
Comparative Study of Batch and Continuous Flow Reactors in Selective Hydrogenation of Functional Groups in Organic Compounds: What Is More Effective?


Abstract
:1. Introduction
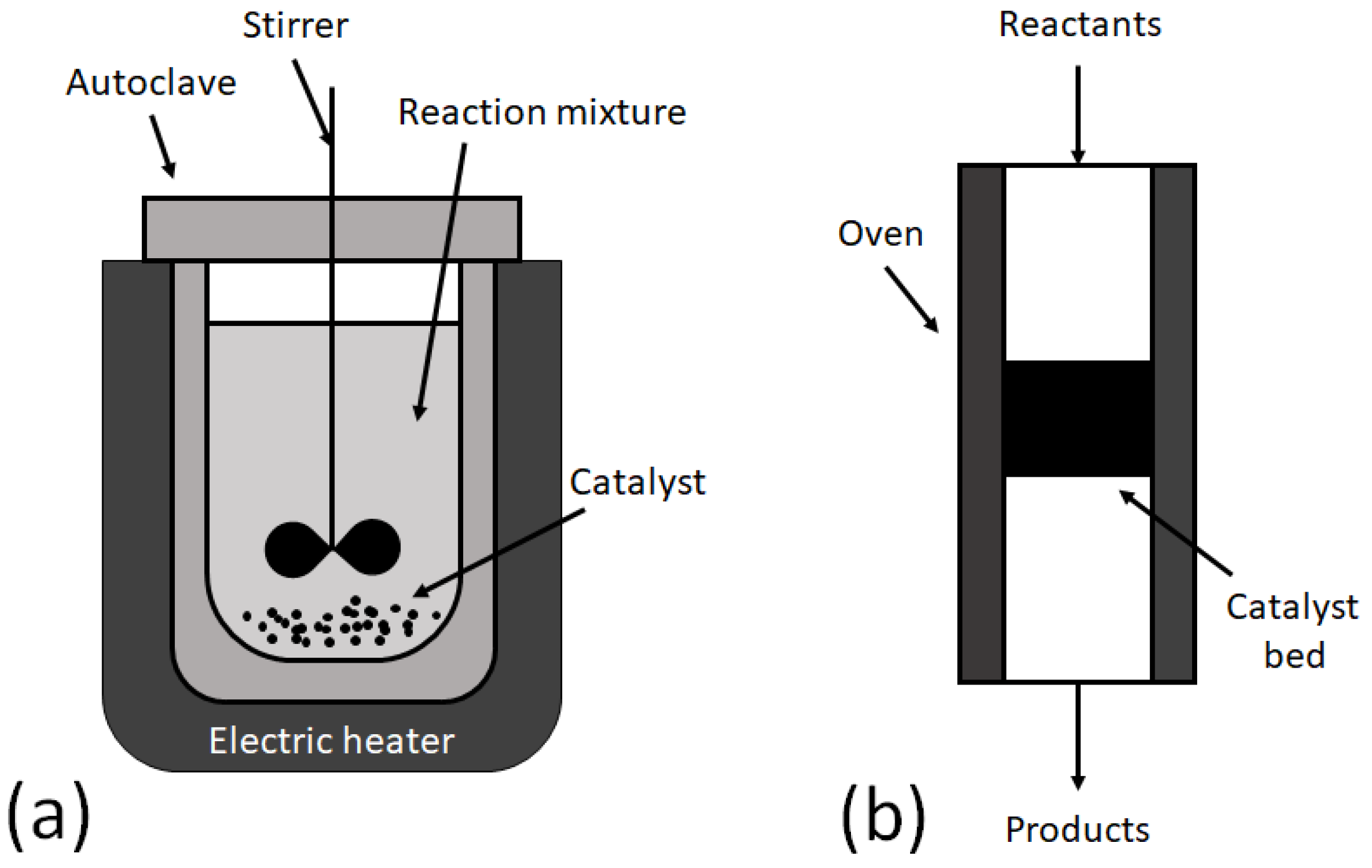
- the reaction is performed in the liquid phase;
- temperature control and vigorous stirring is needed to make sure that the temperature and composition are uniform in the whole volume of the reactor;
- concentrations of reactants and products are changed with the clock time, meaning that the longer the reaction time, the higher the product yield is;
- long synthesis can result in catalyst deactivation without knowing it has happened;
- catalyst deactivation is determined by reactivation of the catalyst and repeating the catalytic run with a washed catalyst;
- catalyst particles disturb the sampling procedure by possibly blocking the sampling port, if it is present in the autoclave;
- batch synthesis should be repeated several times to produce a high amount of the desired product.
- the reaction is performed in the gas phase;
- the composition of the gas at the outlet does not change with the clock time;
- precise control of the molar ratio of reactants is possible by controlling the flow rates of reactants;
- the change of residence time without changing the catalyst in the reactor;
- catalyst deactivation is determined by the long-term stability test with online measurement of the gas mixture;
- there is no need to start and stop the continuous process for the production of the target product in a high yield.
- there is an acceptable level with respect to yield, scale, and reaction time;
- the existing synthesis route fits the existing batch equipment;
- the immediate goal is optimization of discrete variables;
- there is low market growth (<1 kt/a);
- precipitate drives the reaction to completion.
- one of the reagents is gas;
- the reaction is performed over a heterogeneous catalyst;
- there is high marker volume (>10 kt/a);
- products suffer under catalyst deactivation;
- heating accelerates the reaction.
2. Selective Hydrogenation of Functionalized Nitroarenes
2.1. Halogenated Nitroarenes
2.2. Nitrophenols
3. Selective Hydrogenation of Carbonyl Groups in Acids and Aldehydes Containing Additional Double Bonds
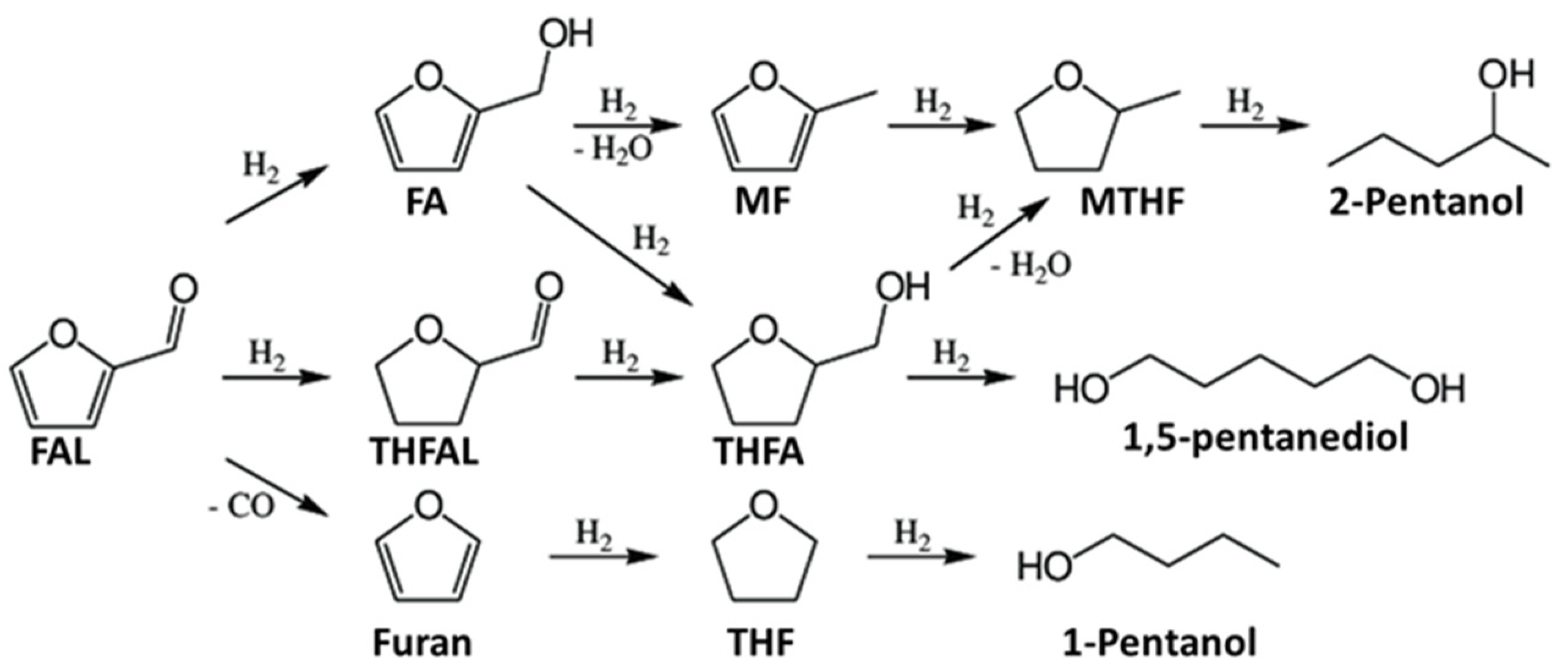
3.1. Hydrogenation of Furfural or 5-Hydroxymethylfurfural
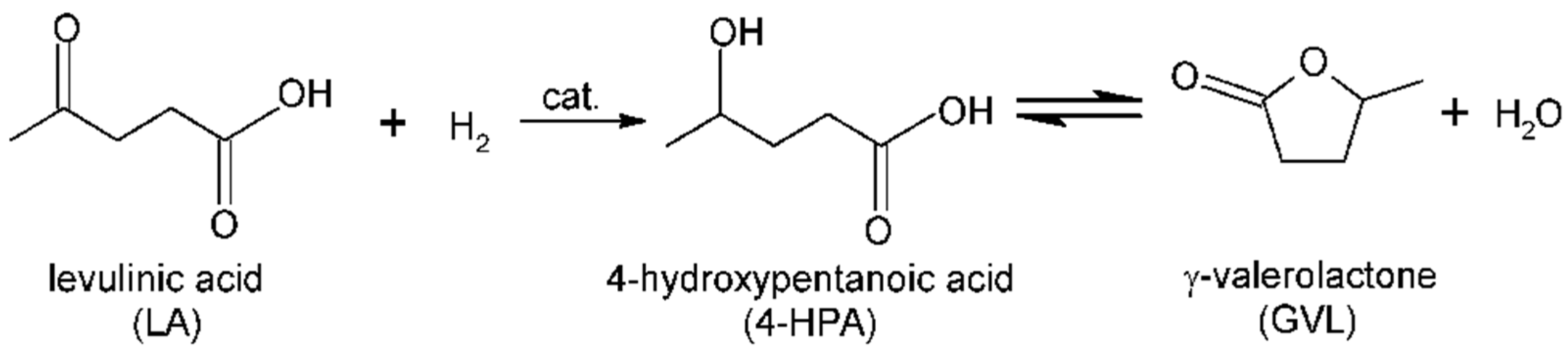
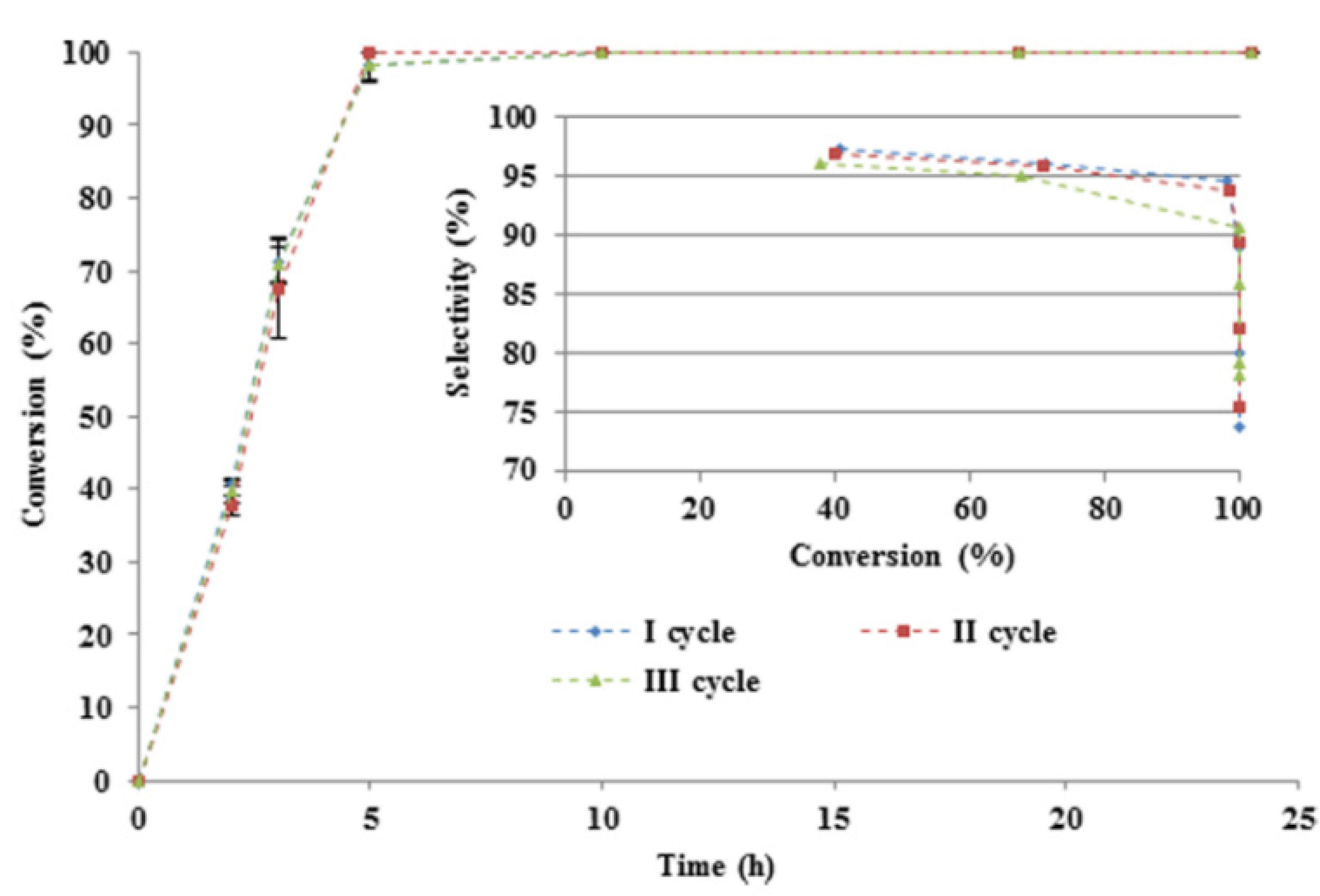
3.2. Hydrogenation of Levulinic Acid to γ-Valerolactone
3.3. Hydrogenation of Cinnamaldehyde
4. Hydrogenation of Unsaturated Carbon–Carbon Bonds
4.1. Semihydrogenation of Carbon Triple Bonds
4.2. Selective Hydrogenation of Double Carbon Bonds
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stitt, E.H.; Rooney, D.W. Switching from Batch to Continuous Processing for Fine and Intermediate-Scale Chemicals Manufacture. In Novel Concepts in Catalysis and Chemical Reactors: Improving the Efficiency for the Future; Cybulski, A., Moulijn, J.A., Stankiewicz, A., Eds.; WILEY-VCH Verlag GmbH & Co.: Weinheim, Germany, 2010; pp. 309–330. [Google Scholar]
- Costandy, J.G.; Edgar, T.F.; Baldea, M. Switching from Batch to Continuous Reactors Is a Trajectory Optimization Problem. Ind. Eng. Chem. Res. 2019, 58, 13718–13736. [Google Scholar] [CrossRef]
- Plutschack, M.B.; Pieber, B.; Gilmore, K.; Seeberger, P.H. The Hitchhiker’s Guide to Flow Chemistry. Chem. Rev. 2017, 117, 11796–11893. [Google Scholar] [CrossRef] [PubMed]
- Holtze, C.; Boehling, R. Batch or flow chemistry?—A current industrial opinion on process selection. Curr. Opin. Chem. Eng. 2022, 36, 100798. [Google Scholar] [CrossRef]
- Ouyang, W.; Yepez, A.; Romero, A.A.; Luque, R. Towards industrial furfural conversion: Selectivity and stability of palladium and platinum catalysts under continuous flow regime. Catal. Today 2018, 308, 32–37. [Google Scholar] [CrossRef]
- Saito, Y.; Ishitani, H.; Ueno, M.; Kobayashi, S. Selective Hydrogenation of Nitriles to Primary Amines Catalyzed by a Polysilane/SiO2-Supported Palladium Catalyst under Continuous-Flow Conditions. Chem. Open 2017, 6, 211–215. [Google Scholar] [CrossRef]
- Mhadmhan, S.; Franco, A.; Pineda, A.; Reubroycharoen, P.; Luque, R. Continuous Flow Selective Hydrogenation of 5-Hydroxymethylfurfural to 2,5-Dimethylfuran Using Highly Active and Stable Cu–Pd/Reduced Graphene Oxide. ACS Sustain. Chem. Eng. 2019, 7, 14210–14216. [Google Scholar] [CrossRef]
- Romero, A.H. Reduction of Nitroarenes via Catalytic Transfer Hydrogenation Using Formic Acid as Hydrogen Source: A Comprehensive Review. ChemistrySelect 2020, 5, 13054–13075. [Google Scholar] [CrossRef]
- Orlandi, M.; Brenna, D.; Harms, R.; Jost, S.; Benaglia, M. Recent developments in the reduction of aromatic and aliphatic nitro compounds to amines. Org. Process. Res. Dev. 2018, 22, 430–445. [Google Scholar] [CrossRef]
- Dell’Anna, M.M.; Gallo, V.; Mastrorilli, P.; Romanazzi, G. A Recyclable Nanoparticle-Supported Rhodium Catalyst for Hydrogenation Reactions. Molecules 2010, 15, 3311–3318. [Google Scholar] [CrossRef]
- Li, J.; Ding, S.; Wang, F.; Zhao, H.; Kou, J.; Akram, M.; Xu, M.; Gao, W.; Liu, C.; Yang, H.; et al. Platinum clusters anchored on sulfur-doped ordered mesoporous carbon for chemoselective hydrogenation of halogenated nitroarenes. J. Colloid Interface Sci. 2022, 625, 640–650. [Google Scholar] [CrossRef]
- Cárdenas-Lizana, F.; Lamey, D.; Perret, N.; Gómez-Quero, S.; Kiwi-Minsker, L.; Keane, M.A. Au/Mo2N as a new catalyst formulation for the hydrogenation of p-chloronitrobenzene in both liquid and gas phases. Catal. Commun. 2012, 21, 46–51. [Google Scholar] [CrossRef]
- Wang, X.; Cardenas-Lizana, F.; Keane, M.A. Toward Sustainable Chemoselective Nitroarene Hydrogenation Using Supported Gold as Catalyst. ACS Sustain. Chem. Eng. 2014, 2, 2781–2789. [Google Scholar] [CrossRef]
- Madon, R.J.; Boudart, M. Experimental criterion for the absence of artifacts in the measurement of rates of heterogeneous catalytic reactions. Ind. Eng. Chem. Fundam. 1982, 21, 438–447. [Google Scholar] [CrossRef]
- Perret, N.; Wang, X.; Onfroy, T.; Calers, C.; Keane, M.A. Selectivity in the gas-phase hydrogenation of 4-nitrobenzaldehyde over supported Au catalysts. J. Catal. 2014, 309, 333–342. [Google Scholar] [CrossRef]
- Coq, B.; Tijani, A.; Dutartre, R.; Figuéras, F. Influence of support and metallic precursor on the hydrogenation of p-chloronitrobenzene over supported platinum catalysts. J. Mol. Catal. 1993, 79, 253–264. [Google Scholar] [CrossRef]
- Cárdenas-Lizana, F.; Berguerand, C.; Yuranov, I.; Kiwi-Minsker, L. Chemoselective hydrogenation of nitroarenes: Boosting nanoparticle efficiency by confinement within highly porous polymericm framework. J. Catal. 2013, 301, 103–111. [Google Scholar] [CrossRef]
- Wang, X.D.; Liang, M.H.; Zhang, J.L.; Wang, Y. Selective Hydrogenation of Aromatic Chloronitro Compounds. Curr. Org. Chem. 2007, 11, 299–314. [Google Scholar] [CrossRef]
- Li, J.; Shi, X.-Y.; Bi, Y.-Y.; Wei, J.-F.; Chen, Z.-G. Pd Nanoparticles in Ionic Liquid Brush: A Highly Active and Reusable Heterogeneous Catalytic Assembly for Solvent-Free or On-Water Hydrogenation of Nitroarene under Mild Conditions. ACS Catal. 2011, 1, 657–664. [Google Scholar] [CrossRef]
- Cárdenas-Lizana, F.; Gómez-Quero, S.; Keane, M.A. Ultra-selective gas phase catalytic hydrogenation of aromatic nitro compounds over Au/Al2O3. Catal. Commun. 2008, 9, 475–481. [Google Scholar] [CrossRef]
- Alex, H.; Loos, P.; Baramov, T.; Barry, J.; Godiawala, T.; Hassfeld, J.; Steinfeldt, N. Polymer Encapsulated Cobalt-Based Catalysts (Co EnCatTM) for Selective Continuous Hydrogenation of 1-Iodo-4-nitrobenzene. ChemCatChem 2017, 9, 3210–3217. [Google Scholar] [CrossRef]
- Loos, P.; Alex, H.; Hassfeld, J.; Lovis, K.; Platzek, J.; Steinfeldt, N.; Hübner, S. Selective Hydrogenation of Halogenated Nitroaromatics to Haloanilines in Batch and Flow. Org. Process Res. Dev. 2016, 20, 452–464. [Google Scholar] [CrossRef]
- Pentsak, E.O.; Eremin, D.B.; Gordeev, E.G.; Ananikov, V.P. Phantom Reactivity in Organic and Catalytic Reactions as a Consequence of Microscale Destruction and Contamination-Trapping Effects of Magnetic Stir Bars. ACS Catal. 2019, 9, 3070–3081. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Mondal, K.; Mukhopadhyay, K.; Prasada, N.E.; Sharma, A. Facile reduction of para-nitrophenols: Catalytic efficiency of silver nanoferns in batch and continuous flow reactors. RSC Adv. 2016, 6, 113981. [Google Scholar] [CrossRef]
- Li, Y.; Xu, H.; Zhang, G. Porous carbon-encapsulated CuxO/Cu catalyst derived from N-coordinated MOF for ultrafast 4-nitrophenol reduction in batch and continuous flow reactors. J. Environ. Chem. Eng. 2022, 10, 108677. [Google Scholar] [CrossRef]
- Zhang, W.; Tan, F.; Wang, W.; Qiu, X.; Qiao, X.; Chen, J. Facile, template-free synthesis of silver nanodendrites with high catalytic activity for the reduction of p-nitrophenol. J. Hazard. Mater. 2012, 217–218, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Labana, S.; Pandey, G.; Paul, D.; Sharma, N.K.; Basu, A.; Jain, R.K. Pot and Field Studies on Bioremediation of p-Nitrophenol Contaminated Soil Using Arthrobacterprotophormiae RKJ100. Environ. Sci. Technol. 2005, 39, 3330–3337. [Google Scholar] [CrossRef] [PubMed]
- Melataguia Tchieno, F.M.; Kenfack Tonle, I. p-Nitrophenol determination and remediation: An overview. Rev. Anal. Chem. 2018, 37, 20170019. [Google Scholar] [CrossRef]
- Mitchell, S. Kirk-Othmer Encyclopaedia of Chemical Technology, 4th ed.; Wiley-Interscience: New York, NY, USA, 1992; pp. 481–580. [Google Scholar]
- Mondal, K.; Kumar, J.; Sharma, A. Self-organized macroporous thin carbon films for supported metal catalysis. Colloids Surf. A 2013, 427, 83–94. [Google Scholar] [CrossRef]
- Jiang, S.; Ni, H.; Li, P.; Wang, J.; Ren, H. Metal/N-doped carbon (Metal = Ag, Cu, Ni) nanocatalysts for selective hydrogenation of 4-nitrophenol. Catal. Commun. 2021, 151, 106280. [Google Scholar] [CrossRef]
- Sun, X.; He, P.; Gao, Z.; Liao, Y.; Weng, S.; Zhao, Z.; Song, H.; Zhao, Z. Multi-crystalline N-doped Cu/CuxO/C foam catalyst derived from alkaline N-coordinated HKUST-1/ CMC for enhanced 4-nitrophenol reduction. J. Colloid Interface Sci. 2019, 553, 1–13. [Google Scholar] [CrossRef]
- Šivec, R.; Grilc, M.; Huš, M.; Likozar, B. Multiscale Modeling of (Hemi)cellulose Hydrolysis and Cascade Hydrotreatment of 5-Hydroxymethylfurfural, Furfural, and Levulinic Acid. Ind. Eng. Chem. Res. 2019, 58, 16018–16032. [Google Scholar] [CrossRef]
- Bonacci, S.; Nardi, M.; Costanzo, P.; De Nino, A.; Di Gioia, M.; Oliverio, M.; Procopio, A. Montmorillonite K10-Catalyzed Solvent-Free Conversion of Furfural into Cyclopentenones. Catalysts 2019, 9, 301. [Google Scholar] [CrossRef]
- Jiang, Y.; Woortman, A.J.; Alberda van Ekenstein, G.R.; Petrović, D.M.; Loos, K. Enzymatic Synthesis of Biobased Polyesters Using 2,5-Bis(hydroxymethyl)furan as the Building Block. Micromolecules 2014, 15, 2482–2493. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Andrioletti, B.; Queneau, Y. Furfural and 5-(hydroxymethyl)furfural: Two pivotal intermediates for bio-based chemistry. Curr. Opin. Green Sustain. Chem. 2020, 26, 100384. [Google Scholar] [CrossRef]
- Audemar, M.; Wang, Y.; Zhao, D.; Royer, S.; Jérôme, F.; Len, C.; De Oliveira Vigier, K. Synthesis of Furfuryl Alcohol from Furfural: A Comparison between Batch and Continuous Flow Reactors. Energies 2020, 13, 1002. [Google Scholar] [CrossRef]
- Wei, H.; Feng, D.; Shu, G.; Pan, M.; Guo, Y.; Gao, D.; Li, W. Experimental investigation on the combustion and emissions characteristics of 2-methylfuran gasoline blend fuel in spark-ignition engine. Appl. Energy 2014, 132, 317–324. [Google Scholar] [CrossRef]
- Mishra, D.K.; Lee, H.J.; Truong, C.C.; Kim, J.; Suh, Y.-W.; Baek, J.; Kim, Y.J. Ru/MnCo2O4 as a catalyst for tunable synthesis of 2,5-bis(hydroxymethyl)furan or 2,5-bis(hydroxymethyl)tetrahydrofuran from hydrogenation of 5-hydroxymethylfurfural. Mol. Catal. 2019, 484, 110722. [Google Scholar] [CrossRef]
- Morozov, E. Furfural Production, 2nd ed.; Forest Industry: Moscow, Russia, 1988; pp. 32–56. [Google Scholar]
- Wang, Y.; Prinsen, P.; Triantafyllidis, K.S.; Karakoulia, S.A.; Trikalitis, P.N.; Yepez, A.; Len, C.; Luque, R. Comparative Study of Supported Monometallic Catalysts in the Liquid-Phase Hydrogenation of Furfural: Batch Versus Continuous Flow. ACS Sustain. Chem. Eng. 2018, 6, 9831–9844. [Google Scholar] [CrossRef]
- Selishcheva, S.A.; Smirnov, A.A.; Fedorov, A.V.; Ermakov, D.Y.; Gulyaeva, Y.K.; Yakovlev, V.A. Production of Furfuryl Alcohol in the Presence of Copper-Containing Catalysts in the Selective Hydrogenation of Furfural. Catal. Ind. 2019, 11, 216–223. [Google Scholar] [CrossRef]
- Wang, Y.; Prinsen, P.; Triantafyllidis, K.S.; Karakoulia, S.A.; Yepez, A.; Len, C.; Luque, R. Batch versus Continuous Flow Performance of Supported Mono- and Bimetallic Nickel Catalysts for Catalytic Transfer Hydrogenation of Furfural in Isopropanol. ChemCatChem 2018, 10, 3459–3468. [Google Scholar] [CrossRef]
- Yang, X.; Meng, Q.; Ding, G.; Wang, Y.; Chen, H.; Zhu, Y.L.; Li, Y.W. Construction of novel Cu/ZnO-Al2O3 composites for furfural hydrogenation: The role of Al components. Appl. Catal. A 2018, 561, 78–86. [Google Scholar] [CrossRef]
- Ghashghaee, M.; Shirvani, S.; Ghambarian, M. Kinetic models for hydroconversion of furfural over the ecofriendly Cu-MgO catalyst: An experimental and theoretical study. Appl. Catal. A 2017, 545, 134–147. [Google Scholar] [CrossRef]
- Gilkey, M.J.; Panagiotopoulou, P.; Mironenko, A.V.; Jenness, G.R.; Vlachos, D.G.; Xu, B. Mechanistic Insights into Metal Lewis Acid-Mediated Catalytic Transfer Hydrogenation of Furfural to 2-Methylfuran. ACS Catal. 2015, 5, 3988–3994. [Google Scholar] [CrossRef]
- Koehle, M.; Lobo, R.F. Lewis acidic zeolite Beta catalyst for the Meerwein–Ponndorf–Verley reduction of furfural. Catal. Sci. Technol. 2016, 6, 3018–3026. [Google Scholar] [CrossRef]
- Xinghua, Z.; Tiejun, W.; Longlong, M.; Chuangzhi, W. Aqueous-phase catalytic process for production of pentane from furfural over nickel-based catalysts. Fuel 2010, 89, 2697–2702. [Google Scholar] [CrossRef]
- Gong, W.; Chen, C.; Zhang, H.; Zhang, Y.; Zhang, Y.; Wang, G.; Zhao, H. Highly selective liquid-phase hydrogenation of furfural over N-doped carbon supported metallic nickel catalyst under mild conditions. Mol. Catal. 2017, 429, 51–59. [Google Scholar] [CrossRef]
- Taylor, M.J.; Jiang, L.; Reichert, J.; Papageorgiou, A.C.; Beaumont, S.K.; Wilson, K.; Lee, A.F.; Barth, J.V.; Kyriakou, G. Catalytic Hydrogenation and Hydrodeoxygenation of Furfural over Pt(111): A Model System for the Rational Design and Operation of Practical Biomass Conversion Catalysts. J. Phys. Chem. C Nanomater. Interfaces 2017, 121, 8490–8497. [Google Scholar] [CrossRef]
- Kim, J.; Bathula, H.B.; Yun, S.; Jo, Y.; Lee, S.; Baik, J.H.; Suh, Y.-W. Hydrogenation of 5-hydroxymethylfurfural into 2,5-bis(hydroxymethyl)furan over mesoporous Cu–Al2O3 catalyst: From batch to continuous processing. J. Ind. Eng. Chem. 2021, 102, 186–194. [Google Scholar] [CrossRef]
- Kang, S.; Fu, J.; Zhang, G. From lignocellulosic biomass to levulinic acid: A review on acid-catalyzed hydrolysis. Renew. Sustain. Energy Rev. 2018, 94, 340–362. [Google Scholar] [CrossRef]
- Alonso, D.M.; Wettstein, S.G.; Dumesic, J.A. Gammavalerolactone, a sustainable platform molecule derived from lignocellulosic biomass. Green Chem. 2013, 15, 584–595. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, Y.; Luo, X. The Relationship between Structure and Catalytic Activity-Stability of Non-Precious Metal-Based Catalysts towards Levulinic Acid Hydrogenation to γ-Valerolactone: A Review. Energies 2022, 15, 8093. [Google Scholar] [CrossRef]
- Bereczky, Á.; Lukács, K.; Farkas, M.; Dóbé, S. Effect of γ-Valerolactone Blending on Engine Performance, Combustion Characteristics and Exhaust Emissions in a Diesel Engine. Nat. Resour. 2014, 5, 177–191. [Google Scholar]
- Dutta, S.; Yu IK, M.; Tsang DC, W.; Ng, Y.H.; Ok, Y.S.; Sherwood, J.; Clark, J.H. Green synthesis of gamma-valerolactone (GVL) through hydrogenation of biomass-derived levulinic acid using non-noble metal catalysts: A critical review. Chem. Eng. J. 2019, 372, 992–1006. [Google Scholar] [CrossRef]
- Genuino, H.C.; van de Bovenkamp, H.H.; Wilbers, E.; Winkelman, J.G.M.; Goryachev, A.; Hofmann, J.P.; Hensen, E.J.M.; Weckhuysen, B.M.; Bruijnincx, P.C.A.; Heeres, H.J. Catalytic Hydrogenation of Renewable Levulinic Acid to γ-Valerolactone: Insights into the Influence of Feed Impurities on Catalyst Performance in Batch and Flow Reactors. ACS Sustain. Chem. Eng. 2020, 8, 5903–5919. [Google Scholar] [CrossRef]
- Ruiz-López, E.; Ribota Peláez, M.; Blasco Ruz, M.; Domínguez Leal, M.I.; Martínez Tejada, M.; Ivanova, S.; Ángel Centeno, M. Formic Acid Dehydrogenation over Ru- and Pd-Based Catalysts: Gas- vs. Liquid-Phase Reactions. Materials 2023, 16, 472. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, A.M.; Jędrzejczyk, M.; Sneka-Płatek, O.; Keller, N.; Dumon, A.S.; Michel, C.; Sautet, P.; Grams, J. Ru catalysts for levulinic acid hydrogenation with formic acid as a hydrogen source. Green Chem. 2016, 18, 2014–2028. [Google Scholar] [CrossRef]
- Grillo, G.; Manzoli, M.; Bucciol, F.; Tabasso, S.; Tabanelli, T.; Cavani, F.; Cravotto, G. Hydrogenation of Levulinic Acid to γ-Valerolactone via Green Microwave-Assisted Reactions Either in Continuous Flow or Solvent-Free Batch Processes. Ind. Eng. Chem. Res. 2021, 60, 16756–16768. [Google Scholar] [CrossRef]
- Tang, X.; Hu, L.; Sun, Y.; Zhao, G.; Hao, W.; Lin, L. Conversion of Biomass-Derived Ethyl Levulinate into γ-Valerolactone via Hydrogen Transfer from Supercritical Ethanol over a ZrO2 Catalyst. RSC Adv. 2013, 3, 10277–10284. [Google Scholar] [CrossRef]
- Komanoya, T.; Nakajima, K.; Kitano, M.; Hara, M. Synergistic Catalysis by Lewis Acid and Base Sites on ZrO2 for Meerwein- Ponndorf-Verley Reduction. J. Phys. Chem. C 2015, 119, 26540–26546. [Google Scholar] [CrossRef]
- Tabanelli, T.; Paone, E.; Vásquez, P.B.; Pietropaolo, R.; Cavani, F.; Mauriello, F. Transfer Hydrogenation of Methyl and Ethyl Levulinate Promoted by a ZrO2 Catalyst: Comparison of Batch vs Continuous Gas-Flow Conditions. ACS Sustain. Chem. Eng. 2019, 7, 9937–9947. [Google Scholar] [CrossRef]
- Mauriello, F.; Ariga, H.; Musolino, M.G.; Pietropaolo, R.; Takakusagi, S.; Asakura, K. Exploring the catalytic properties of supported palladium catalysts in the transfer hydrogenolysis of glycerol. Appl. Catal. B 2015, 166–167, 121–131. [Google Scholar] [CrossRef]
- Letizia, C.S.; Cocchiara, J.; Lalko, J.; Lapczynski, A.; Api, A.M. Fragrance material review on cinnamyl alcohol. Food Chem. Toxicol. 2005, 43, 837–866. [Google Scholar] [CrossRef] [PubMed]
- Durndell, L.J.; Wilson, K.; Lee, A.F. Platinum-catalysed cinnamaldehyde hydrogenation in continuous flow. RSC Adv. 2015, 5, 80022. [Google Scholar] [CrossRef]
- Durndell, L.J.; Parlett CM, A.; Hondow, N.S.; Isaacs, M.A.; Wilson, K.; Lee, A.F. Selectivity control in Pt-catalyzed cinnamaldehyde hydrogenation. Sci. Rep. 2015, 5, 9425. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.M.; Punji, B. 3d Transition Metal-Catalyzed Hydrogenation of Nitriles and Alkynes. Chem. Asian J. 2020, 15, 690–708. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Yamamoto, H.; Kawai, K.; Park, K.; Aono, N.; Sajiki, H. Development of Silicon Carbide-Supported Palladium Catalysts and Their Application as Semihydrogenation Catalysts for Alkynes under Batch- and Continuous-Flow Conditions. Catalysts 2022, 12, 1253. [Google Scholar] [CrossRef]
- Numwong, N.; Luengnaruemitchai, A.; Chollacoop, N.; Yoshimura, Y. Partial hydrogenation of polyunsaturated fatty acid methyl esters over Pd/activated carbon: Effect of type of reactor. Chem. Eng. J. 2012, 210, 173–181. [Google Scholar] [CrossRef]
- Amin, A. Review of diesel production from renewable resources: Catalysis, process kinetics and technologies. Ain Shams Eng. J. 2019, 10, 821–839. [Google Scholar] [CrossRef]
- Ershov, M.A.; Savelenko, V.D.; Makhova, U.A.; Makhmudova, A.E.; Zuikov, A.V.; Kapustin, V.M.; Abdellatief TM, M.; Burov, N.O.; Geng, T.; Ali Abdelkareem, M.; et al. Current Challenge and Innovative Progress for Producing HVO and FAME Biodiesel Fuels and Their Applications. Waste Biomass Valorization 2023, 14, 505–521. [Google Scholar] [CrossRef]
- Verma, P.; Sharma, M.P.; Dwivedi, G. Evaluation and enhancement of cold flow properties of palm oil and its biodiesel. Energy Rep. 2016, 2, 8–13. [Google Scholar] [CrossRef]
- Lv, P.; Cheng, Y.; Yang, L.; Yuan, Z.; Li, H.; Luo, W. Improving the low temperature flow properties of palm oil biodiesel: Addition of cold flow improver. Fuel Process. Technol. 2013, 110, 61–64. [Google Scholar] [CrossRef]
- Selifonov, S. Methods for Making (−)-Menthol and Oxygenated Menthane Compounds. U.S. Patent US20060155153A1, 13 July 2006. [Google Scholar]
- Liu, Q.; Yu, Y.; Wang, P.; Li, Y. Synthesis of analogues of linckoside B, a new neuritogenic steroid glycoside. New J. Chem. 2013, 37, 3647–3661. [Google Scholar] [CrossRef]
- Rubulotta, G.; Luska, K.L.; Urbina-Blanco, C.A.; Eifert, T.; Palkovits, R.; Quadrelli, E.A.; Thieuleux, C.; Leitner, W. Highly Selective Hydrogenation of R-(+)-Limonene to (+)-p-1-Menthene in Batch and Continuous Flow Reactors. ACS Sustain. Chem. Eng. 2017, 5, 3762–3767. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Operation Mode | Solvent | Substrate | PH2, atm | T, °C | SCAN/SAN/SNB b | r c | Reference |
|---|---|---|---|---|---|---|---|---|
| Pd/C | Batch liquid | Ethanol | o-CNB | 5 | 150 | 79/20/1 | 2290 | [13] |
| Pd/C | Ethanol | o-CNB | 10 | 150 | 81/18/1 | 2650 | [13] | |
| Pd/C | Ethanol | o-CNB | 12 | 150 | 86/13/1 | 2910 | [13] | |
| Au/TiO2 | Ethanol | o-CNB | 5 | 150 | 100/0/0 | 10 | [13] | |
| Au/TiO2 | Ethanol | o-CNB | 12 | 150 | 100/0/0 | 167 | [13] | |
| Au/Mo2N | n.m. a | p-CNB | 11 | 150 | 100/0/0 | - | [12] | |
| Au/Mo2N | Continuous gas | Ethanol | o-CNB | 1 | 220 | 100/0/0 | 42 | [12] |
| Au/TiO2 | Ethanol | o-CNB | 1 | 150 | 100/0/0 | 12 | [13] | |
| Au/TiO2 | Ethanol | p-CNB | 1 | 150 | 100/0/0 | 12 | [13] | |
| Au/Al2O3 | Ethanol | p-CNB | 1 | 220 | 100/0/0 | 12 | [12] | |
| Pd/Mo2N | Ethanol | p-CNB | 1 | 220 | ~55/5/45 | 66 | [12] |
| Reaction Conditions | 110 °C, 20 Bar of H2, 10 mL of THF, 80 min, Raney Co | 100 °C, 85 Bar of H2, 10 mL of THF:H2O (95:5), 40 min, Co EnCatTM | ||||
|---|---|---|---|---|---|---|
| C Substrate, mol/L | INB Conversion, % | IAN yield, % | Yield (Aniline), % | INB Conversion, % | IAN Yield, % | Yield (Aniline), % |
| 0.05 | 82 | 48 | <2 | 91.6 | 70.4 | 3.1 |
| 0.1 | 77 | 50 | <2 | 90.9 | 71.8 | 3.1 |
| 0.2 * | 81 | 42 | <2 | 79.0 | 60.7 | 3.3 |
| Catalyst | Amount, mg | Temperature, °C | 4-Nitrophenol Amount | NaBH4 Amount | Reference | |
|---|---|---|---|---|---|---|
| mL | M | mL | ||||
| CuxO/Cu/NC | 0.075–0.125 | 25 | 0.027 | 0.01 | 0.27 | [25] |
| cAg-NF | 0.450 (Ag-NF) | 0.07 | 7 | [24] | ||
| Catalyst | T, °C | P(H2), bar | Conversion, % | Yield, % | Selectivity MF, % | |
|---|---|---|---|---|---|---|
| FA | MF | |||||
| 5%Ni/AC | 200 | 0 | 10 | 10 | 0 | 0 |
| 200 | 30 | 85 | 6 | 66 | 78 | |
| 260 | 0 | 95 | 20 | 50 | 53 | |
| 5%Ni-15%W/AC | 200 | 30 | 50 | 32 | 15 | 30 |
| 260 | 0 | 83 | 16 | 6 | 7 | |
| 10%Ni-15%W/AC | 200 | 30 | 51 | 8 | 20 | 39 |
| 260 | 0 | 83 | 25 | 6 | 7 | |
| Catalyst | XPS, wt.% | ICP-MS, wt.% | Selectivity MF, % | |||
|---|---|---|---|---|---|---|
| NiO | Ni0 | W | Ni | W | ||
| 5% Ni/AC, fresh | 0.5 | 0.4 | - | 4.1 | - | - |
| 5%Ni/AC, used (batch, 200 °C, 30 bar H2) | 0.3 | 0.2 | - | 2.3 | - | 78 |
| 5%Ni/AC, used (batch, 260 °C, no H2) | 0.6 | 0.6 | - | 2.4 | - | 53 |
| 5%Ni/AC, used (continuous flow, 260 °C, 30 bar, no H2) | 0.7 | 0.2 | - | 3.9 | - | 40 b |
| 5%Ni–15%W/AC, fresh | 0.5 | 0.1 | 17.4 | 4.3 | 0.1 a | - |
| 5%Ni–15%W/AC, used (batch, 260 °C, no H2) | 0.2 | 0.0 | 5.6 | 2.6 | 0.1 a | 7 |
| 5%Ni–15%W/AC, used (continuous flow, 260 °C, 30 bar, no H2) | 0.7 | 0.1 | 15.4 | 3.2 | 0.1 a | 85 b |
| H2 Pressure, bar | Temperature, °C | HMF Conversion, % | BHMF Selectivity, % |
|---|---|---|---|
| 50 | 60 | 95.0 | 98.0 |
| 50 | 70 | 99.1 | 99.5 |
| 50 | 100 | 98.7 | 99.1 |
| 30 | 70 | 97.5 | 98.1 |
| 20 | 70 | 97.0 | 97.9 |
| 10 | 70 | 24.5 | 98.0 |
| HCOOH:LA Ratio | Induction Period, Minutes | GVL+4-HPA Yield, % | |||
|---|---|---|---|---|---|
| 60 min | 120 min | 180 min | 300 min | ||
| 1:20 | 60 | 5 | 100 | 100 | - |
| 1:10 | 120 | ~2 | 5 | 100 | 95 |
| 1:8 | 180 | ~2 | ~2 | 12 | 100 |
| 1:5 | - | ~2 | ~2 | ~2 | - |
| 1:1 | - | 0 | 0 | 0 | 2 |
| Reaction Time, h | C18:2 | t-C18:1 | c-C18:1 | C18:0 |
|---|---|---|---|---|
| 0 | 5 | 0 | 30.5 | 6 |
| 0.5 | 2.5 | 5 | 27.5 | 7 |
| 1 | 1 | 12.5 | 21 | 7 |
| 1.5 | 0 | 18 | 14 | 9.5 |
| 2 | 0 | 19.5 | 12 | 12 |
| 3 | 0 | 19 | 10 | 14 |
| 4 | 0 | 9 | 16 | 17.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bukhtiyarova, M.V.; Nuzhdin, A.L.; Bukhtiyarova, G.A. Comparative Study of Batch and Continuous Flow Reactors in Selective Hydrogenation of Functional Groups in Organic Compounds: What Is More Effective? Int. J. Mol. Sci. 2023, 24, 14136. https://doi.org/10.3390/ijms241814136
Bukhtiyarova MV, Nuzhdin AL, Bukhtiyarova GA. Comparative Study of Batch and Continuous Flow Reactors in Selective Hydrogenation of Functional Groups in Organic Compounds: What Is More Effective? International Journal of Molecular Sciences. 2023; 24(18):14136. https://doi.org/10.3390/ijms241814136
Chicago/Turabian StyleBukhtiyarova, Marina V., Alexey L. Nuzhdin, and Galina A. Bukhtiyarova. 2023. "Comparative Study of Batch and Continuous Flow Reactors in Selective Hydrogenation of Functional Groups in Organic Compounds: What Is More Effective?" International Journal of Molecular Sciences 24, no. 18: 14136. https://doi.org/10.3390/ijms241814136
APA StyleBukhtiyarova, M. V., Nuzhdin, A. L., & Bukhtiyarova, G. A. (2023). Comparative Study of Batch and Continuous Flow Reactors in Selective Hydrogenation of Functional Groups in Organic Compounds: What Is More Effective? International Journal of Molecular Sciences, 24(18), 14136. https://doi.org/10.3390/ijms241814136