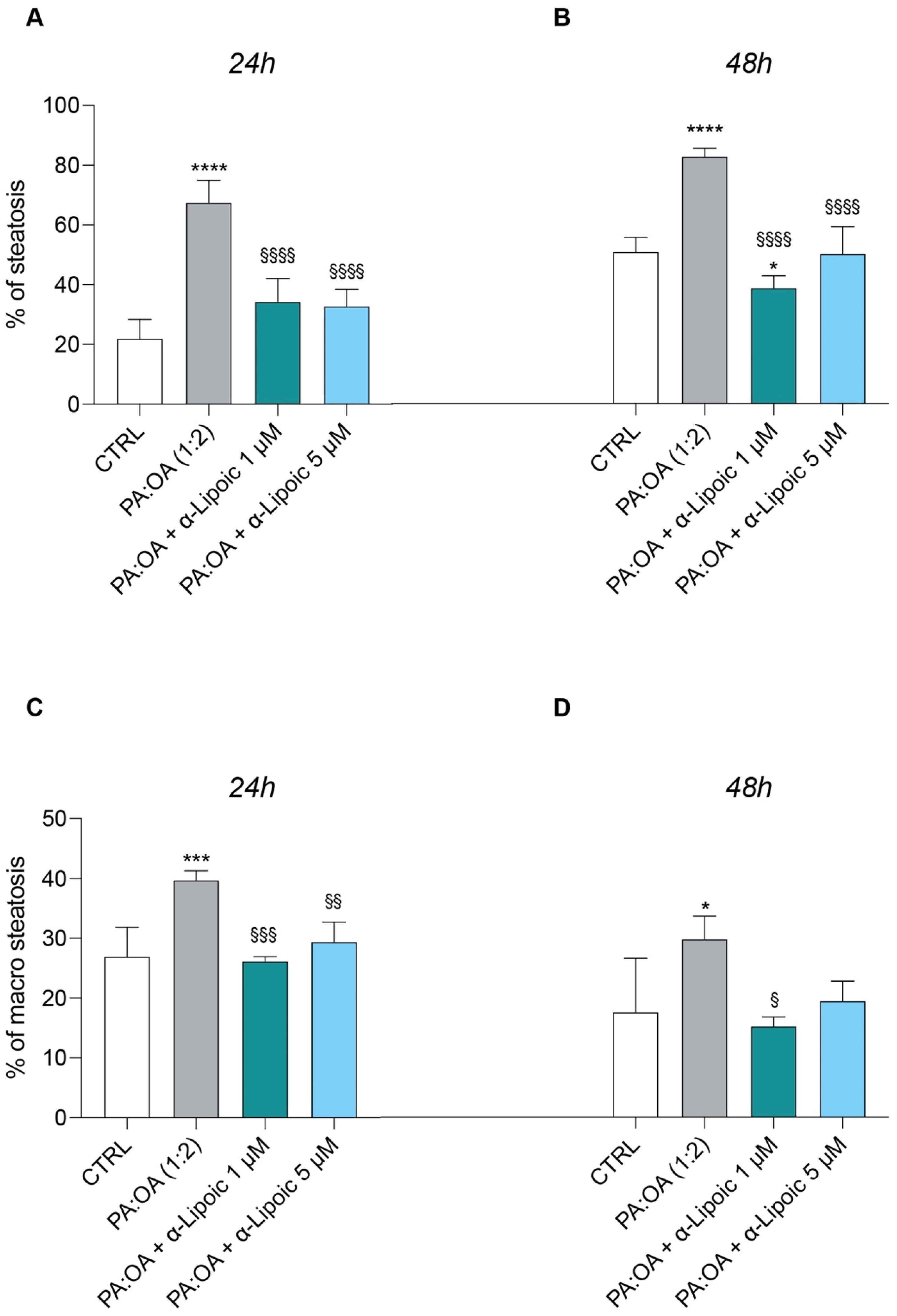
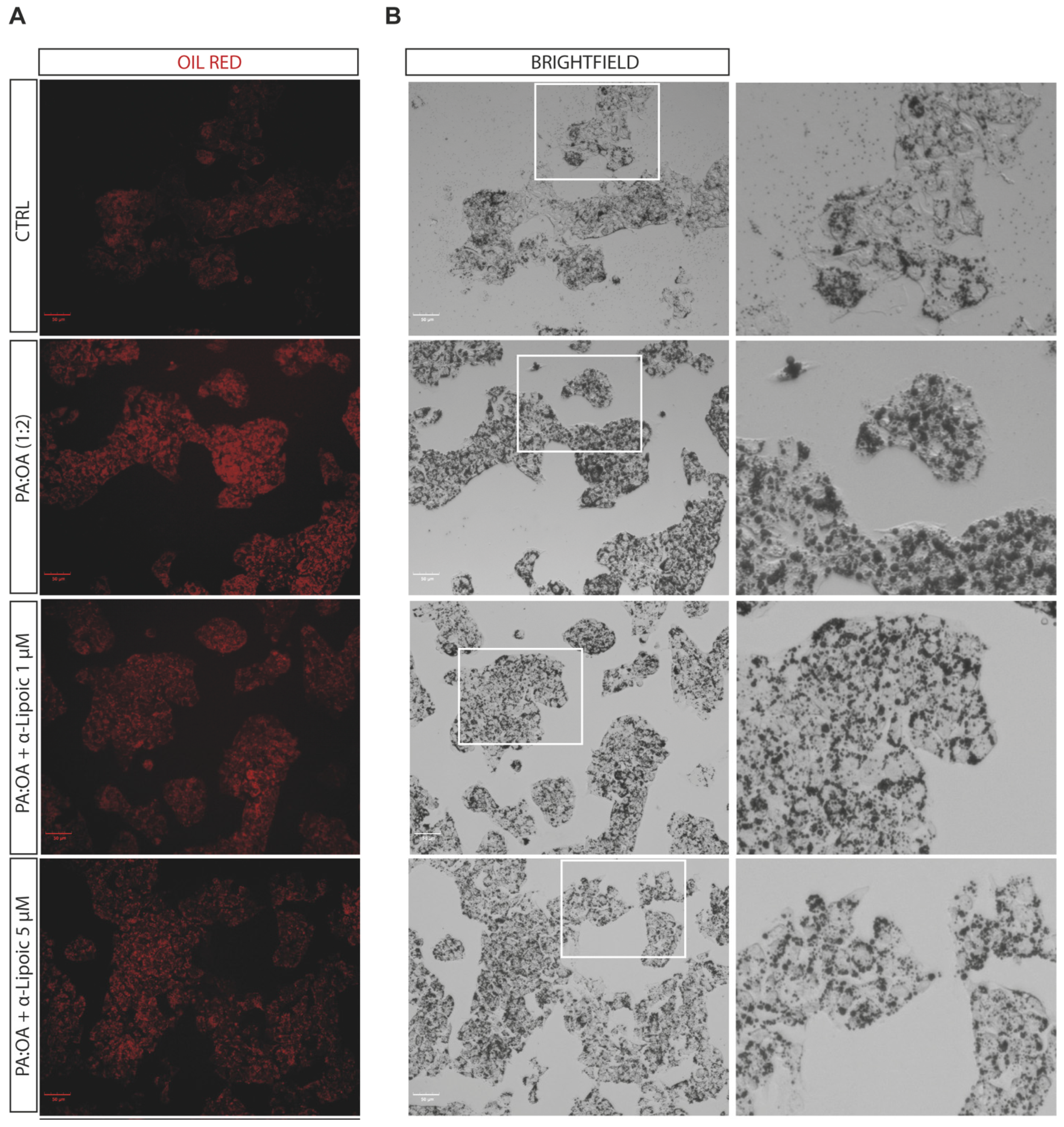
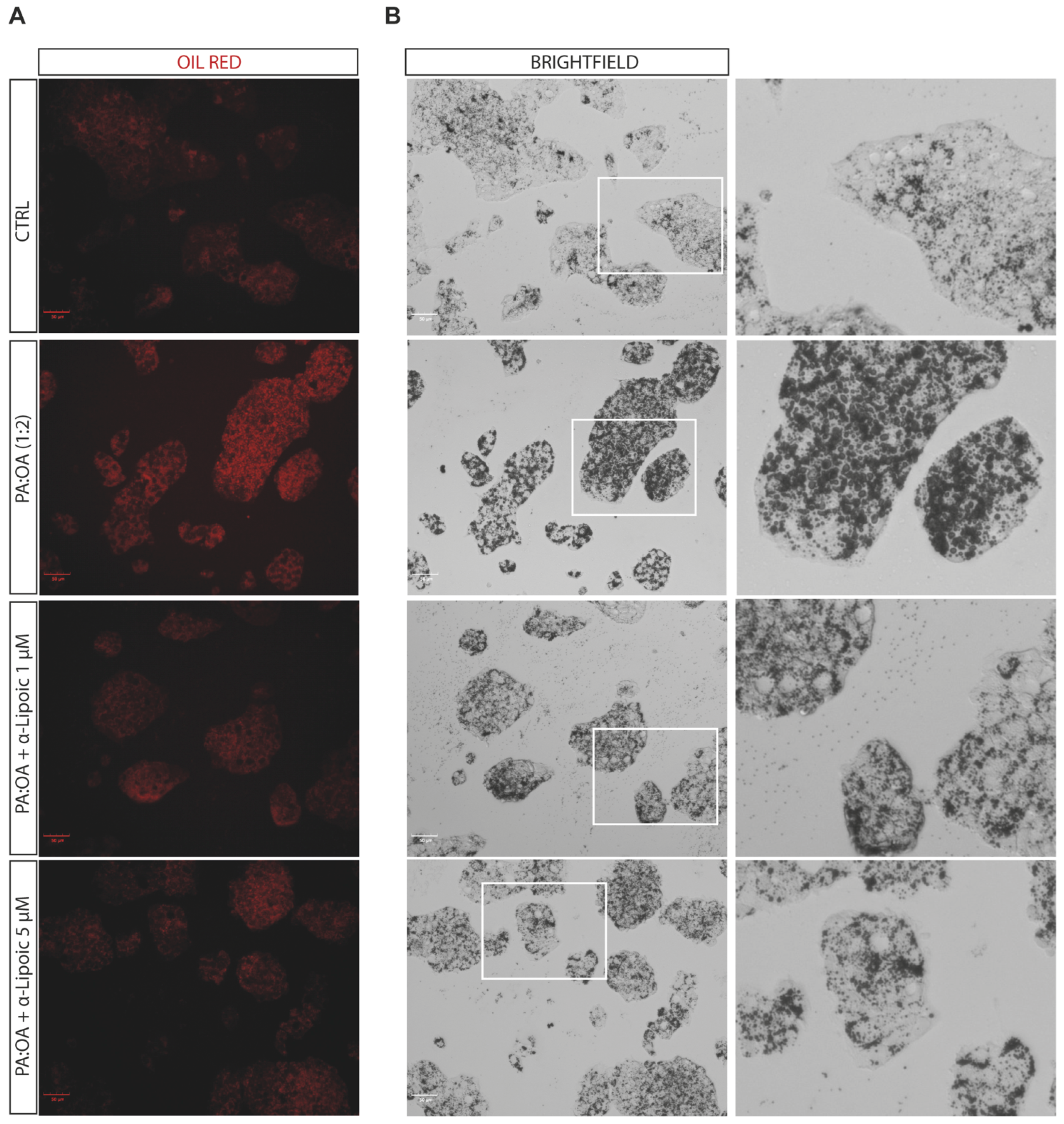
2.2. Lipoic Acid Improves Steatosis and Reduces the Area of Lipid Droplets in PA:OA-Treated HepG2 Cells
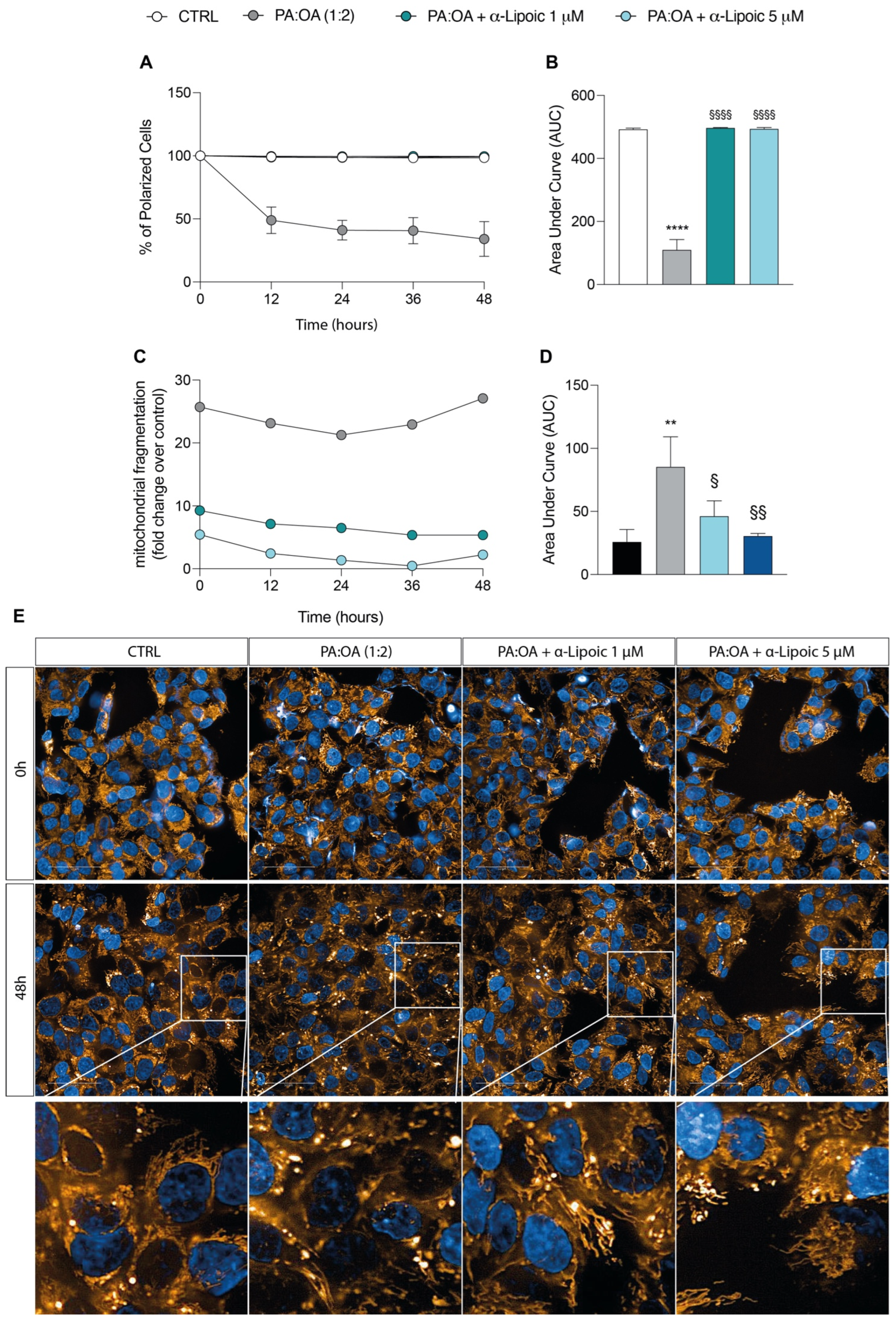
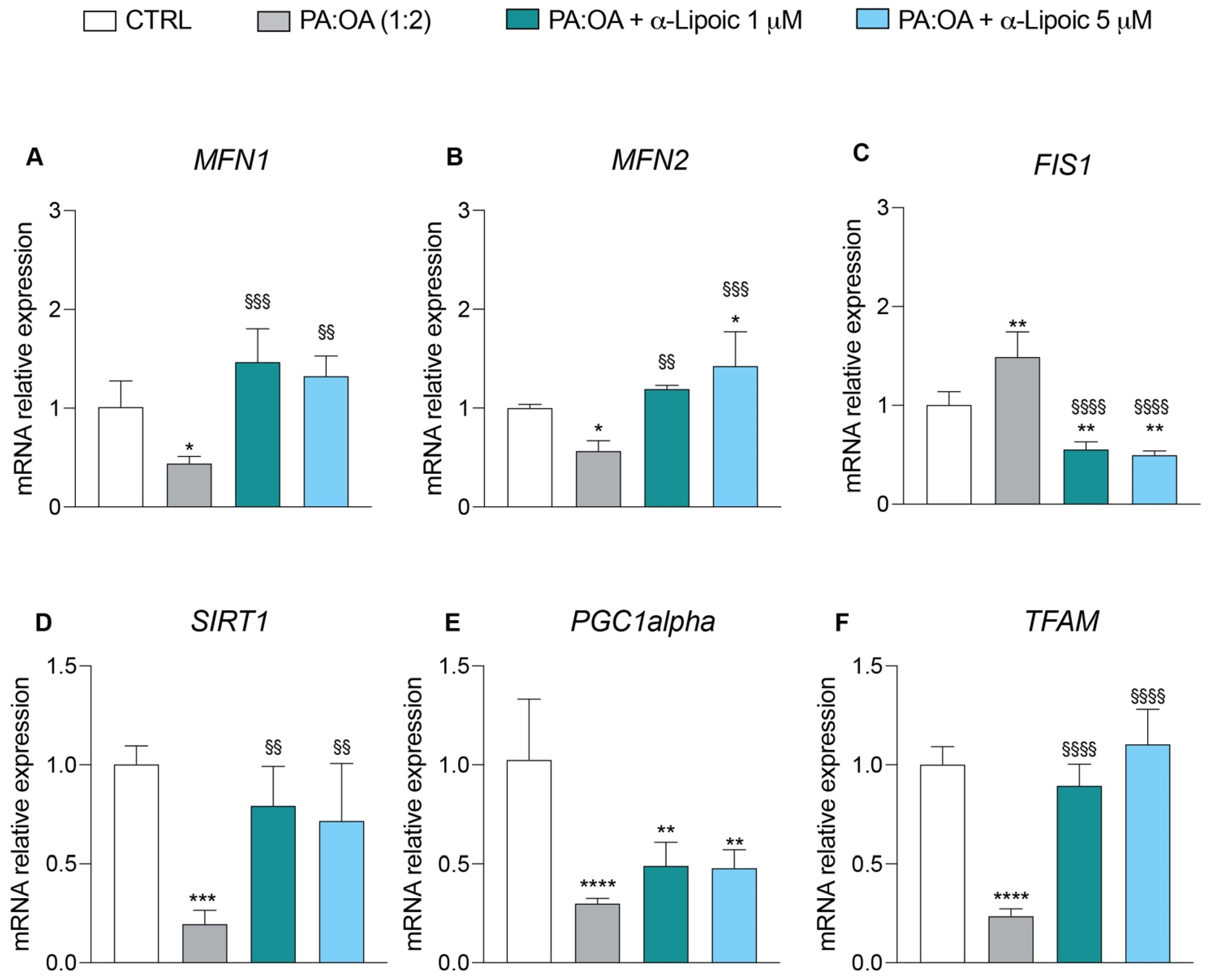
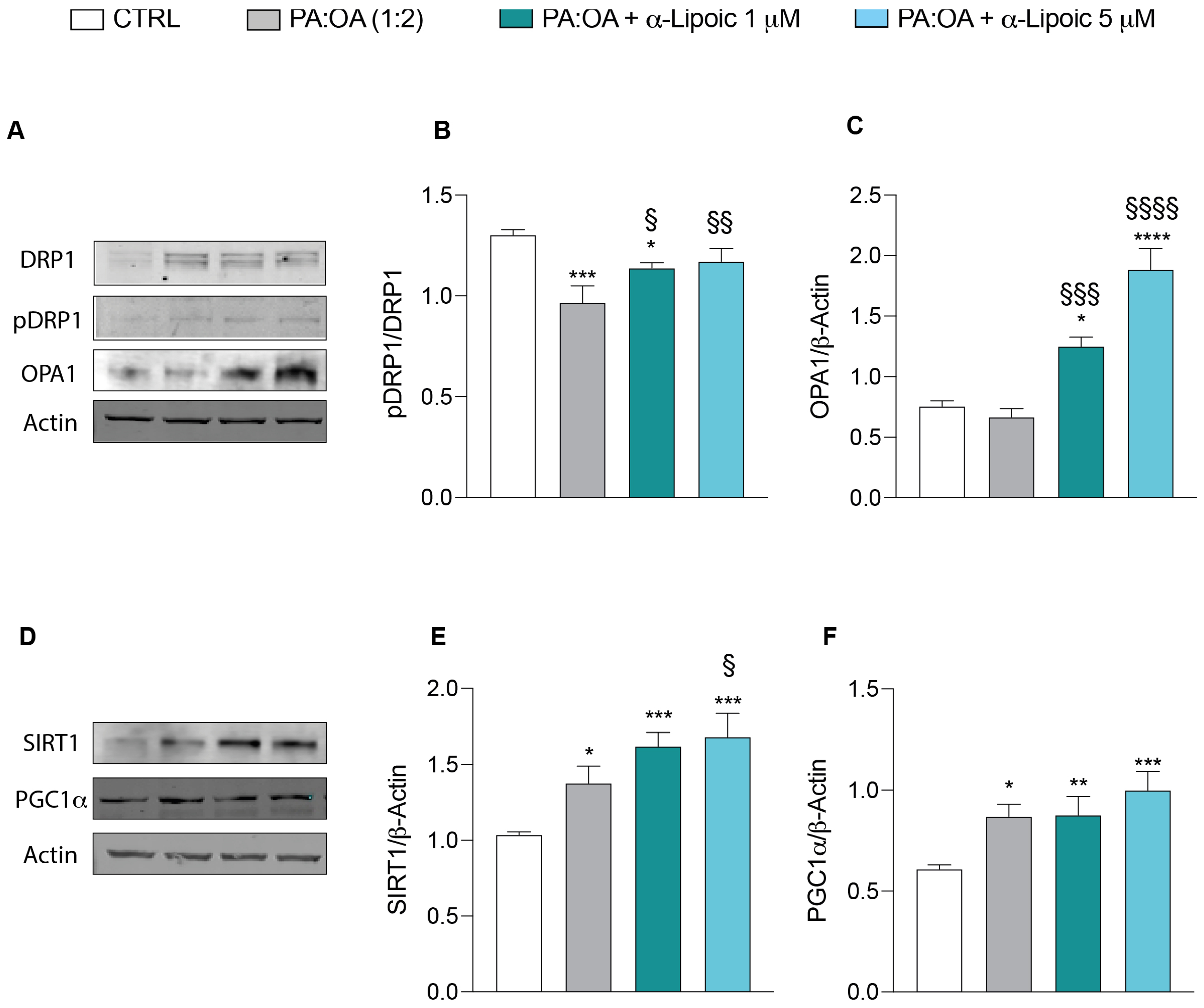
2.3. Lipoic Acid Restores Mitochondrial Membrane Potential and Inhibits Fragmentation
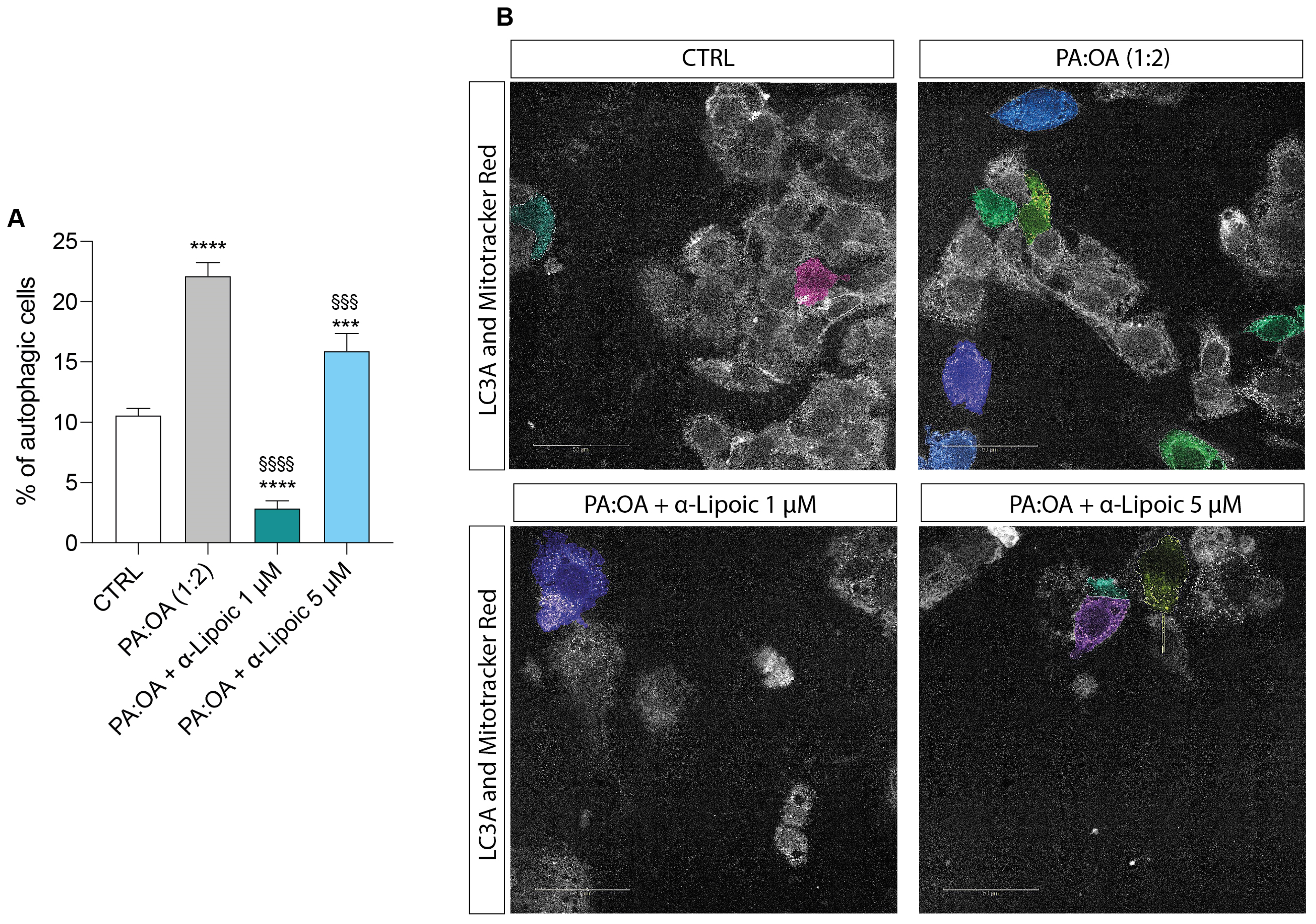
2.4. Lipoic Acid Restores the Effect of PA:OA on Autophagy
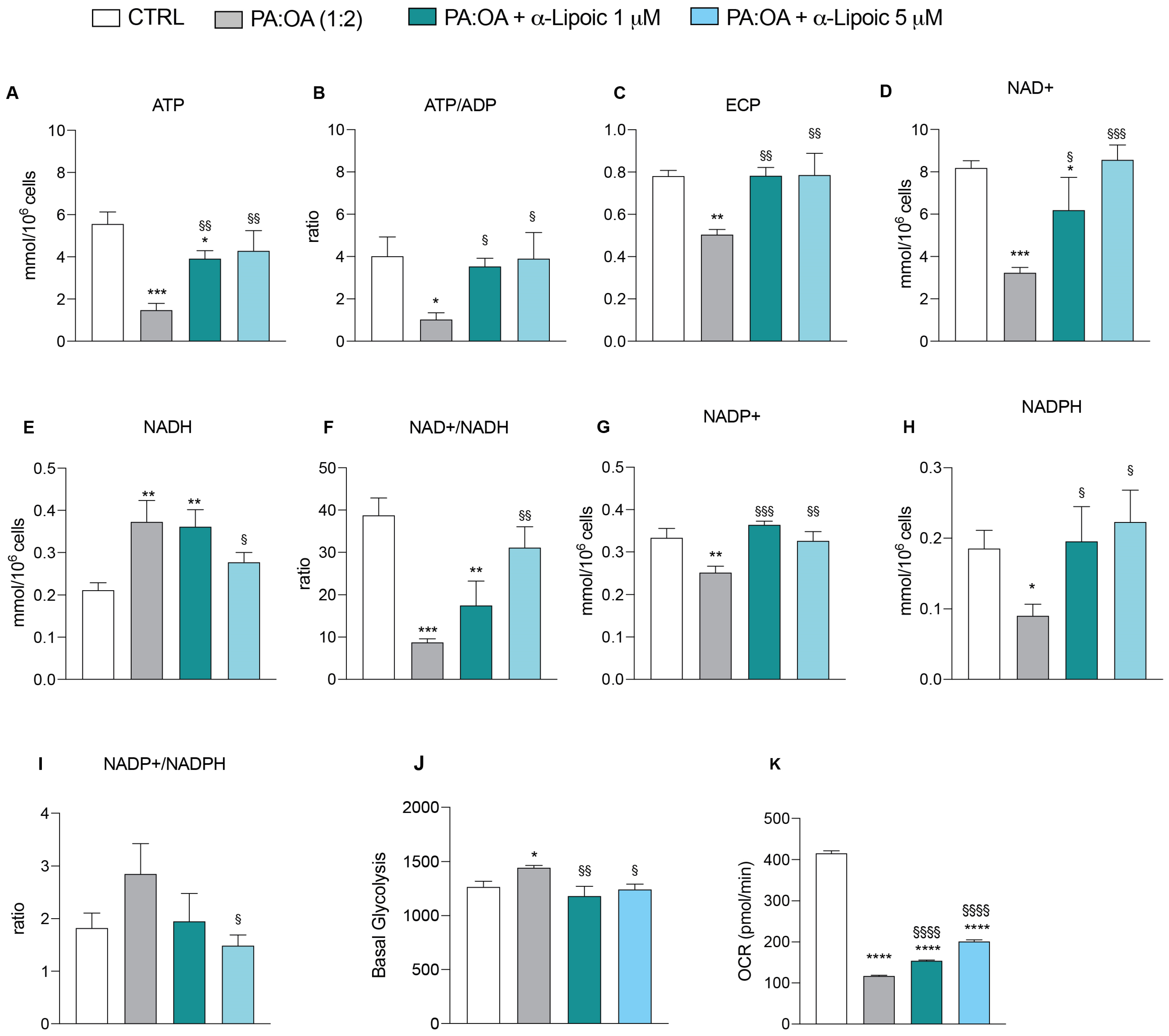
2.5. α-Lipoic Acid Restores Energy Metabolism of Steatotic HepG2 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Pharmacological Treatments
4.2. Real-Time Monitoring of Cell Viability
4.3. Oil Red O Staining
4.4. Evaluation of Mitochondrial Fragmentation
4.5. Evaluation of Mitochondrial Membrane Potential (Δψ)
4.6. Real-Time PCR for Gene Expression Analysis
4.7. Western Blot Analysis
4.8. Immunocytochemical Analysis
4.9. Metabolomic Profile by HPLC Analysis
4.10. Quantification of Cellular Glycolytic Rate Using Agilent Seahorse XF Technology
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Shay, K.P.; Moreau, R.F.; Smith, E.J.; Smith, A.R.; Hagen, T.M. Alpha-lipoic acid as a dietary supplement: Molecular mechanisms and therapeutic potential. Biochim. Biophys. Acta 2009, 1790, 1149–1160. [Google Scholar] [CrossRef]
- Golbidi, S.; Badran, M.; Laher, I. Diabetes and alpha lipoic Acid. Front. Pharmacol. 2011, 2, 69. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J. From lipoic acid to multi-enzyme complexes. Protein Sci. 1998, 7, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Valdecantos, M.P.; Perez-Matute, P.; Gonzalez-Muniesa, P.; Prieto-Hontoria, P.L.; Moreno-Aliaga, M.J.; Martinez, J.A. Lipoic acid administration prevents nonalcoholic steatosis linked to long-term high-fat feeding by modulating mitochondrial function. J. Nutr. Biochem. 2012, 23, 1676–1684. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.S.; Kim, S.K.; Shin, H.J.; Jeon, B.T.; Hahm, J.R.; Roh, G.S. alpha-lipoic acid prevents non-alcoholic fatty liver disease in OLETF rats. Liver Int. 2012, 32, 1565–1573. [Google Scholar] [CrossRef]
- Wang, Y.; Li, X.; Guo, Y.; Chan, L.; Guan, X. alpha-Lipoic acid increases energy expenditure by enhancing adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor-gamma coactivator-1alpha signaling in the skeletal muscle of aged mice. Metabolism 2010, 59, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wang, M.; Gan, L.; Zhang, B.; Desnick, R.J.; Yasuda, M. Characterization of the hepatic transcriptome following phenobarbital induction in mice with AIP. Mol. Genet. Metab. 2019, 128, 382–390. [Google Scholar] [CrossRef]
- Ong, S.L.; Vohra, H.; Zhang, Y.; Sutton, M.; Whitworth, J.A. The effect of alpha-lipoic acid on mitochondrial superoxide and glucocorticoid-induced hypertension. Oxid. Med. Cell. Longev. 2013, 2013, 517045. [Google Scholar] [CrossRef]
- Moreira, P.I.; Harris, P.L.; Zhu, X.; Santos, M.S.; Oliveira, C.R.; Smith, M.A.; Perry, G. Lipoic acid and N-acetyl cysteine decrease mitochondrial-related oxidative stress in Alzheimer disease patient fibroblasts. J. Alzheimers Dis. 2007, 12, 195–206. [Google Scholar] [CrossRef]
- Valdecantos, M.P.; Perez-Matute, P.; Gonzalez-Muniesa, P.; Prieto-Hontoria, P.L.; Moreno-Aliaga, M.J.; Martinez, J.A. Lipoic acid improves mitochondrial function in nonalcoholic steatosis through the stimulation of sirtuin 1 and sirtuin 3. Obesity 2012, 20, 1974–1983. [Google Scholar] [CrossRef]
- Padmalayam, I.; Hasham, S.; Saxena, U.; Pillarisetti, S. Lipoic acid synthase (LASY): A novel role in inflammation, mitochondrial function, and insulin resistance. Diabetes 2009, 58, 600–608. [Google Scholar] [CrossRef][Green Version]
- Dos Santos, S.M.; Romeiro, C.F.R.; Rodrigues, C.A.; Cerqueira, A.R.L.; Monteiro, M.C. Mitochondrial Dysfunction and Alpha-Lipoic Acid: Beneficial or Harmful in Alzheimer’s Disease? Oxid. Med. Cell. Longev. 2019, 2019, 8409329. [Google Scholar] [CrossRef]
- Dieter, F.; Esselun, C.; Eckert, G.P. Redox Active alpha-Lipoic Acid Differentially Improves Mitochondrial Dysfunction in a Cellular Model of Alzheimer and Its Control Cells. Int. J. Mol. Sci. 2022, 23, 9186. [Google Scholar] [CrossRef]
- Ren, L.; Chen, X.; Chen, X.; Li, J.; Cheng, B.; Xia, J. Mitochondrial Dynamics: Fission and Fusion in Fate Determination of Mesenchymal Stem Cells. Front. Cell Dev. Biol. 2020, 8, 580070. [Google Scholar] [CrossRef]
- Scott, I.; Youle, R.J. Mitochondrial fission and fusion. Essays Biochem. 2010, 47, 85–98. [Google Scholar] [CrossRef]
- Liu, Y.J.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Shen, S. Lipoic acid alleviates schistosomiasis-induced liver fibrosis by upregulating Drp1 phosphorylation. Acta Trop. 2020, 206, 105449. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.; Paolini, E.; Meroni, M.; Duca, L.; Motta, I.; Fracanzani, A.L.; Di Pierro, E.; Dongiovanni, P. alpha-Lipoic Acid Improves Hepatic Metabolic Dysfunctions in Acute Intermittent Porphyria: A Proof-of-Concept Study. Diagnostics 2021, 11, 1628. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G.; Kowluru, R.A. Mitochondrial Dynamics in the Metabolic Memory of Diabetic Retinopathy. J. Diabetes Res. 2022, 2022, 3555889. [Google Scholar] [CrossRef]
- Brunt, E.M.; Wong, V.W.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Primers 2015, 1, 15080. [Google Scholar] [CrossRef]
- Ramanathan, R.; Ali, A.H.; Ibdah, J.A. Mitochondrial Dysfunction Plays Central Role in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 7280. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Toan, S.; Zhou, H. Role of mitochondrial quality control in the pathogenesis of nonalcoholic fatty liver disease. Aging 2020, 12, 6467–6485. [Google Scholar] [CrossRef] [PubMed]
- Galloway, C.A.; Lee, H.; Brookes, P.S.; Yoon, Y. Decreasing mitochondrial fission alleviates hepatic steatosis in a murine model of nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G632–G641. [Google Scholar] [CrossRef] [PubMed]
- Legaki, A.I.; Moustakas, I.I.; Sikorska, M.; Papadopoulos, G.; Velliou, R.I.; Chatzigeorgiou, A. Hepatocyte Mitochondrial Dynamics and Bioenergetics in Obesity-Related Non-Alcoholic Fatty Liver Disease. Curr. Obes. Rep. 2022, 11, 126–143. [Google Scholar] [CrossRef] [PubMed]
- Melli, G.; Taiana, M.; Camozzi, F.; Triolo, D.; Podini, P.; Quattrini, A.; Taroni, F.; Lauria, G. Alpha-lipoic acid prevents mitochondrial damage and neurotoxicity in experimental chemotherapy neuropathy. Exp. Neurol. 2008, 214, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Solmonson, A.; DeBerardinis, R.J. Lipoic acid metabolism and mitochondrial redox regulation. J. Biol. Chem. 2018, 293, 7522–7530. [Google Scholar] [CrossRef] [PubMed]
- Walgren, J.L.; Amani, Z.; McMillan, J.M.; Locher, M.; Buse, M.G. Effect of R(+)alpha-lipoic acid on pyruvate metabolism and fatty acid oxidation in rat hepatocytes. Metabolism 2004, 53, 165–173. [Google Scholar] [CrossRef]
- Ambrus, A.; Tretter, L.; Adam-Vizi, V. Inhibition of the alpha-ketoglutarate dehydrogenase-mediated reactive oxygen species generation by lipoic acid. J. Neurochem. 2009, 109 (Suppl. S1), 222–229. [Google Scholar] [CrossRef]
- Palaniappan, A.R.; Dai, A. Mitochondrial ageing and the beneficial role of alpha-lipoic acid. Neurochem. Res. 2007, 32, 1552–1558. [Google Scholar] [CrossRef]
- Barbato, A.; Scandura, G.; Puglisi, F.; Cambria, D.; La Spina, E.; Palumbo, G.A.; Lazzarino, G.; Tibullo, D.; Di Raimondo, F.; Giallongo, C.; et al. Mitochondrial Bioenergetics at the Onset of Drug Resistance in Hematological Malignancies: An Overview. Front. Oncol. 2020, 10, 604143. [Google Scholar] [CrossRef]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518. [Google Scholar] [CrossRef]
- Kakimoto, P.A.; Serna, J.D.C.; de Miranda Ramos, V.; Zorzano, A.; Kowaltowski, A.J. Increased glycolysis is an early consequence of palmitate lipotoxicity mediated by redox signaling. Redox Biol. 2021, 45, 102026. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lechon, M.J.; Donato, M.T.; Martinez-Romero, A.; Jimenez, N.; Castell, J.V.; O’Connor, J.E. A human hepatocellular in vitro model to investigate steatosis. Chem. Biol. Interact. 2007, 165, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Giallongo, C.; Tibullo, D.; La Cava, P.; Branca, A.; Parrinello, N.; Spina, P.; Stagno, F.; Conticello, C.; Chiarenza, A.; Vigneri, P.; et al. BRIT1/MCPH1 expression in chronic myeloid leukemia and its regulation of the G2/M checkpoint. Acta Haematol. 2011, 126, 205–210. [Google Scholar] [CrossRef]
- Tibullo, D.; Caporarello, N.; Giallongo, C.; Anfuso, C.D.; Genovese, C.; Arlotta, C.; Puglisi, F.; Parrinello, N.L.; Bramanti, V.; Romano, A.; et al. Antiproliferative and Antiangiogenic Effects of Punica granatum Juice (PGJ) in Multiple Myeloma (MM). Nutrients 2016, 8, 611. [Google Scholar] [CrossRef] [PubMed]
- Lazzarino, G.; Amorini, A.M.; Fazzina, G.; Vagnozzi, R.; Signoretti, S.; Donzelli, S.; Di Stasio, E.; Giardina, B.; Tavazzi, B. Single-sample preparation for simultaneous cellular redox and energy state determination. Anal. Biochem. 2003, 322, 51–59. [Google Scholar] [CrossRef]
- Romitelli, F.; Santini, S.A.; Chierici, E.; Pitocco, D.; Tavazzi, B.; Amorini, A.M.; Lazzarino, G.; Di Stasio, E. Comparison of nitrite/nitrate concentration in human plasma and serum samples measured by the enzymatic batch Griess assay, ion-pairing HPLC and ion-trap GC-MS: The importance of a correct removal of proteins in the Griess assay. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 851, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Tibullo, D.; Giallongo, C.; Romano, A.; Vicario, N.; Barbato, A.; Puglisi, F.; Parenti, R.; Amorini, A.M.; Wissam Saab, M.; Tavazzi, B.; et al. Mitochondrial Functions, Energy Metabolism and Protein Glycosylation are Interconnected Processes Mediating Resistance to Bortezomib in Multiple Myeloma Cells. Biomolecules 2020, 10, 696. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 12 h | 24 h | 36 h | 48 h | 72 h | AUC | |
|---|---|---|---|---|---|---|
| CTRL | 3.41 ± 0.9 | 4.81 ± 1.06 | 6.47 ± 1.6 | 8.26 ± 1.9 | 11.35 ± 2.7 | 21.64 ± 4.5 |
| PA:OA (1:2) | 1.50 ± 0.8 * | 2.67 ± 0.8 * | 3.81 ± 1.1 | 5.14 ± 1.4 | 7.18 ± 2.04 | 11.98 ± 3.5 |
| PA:OA + α-Lipoic 1 μM | 2.71 ± 1.7 | 4.88 ± 2.3 § | 6.88 ± 2.6 | 9.33 ± 3.4 | 13.40 ± 4.3 § | 21.69 ± 8.4 |
| PA:OA + α-Lipoic 5 μM | 3.34 ± 0.32 | 4.88 ± 0.1 § | 6.74 ± 0.1 | 8.72 ± 0.5 | 12.17 ± 1.0 | 21.08 ± 0.7 |
| CTRL | PA:OA (1:2) | PA:OA + α-lipoico 1 µM | PA:OA + α-lipoico 5 µM | ||
|---|---|---|---|---|---|
| Steatotic cells—Total Lipid droplets Area | 12 h | 178.5 ± 14.3 | 224.95 ±7.4 *** | 176.6 ± 2.5 §§§ | 188.3 ±11.6 §§ |
| 48 h | 144.7 ± 31.2 | 190.1 ± 10.8 * | 140.2 ± 5.4 § | 154.6 ± 9.0 | |
| Steatotic cells—Number of Lipid droplets | 12 h | 6.99 ± 0.48 | 6.7 ± 0.04 | 6.2 ± 0.1 | 6.5 ± 0.3 |
| 48 h | 6.2 ± 1.4 | 5.7 ± 0.3 | 5.6 ± 0.1 | 5.9 ± 0.2 | |
| Steatotic cells—Number of Lipid droplets per Area of Cytoplasm | 12 h | 0.020 ± 0.0006 | 0.017 ± 0.0002 ** | 0.019 ± 0.0006 §§ | 0.019 ± 0.0003 §§ |
| 48 h | 0.019 ± 0.0009 | 0.017 ± 0.0003 * | 0.018 ± 0.0003 | 0.019 ± 0.0003 §§ | |
| Steatotic cells—Lipid droplets Area (µm²) | 12 h | 63.7 ± 5.1 | 80.4 ± 2.7 *** | 63.1 ± 0.9 §§§ | 67.2 ± 4.1 §§ |
| 48 h | 51.7 ± 11.2 | 67.9 ± 3.8 * | 50.1 ± 1.9 § | 55.19 ± 3.2 | |
| Total cells—Total Lipid droplets Area | 12 h | 158.75 ± 8.2 | 217.2 ± 7.2 **** | 164.1 ± 4.3 §§§§ | 172 ± 11.3 §§§§ |
| 48 h | 126.3 ± 18.9 | 177.9 ±13.1 *** | 112.8 ± 2.5 §§§ | 140.0 ± 7.2 §§ | |
| Total cells—Number of Lipid droplets | 12 h | 6.8 ± 0.2 | 6.52 ± 0.05 | 6.03 ± 0.1 ** | 6.2 ± 0.3 * |
| 48 h | 5.7 ± 1.0 | 5.4 ± 0.3 | 5.19 ± 0.07 | 5.6 ± 0.2 | |
| Total cells—Number of Lipid droplets per Area of Cytoplasm | 12 h | 0.02 ± 0.0006 | 0.018 ± 0.0003 **** | 0.02 ± 0.0007 §§ | 0.02 ± 0.0003 §§§ |
| 48 h | 0.019 ± 0.0006 | 0.017 ± 0.0003 ** | 0.018 ± 0.0004 | 0.019 ± 0.0004 §§§ | |
| Total cells—Lipids droplets Area (µm²) | 12 h | 56.5 ± 2.9 | 77.68 ± 2.6 **** | 58.71 ± 1.5 §§§§ | 61.5 ± 4.0 §§§§ |
| 48 h | 45.2 ± 6.7 | 63.6 ± 4.6 *** | 43.9 ± 0.9 §§§ | 50.1 ± 2.5 §§ | |
| Total cells—Cell Volume (µm³) | 12 h | 3312.87 ± 1036.7 | 4039.5 ± 2256.5 | 2299.37 ± 787.3 | 3741.5 ± 1731 |
| 48 h | 1727 ± 528.8 | 2014.2 ± 986.2 | 1585.7 ± 433.3 | 1903.25 ± 794.1 | |
| Total cells—Cell Surface Area (µm²) | 12 h | 3359.3 ± 473.5 | 4449.8 ± 2846.1 | 3414.1 ± 834.9 | 4444.3 ± 2333.8 |
| 48 h | 1726.8 ± 285.8 | 2162.2 ± 1089.7 | 1753.7 ± 442.3 | 2261 ± 1239.4 |
| Genes | Forward Primer (5′⟶3′) | Reverse Primer (5′⟶3′) | Accessionnumber |
|---|---|---|---|
| MFN1 | TCGGGAAGATGAGGCAGTTT | TGCCATTATGCTAAGTCTCCG | NM_033540.3 |
| MFN2 | CGGGAAGGTGAAGCGCAATG | ACCAGGAAGCTGGTACAACG | NM_001127660.2 |
| OPA1 | GAAAGGAGCTCATCTGTTTGGAGTC | TTCTTCCGGAGAACCAAAATCG | NM_001354663.2 |
| FIS1 | ACTACCGGCTCAAGGAATACG | CATGCCCACGAGTCCATCTT | NM_016068.3 |
| PGC1ɑ | ATGAAGGGTACTTTTCTGCCCC | GGTCTTCACCAACCAGAGCA | NM_001330751.2 |
| TFAM | CCGAGGTGGTTTTCATCTGT | AGTCTTCAGCTTTTCCTGCG | NM_003201.3 |
| SIRT1 | AGGCCACGGATAGGTCCATA | GTGGAGGTATTGTTTCCGGC | NM_012238.5 |
| β-Actin | CCTTTGCCGATCCGCCG | AACATGATCTGGGTCATCTTCTCGC | NM_001101.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Longhitano, L.; Distefano, A.; Amorini, A.M.; Orlando, L.; Giallongo, S.; Tibullo, D.; Lazzarino, G.; Nicolosi, A.; Alanazi, A.M.; Saoca, C.; et al. (+)-Lipoic Acid Reduces Lipotoxicity and Regulates Mitochondrial Homeostasis and Energy Balance in an In Vitro Model of Liver Steatosis. Int. J. Mol. Sci. 2023, 24, 14491. https://doi.org/10.3390/ijms241914491
Longhitano L, Distefano A, Amorini AM, Orlando L, Giallongo S, Tibullo D, Lazzarino G, Nicolosi A, Alanazi AM, Saoca C, et al. (+)-Lipoic Acid Reduces Lipotoxicity and Regulates Mitochondrial Homeostasis and Energy Balance in an In Vitro Model of Liver Steatosis. International Journal of Molecular Sciences. 2023; 24(19):14491. https://doi.org/10.3390/ijms241914491
Chicago/Turabian StyleLonghitano, Lucia, Alfio Distefano, Angela Maria Amorini, Laura Orlando, Sebastiano Giallongo, Daniele Tibullo, Giuseppe Lazzarino, Anna Nicolosi, Amer M. Alanazi, Concetta Saoca, and et al. 2023. "(+)-Lipoic Acid Reduces Lipotoxicity and Regulates Mitochondrial Homeostasis and Energy Balance in an In Vitro Model of Liver Steatosis" International Journal of Molecular Sciences 24, no. 19: 14491. https://doi.org/10.3390/ijms241914491
APA StyleLonghitano, L., Distefano, A., Amorini, A. M., Orlando, L., Giallongo, S., Tibullo, D., Lazzarino, G., Nicolosi, A., Alanazi, A. M., Saoca, C., Macaione, V., Aguennouz, M., Salomone, F., Tropea, E., Barbagallo, I. A., Volti, G. L., & Lazzarino, G. (2023). (+)-Lipoic Acid Reduces Lipotoxicity and Regulates Mitochondrial Homeostasis and Energy Balance in an In Vitro Model of Liver Steatosis. International Journal of Molecular Sciences, 24(19), 14491. https://doi.org/10.3390/ijms241914491