
Anti-Tumor Immunogenicity of the Oncolytic Virus CF33-hNIS-antiPDL1 against Ex Vivo Peritoneal Cells from Gastric Cancer Patients
 , ,
, ,
Abstract
:1. Introduction
2. Results
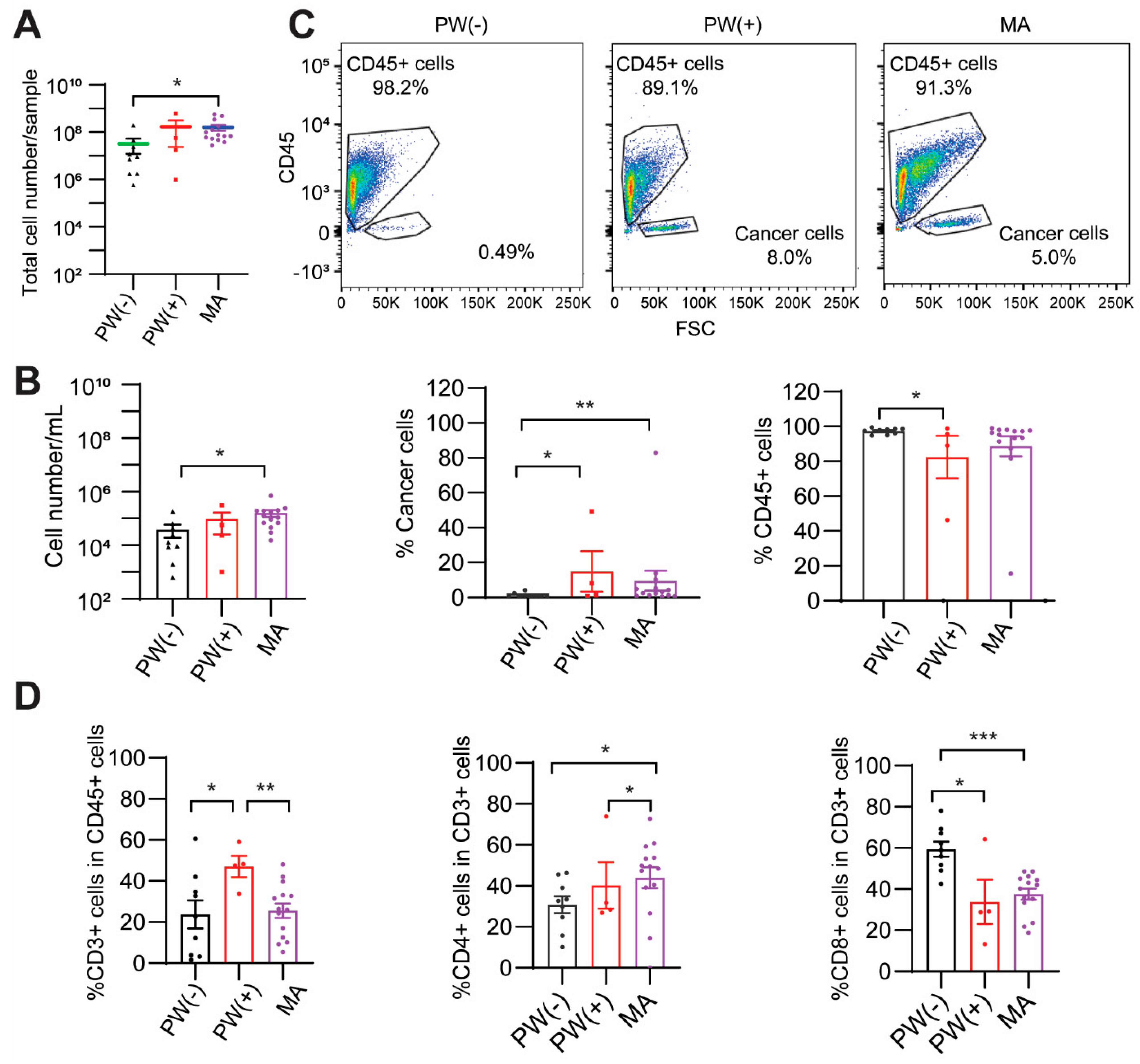
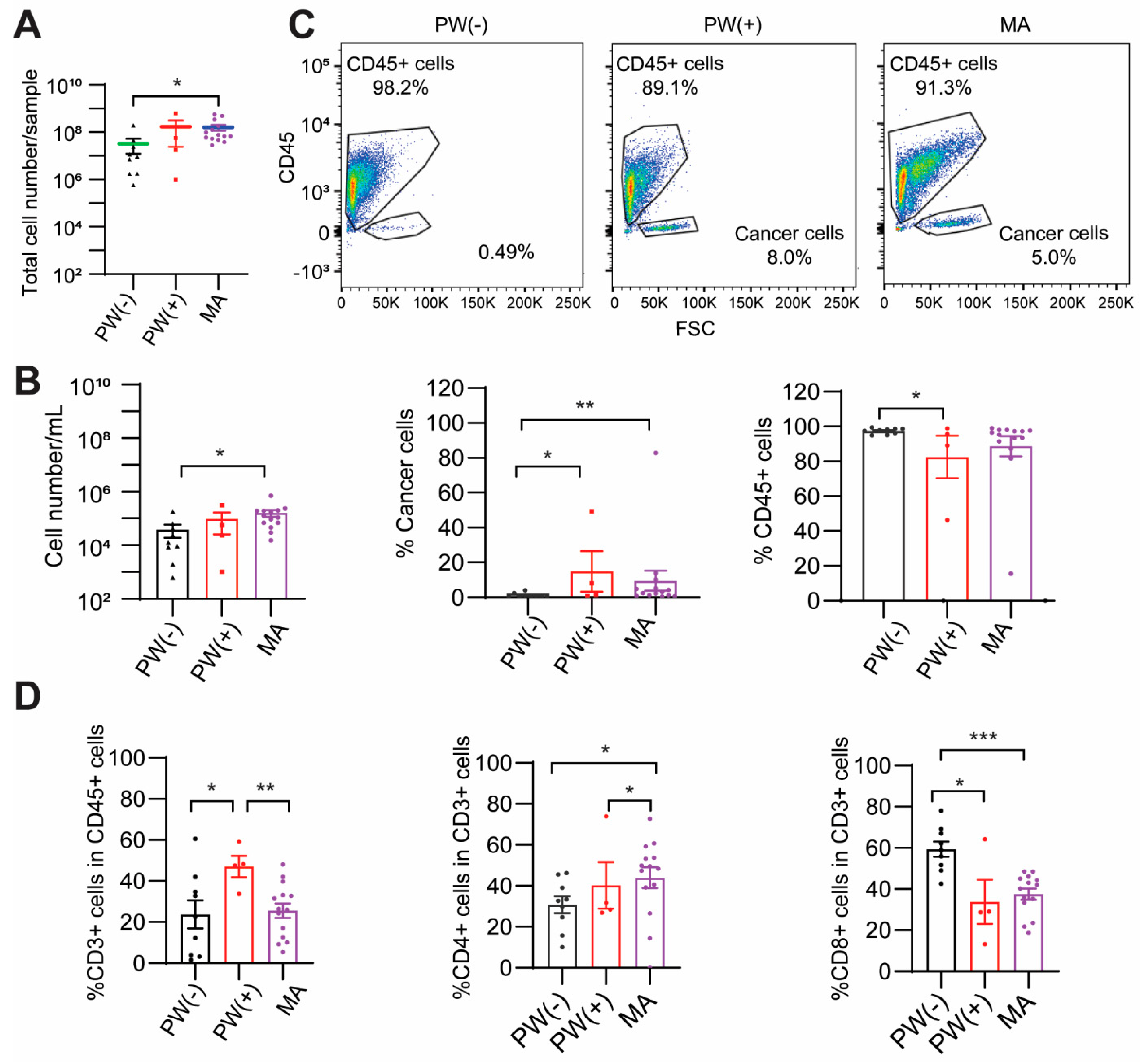
2.1. Baseline Characteristics of Patients
2.2. PW(+) and MA Groups Show a Lower CD8+ T Cell Percentage
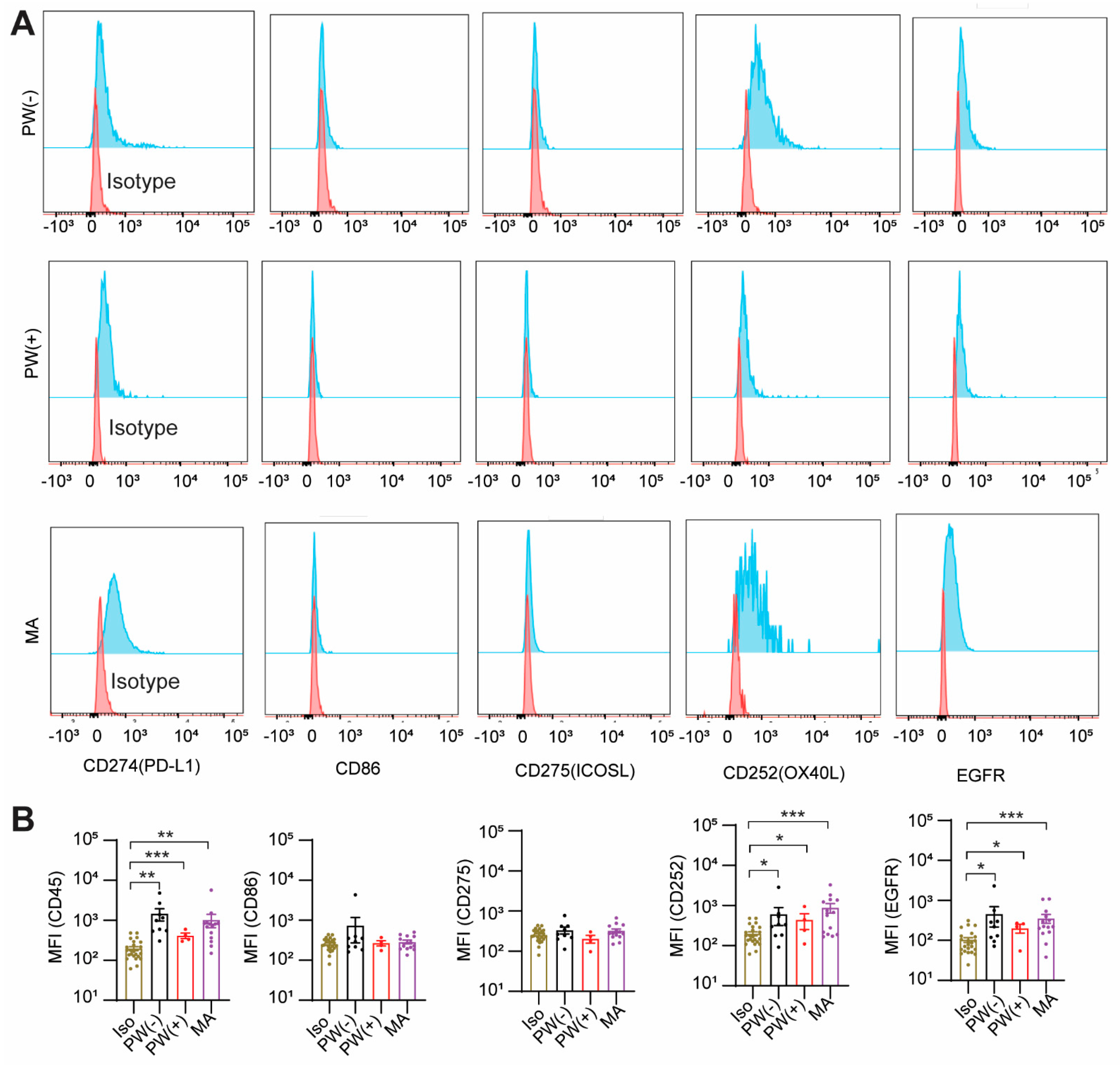
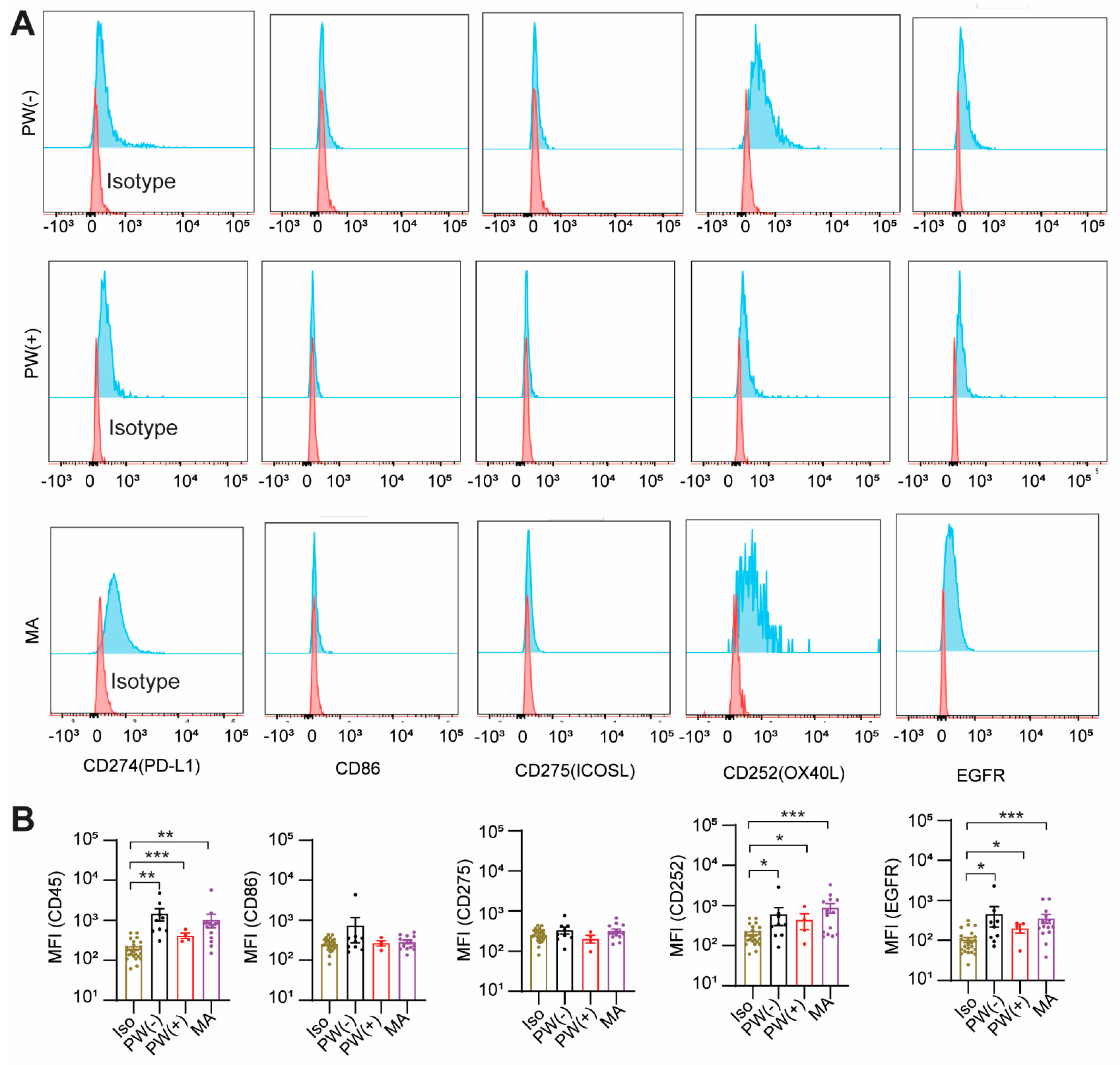
2.3. GCPM Cells Express PD-L1, CD252, and EGFR
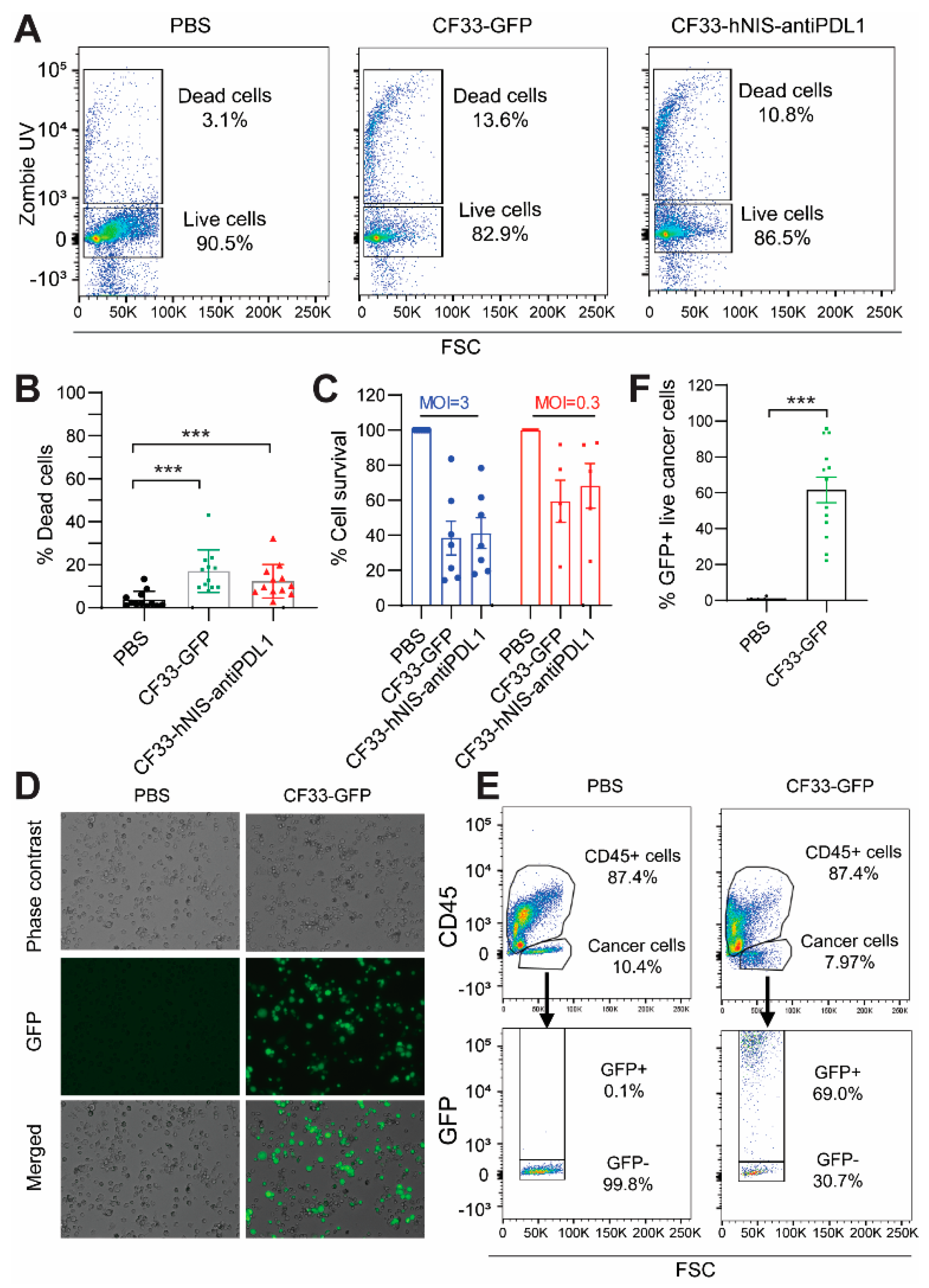
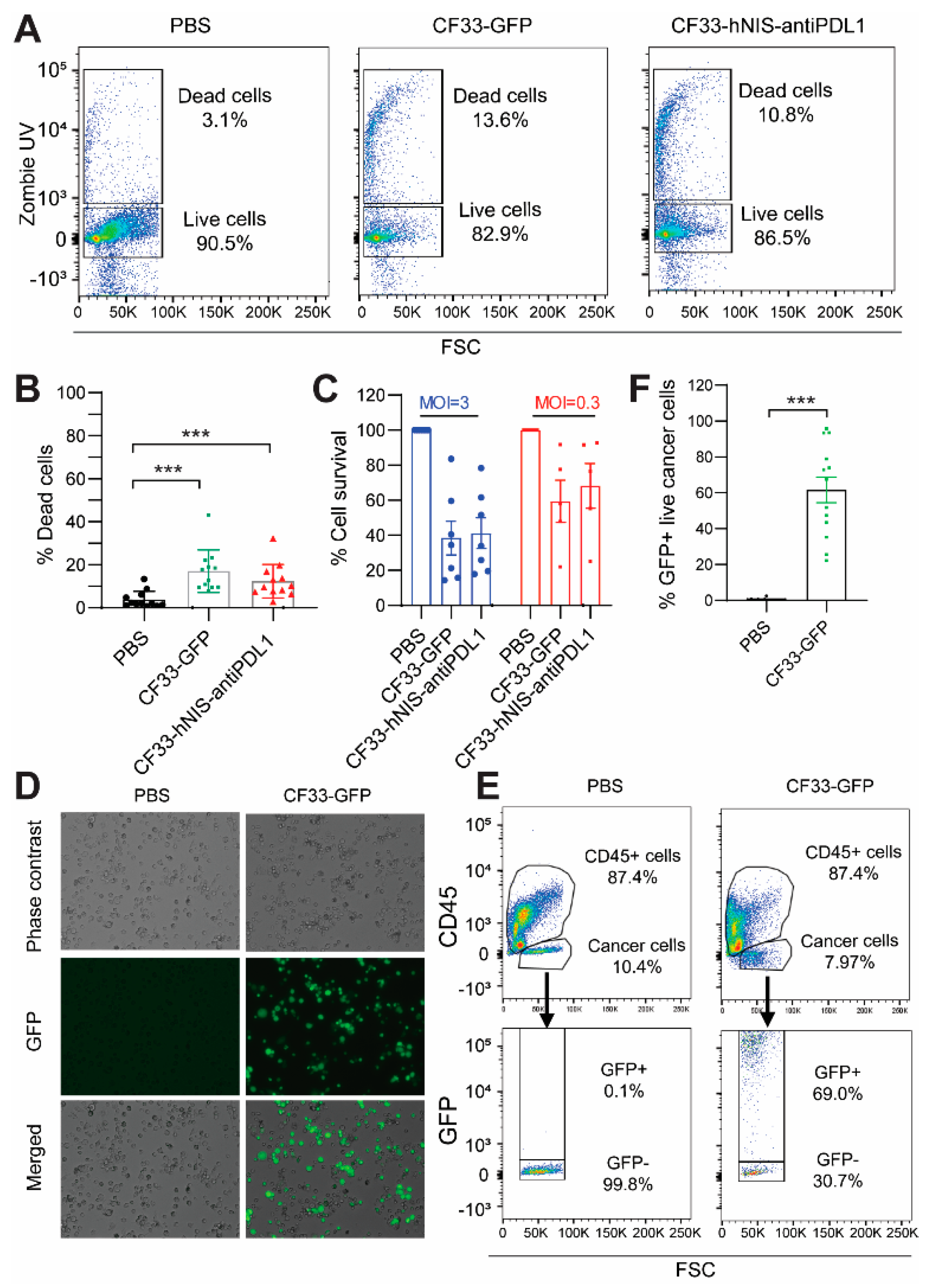
2.4. CF33-hNIS-antiPDL1 Kills GCPM Cells Ex Vivo
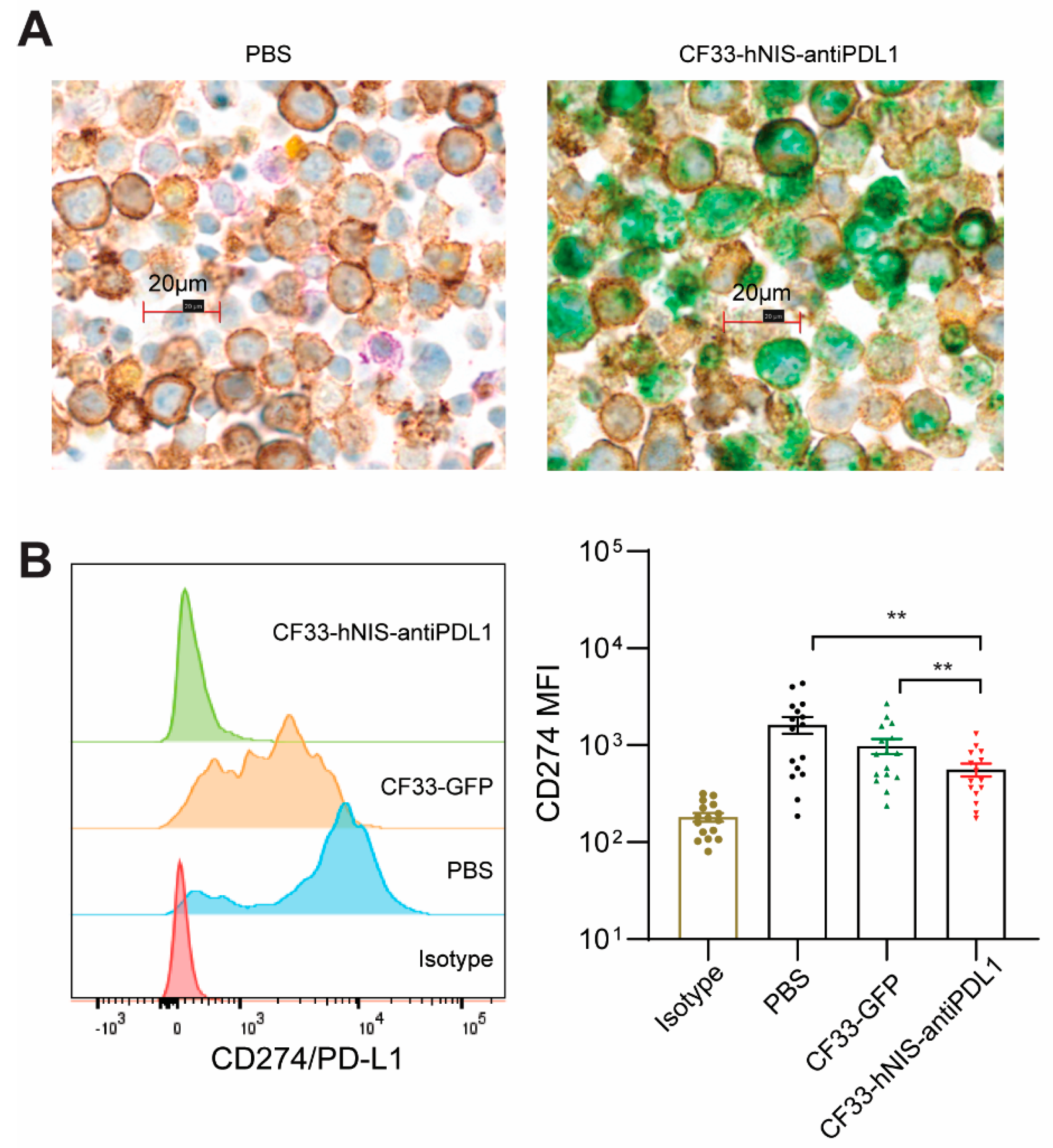
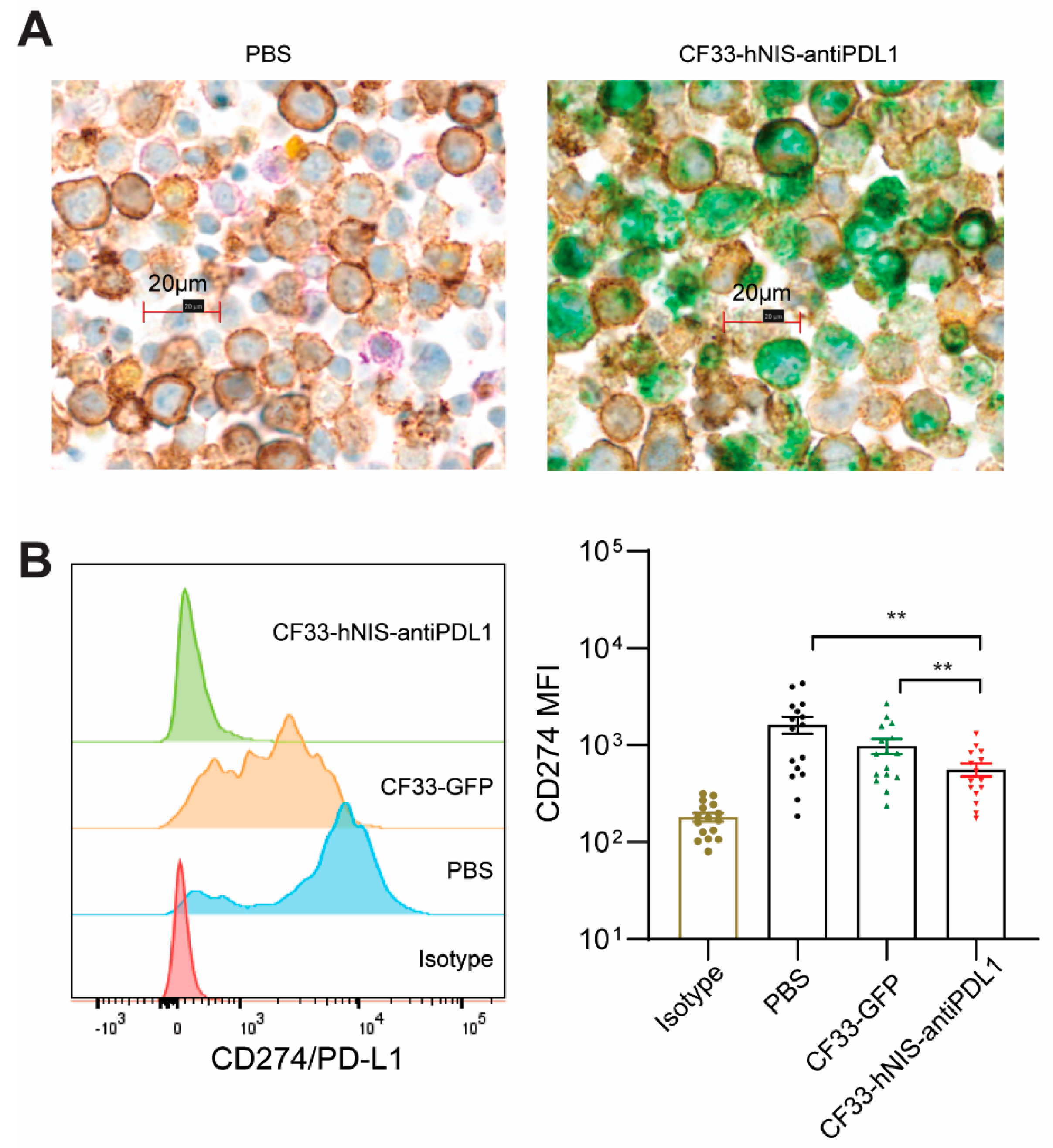
2.5. CF33-hNIS-antiPDL1 Expresses Functional anti-PD-L1 scFv in Ex Vivo Cancer Cells
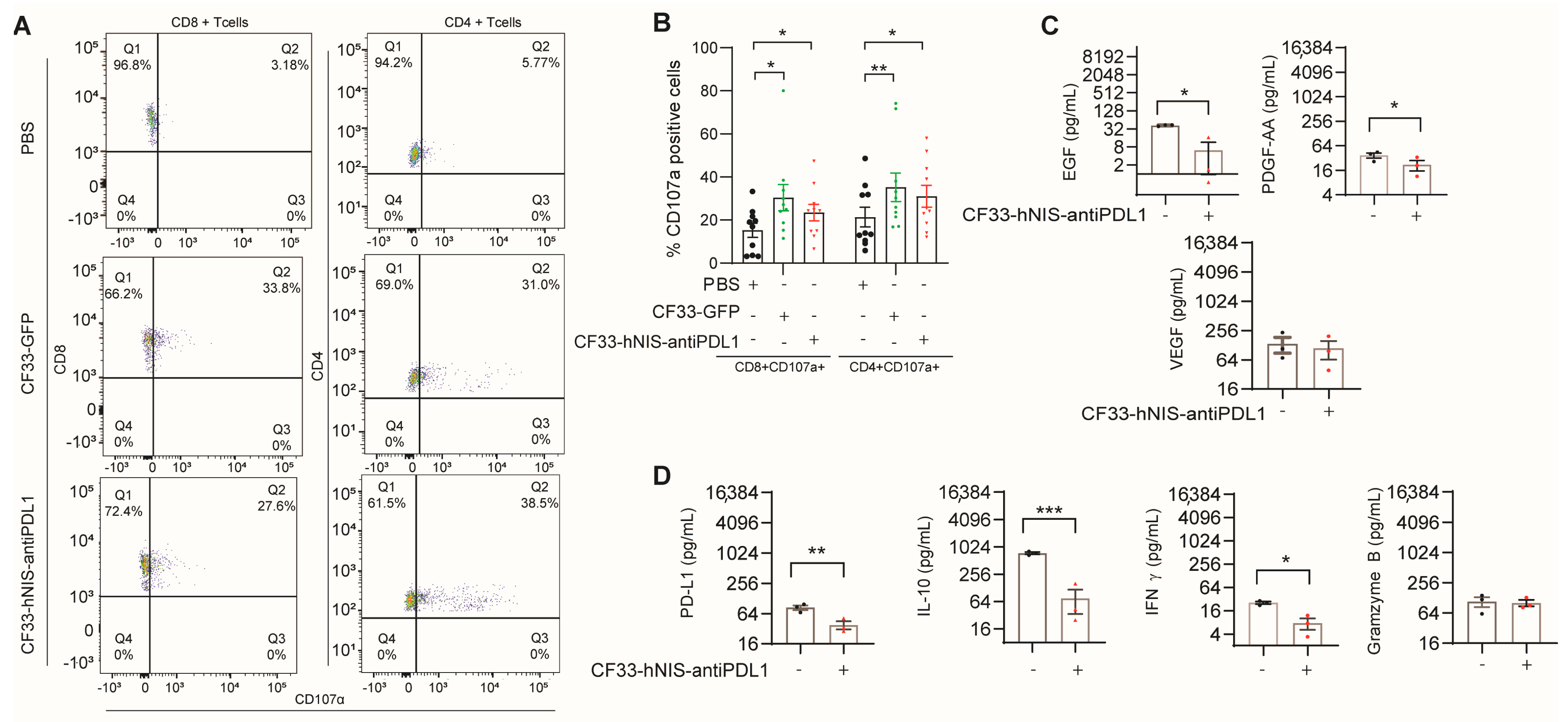
2.6. CF33-hNIS-antiPDL1 Increases T Cell Activation and Decreases Growth Factor Release in Ex Vivo GCPM Cells
3. Discussion
4. Materials and Methods
4.1. Clinical Samples
4.2. Ascites Fractionation
4.3. Antibodies, Reagents, CF33-GFP and CF33-hNIS-antiPDL1 Viruses
4.4. Cytotoxicity Assay
4.5. Cytokine Multiplex Analysis
4.6. Flow Cytometric Analysis
4.7. Fluorescence Microscopy
4.8. Multiplex Immunohistochemistry (mIHC)
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- D’Angelica, M.; Gonen, M.; Brennan, M.F.; Turnbull, A.D.; Bains, M.; Karpeh, M.S. Patterns of initial recurrence in completely resected gastric adenocarcinoma. Ann. Surg. 2004, 240, 808–816. [Google Scholar] [CrossRef]
- Rijken, A.; Lurvink, R.J.; Luyer, M.D.P.; Nieuwenhuijzen, G.A.P.; van Erning, F.N.; van Sandick, J.W.; de Hingh, I. The Burden of Peritoneal Metastases from Gastric Cancer: A Systematic Review on the Incidence, Risk Factors and Survival. J. Clin. Med. 2021, 10, 4882. [Google Scholar] [CrossRef]
- Yarema, R.; Ohorchak, M.; Hyrya, P.; Kovalchuk, Y.; Safiyan, V.; Karelin, I.; Ferneza, S.; Fetsych, M.; Matusyak, M.; Oliynyk, Y.; et al. Gastric cancer with peritoneal metastases: Efficiency of standard treatment methods. World J. Gastrointest. Oncol. 2020, 12, 569–581. [Google Scholar] [CrossRef]
- Kikuchi, H.; Kamiya, K.; Hiramatsu, Y.; Miyazaki, S.; Yamamoto, M.; Ohta, M.; Baba, S.; Konno, H. Laparoscopic narrow-band imaging for the diagnosis of peritoneal metastasis in gastric cancer. Ann. Surg. Oncol. 2014, 21, 3954–3962. [Google Scholar] [CrossRef]
- Riihimaki, M.; Hemminki, A.; Sundquist, K.; Sundquist, J.; Hemminki, K. Metastatic spread in patients with gastric cancer. Oncotarget 2016, 7, 52307–52316. [Google Scholar] [CrossRef]
- Maeda, H.; Kobayashi, M.; Sakamoto, J. Evaluation and treatment of malignant ascites secondary to gastric cancer. World J. Gastroenterol. 2015, 21, 10936–10947. [Google Scholar] [CrossRef]
- Thomassen, I.; van Gestel, Y.R.; van Ramshorst, B.; Luyer, M.D.; Bosscha, K.; Nienhuijs, S.W.; Lemmens, V.E.; de Hingh, I.H. Peritoneal carcinomatosis of gastric origin: A population-based study on incidence, survival and risk factors. Int. J. Cancer 2014, 134, 622–628. [Google Scholar] [CrossRef]
- Sun, B.J.; Lee, B. Review of Regional Therapies for Gastric Cancer with Peritoneal Metastases. Cancers 2022, 14, 570. [Google Scholar] [CrossRef]
- Abdi, E.; Latifi-Navid, S.; Abedi Sarvestani, F.; Esmailnejad, M.H. Emerging therapeutic targets for gastric cancer from a host-Helicobacter pylori interaction perspective. Expert Opin. Ther. Targets 2021, 25, 685–699. [Google Scholar] [CrossRef]
- Chaurasiya, S.; Fong, Y.; Warner, S.G. Oncolytic Virotherapy for Cancer: Clinical Experience. Biomedicines 2021, 9, 419. [Google Scholar] [CrossRef]
- Santos Apolonio, J.; Lima de Souza Goncalves, V.; Cordeiro Santos, M.L.; Silva Luz, M.; Silva Souza, J.V.; Rocha Pinheiro, S.L.; de Souza, W.R.; Sande Loureiro, M.; de Melo, F.F. Oncolytic virus therapy in cancer: A current review. World J. Virol. 2021, 10, 229–255. [Google Scholar] [CrossRef] [PubMed]
- Shalhout, S.Z.; Miller, D.M.; Emerick, K.S.; Kaufman, H.L. Therapy with oncolytic viruses: Progress and challenges. Nat. Rev. Clin. Oncol. 2023, 20, 160–177. [Google Scholar] [CrossRef]
- Lawler, S.E.; Speranza, M.C.; Cho, C.F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017, 3, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Gu, X.; Yu, J.; Ge, S.; Fan, X. Oncolytic Virotherapy: From Bench to Bedside. Front. Cell Dev. Biol. 2021, 9, 790150. [Google Scholar] [CrossRef]
- Gholami, S.; Marano, A.; Chen, N.G.; Aguilar, R.J.; Frentzen, A.; Chen, C.H.; Lou, E.; Fujisawa, S.; Eveno, C.; Belin, L.; et al. A novel vaccinia virus with dual oncolytic and anti-angiogenic therapeutic effects against triple-negative breast cancer. Breast Cancer Res. Treat. 2014, 148, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.K.; Kim, T.S.; Hufford, M.M.; Braciale, T.J. Viral infection of the lung: Host response and sequelae. J. Allergy Clin. Immunol. 2013, 132, 1263–1276, quiz 1277. [Google Scholar] [CrossRef]
- Kato, K.; Omura, H.; Ishitani, R.; Nureki, O. Cyclic GMP-AMP as an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Annu. Rev. Biochem. 2017, 86, 541–566. [Google Scholar] [CrossRef]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, A.; Chaurasiya, S.; Park, A.K.; Lu, J.; Kim, S.I.; Warner, S.G.; Yuan, Y.C.; Liu, Z.; Han, H.; et al. CF33-hNIS-antiPDL1 virus primes pancreatic ductal adenocarcinoma for enhanced anti-PD-L1 therapy. Cancer Gene Ther. 2022, 29, 722–733. [Google Scholar] [CrossRef]
- Chaurasiya, S.; Yang, A.; Kang, S.; Lu, J.; Kim, S.I.; Park, A.K.; Sivanandam, V.; Zhang, Z.; Woo, Y.; Warner, S.G.; et al. Oncolytic poxvirus CF33-hNIS-DeltaF14.5 favorably modulates tumor immune microenvironment and works synergistically with anti-PD-L1 antibody in a triple-negative breast cancer model. Oncoimmunology 2020, 9, 1729300. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Zhang, Z.; Chaurasiya, S.; Park, A.K.; Jung, A.; Lu, J.; Kim, S.I.; Priceman, S.; Fong, Y.; Woo, Y. Development of the oncolytic virus, CF33, and its derivatives for peritoneal-directed treatment of gastric cancer peritoneal metastases. J. Immunother. Cancer 2023, 11, e006280. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.; Zhang, Z.; Yang, A.; Chaurasiya, S.; Park, A.K.; Lu, J.; Kim, S.I.; Warner, S.G.; Von Hoff, D.; Fong, Y. Novel Chimeric Immuno-Oncolytic Virus CF33-hNIS-antiPDL1 for the Treatment of Pancreatic Cancer. J. Am. Coll. Surg. 2020, 230, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Strohlein, M.A.; Heiss, M.M.; Jauch, K.W. The current status of immunotherapy in peritoneal carcinomatosis. Expert. Rev. Anticancer. Ther. 2016, 16, 1019–1027. [Google Scholar] [CrossRef]
- Hotopp, T. HIPEC and CRS in peritoneal metastatic gastric cancer—Who really benefits? Surg. Oncol. 2019, 28, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Braeuer, F.; Fischer, I.; Brammen, L.; Pressl, G.; Fuegger, R.; Rohregger, K.; Wundsam, H. Outcome in Patients Treated With Cytoreductive Surgery and HIPEC for Gastric Cancer With Peritoneal Carcinomatosis. Anticancer. Res. 2020, 40, 2151–2156. [Google Scholar] [CrossRef] [PubMed]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-gamma in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef]
- Burke, J.D.; Young, H.A. IFN-gamma: A cytokine at the right time, is in the right place. Semin. Immunol. 2019, 43, 101280. [Google Scholar] [CrossRef] [PubMed]
- Mell, L.K.; Brumund, K.T.; Daniels, G.A.; Advani, S.J.; Zakeri, K.; Wright, M.E.; Onyeama, S.J.; Weisman, R.A.; Sanghvi, P.R.; Martin, P.J.; et al. Phase I Trial of Intravenous Oncolytic Vaccinia Virus (GL-ONC1) with Cisplatin and Radiotherapy in Patients with Locoregionally Advanced Head and Neck Carcinoma. Clin. Cancer Res. 2017, 23, 5696–5702. [Google Scholar] [CrossRef] [PubMed]
- Lauer, U.M.; Schell, M.; Beil, J.; Berchtold, S.; Koppenhofer, U.; Glatzle, J.; Konigsrainer, A.; Mohle, R.; Nann, D.; Fend, F.; et al. Phase I Study of Oncolytic Vaccinia Virus GL-ONC1 in Patients with Peritoneal Carcinomatosis. Clin. Cancer Res. 2018, 24, 4388–4398. [Google Scholar] [CrossRef]
- Park, S.H.; Breitbach, C.J.; Lee, J.; Park, J.O.; Lim, H.Y.; Kang, W.K.; Moon, A.; Mun, J.H.; Sommermann, E.M.; Maruri Avidal, L.; et al. Phase 1b Trial of Biweekly Intravenous Pexa-Vec (JX-594), an Oncolytic and Immunotherapeutic Vaccinia Virus in Colorectal Cancer. Mol. Ther. 2015, 23, 1532–1540. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Moon, A.; Burke, J.; Hwang, T.H.; Kirn, D.H. A Phase 2, Open-Label, Randomized Study of Pexa-Vec (JX-594) Administered by Intratumoral Injection in Patients with Unresectable Primary Hepatocellular Carcinoma. Methods Mol. Biol. 2015, 1317, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Downs-Canner, S.; Guo, Z.S.; Ravindranathan, R.; Breitbach, C.J.; O’Malley, M.E.; Jones, H.L.; Moon, A.; McCart, J.A.; Shuai, Y.; Zeh, H.J.; et al. Phase 1 Study of Intravenous Oncolytic Poxvirus (vvDD) in Patients with Advanced Solid Cancers. Mol. Ther. 2016, 24, 1492–1501. [Google Scholar] [CrossRef] [PubMed]
- Zeh, H.J.; Downs-Canner, S.; McCart, J.A.; Guo, Z.S.; Rao, U.N.; Ramalingam, L.; Thorne, S.H.; Jones, H.L.; Kalinski, P.; Wieckowski, E.; et al. First-in-man study of western reserve strain oncolytic vaccinia virus: Safety, systemic spread, and antitumor activity. Mol. Ther. 2015, 23, 202–214. [Google Scholar] [CrossRef]
- Husseini, F.; Delord, J.P.; Fournel-Federico, C.; Guitton, J.; Erbs, P.; Homerin, M.; Halluard, C.; Jemming, C.; Orange, C.; Limacher, J.M.; et al. Vectorized gene therapy of liver tumors: Proof-of-concept of TG4023 (MVA-FCU1) in combination with flucytosine. Ann. Oncol. 2017, 28, 169–174. [Google Scholar] [CrossRef]
- Bae, G.E.; Kim, S.H.; Choi, M.K.; Kim, J.M.; Yeo, M.K. Targeted Sequencing of Ascites and Peritoneal Washing Fluid of Patients With Gastrointestinal Cancers and Their Clinical Applications and Limitations. Front. Oncol. 2021, 11, 712754. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Patients | Percentage (%) | |
|---|---|---|
| Total patients | 27 | 100 |
| Age (year) | ||
| 20–39 | 7 | 25.9 |
| 40–59 | 10 | 37 |
| 60+ | 10 | 37 |
| Gender | ||
| Male | 14 | 51.8 |
| Female | 13 | 48.2 |
| Extent of metastatic disease | ||
| No PM | 9 | 33.3 |
| PM only | 14 | 51.8 |
| PM plus | 4 | 14.8 |
| Race/ethnicity | ||
| Asian | 8 | 29.6 |
| Hispanic | 10 | 37 |
| Non-Hispanic White | 8 | 29.6 |
| Black | 1 | 3.7 |
| Clinical T | ||
| T1 | 0 | 0 |
| T2 | 1 | 3.7 |
| T3 | 1 | 3.7 |
| T4 | 25 | 92.6 |
| M-stage | ||
| M0 | 2 | 7.4 |
| M1 | 25 | 92.6 |
| Histology type | ||
| Intestinal | 6 | 22.2 |
| Diffuse | 21 | 77.8 |
| PET/CT | ||
| positive | 22 | 81.5 |
| negative | 5 | 18.5 |
| Grade | ||
| Well-differentiated | 2 | 7.4 |
| Moderately differentiated | 1 | 3.7 |
| Poorly differentiated | 24 | 88.9 |
| Peritoneal cytology | ||
| PW(−) | 9 | 33.3 |
| PW(+) | 4 | 14.8 |
| MA | 14 | 51.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Yang, A.; Chaurasiya, S.; Park, A.K.; Kim, S.-I.; Lu, J.; Valencia, H.; Fong, Y.; Woo, Y. Anti-Tumor Immunogenicity of the Oncolytic Virus CF33-hNIS-antiPDL1 against Ex Vivo Peritoneal Cells from Gastric Cancer Patients. Int. J. Mol. Sci. 2023, 24, 14189. https://doi.org/10.3390/ijms241814189
Zhang Z, Yang A, Chaurasiya S, Park AK, Kim S-I, Lu J, Valencia H, Fong Y, Woo Y. Anti-Tumor Immunogenicity of the Oncolytic Virus CF33-hNIS-antiPDL1 against Ex Vivo Peritoneal Cells from Gastric Cancer Patients. International Journal of Molecular Sciences. 2023; 24(18):14189. https://doi.org/10.3390/ijms241814189
Chicago/Turabian StyleZhang, Zhifang, Annie Yang, Shyambabu Chaurasiya, Anthony K. Park, Sang-In Kim, Jianming Lu, Hannah Valencia, Yuman Fong, and Yanghee Woo. 2023. "Anti-Tumor Immunogenicity of the Oncolytic Virus CF33-hNIS-antiPDL1 against Ex Vivo Peritoneal Cells from Gastric Cancer Patients" International Journal of Molecular Sciences 24, no. 18: 14189. https://doi.org/10.3390/ijms241814189
APA StyleZhang, Z., Yang, A., Chaurasiya, S., Park, A. K., Kim, S.-I., Lu, J., Valencia, H., Fong, Y., & Woo, Y. (2023). Anti-Tumor Immunogenicity of the Oncolytic Virus CF33-hNIS-antiPDL1 against Ex Vivo Peritoneal Cells from Gastric Cancer Patients. International Journal of Molecular Sciences, 24(18), 14189. https://doi.org/10.3390/ijms241814189