
Skin Cancer Microenvironment: What We Can Learn from Skin Aging?

 and
and 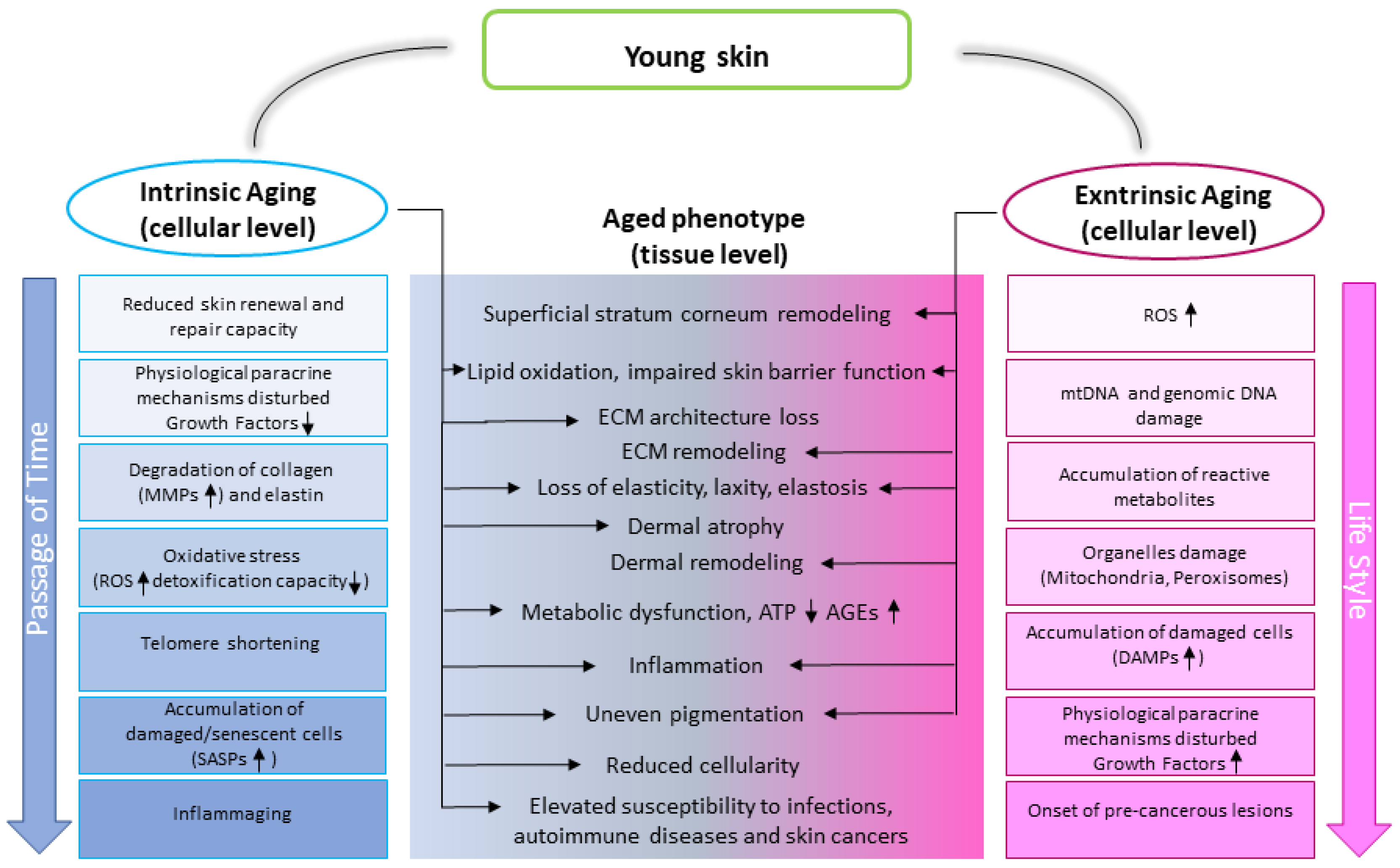{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Skin Aging Characteristics
1.2. Skin Aging Drivers
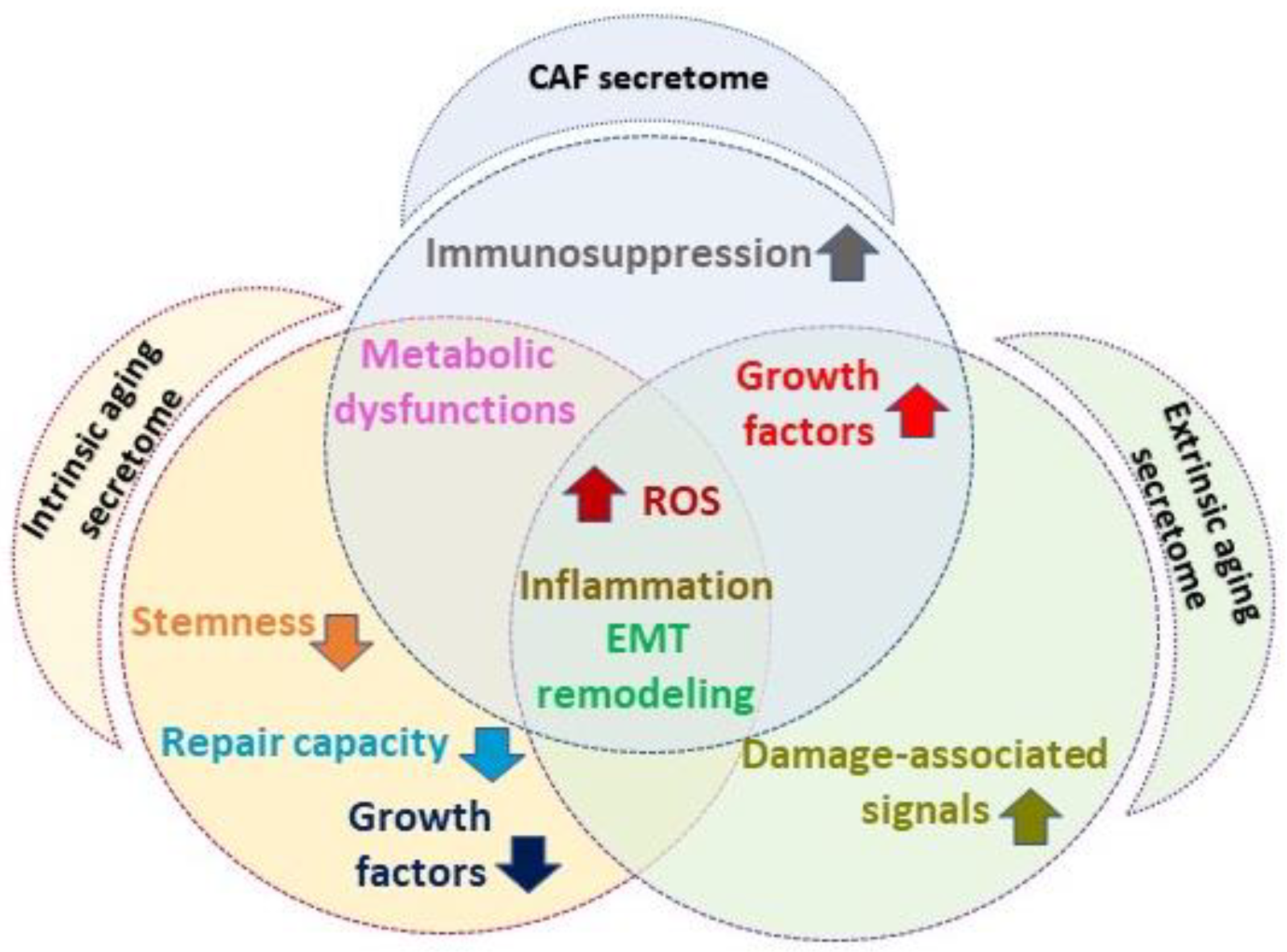
1.3. Senescent Fibroblast and Cancer-Associated Fibroblast Overlapping Features
2. Materials and Methods
3. Skin Aging Phenotype
3.1. Age-Related Modification of Skin Paracrine Network
3.1.1. Growth Factors
3.1.2. The Immune Microenvironment
3.1.3. Metabolic Intercellular Cross-Talk in Skin Aging
3.2. Skin Extracellular Matrix and Aging
4. Melanoma and Non-Melanoma Skin Cancer Microenvironments: Similar and Peculiar Aspects
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Karimkhani, C.; Green, A.C.; Nijsten, T.; Weinstock, M.A.; Dellavalle, R.P.; Naghavi, M.; Fitzmaurice, C. The Global Burden of Melanoma: Results from the Global Burden of Disease Study 2015. Br. J. Dermatol. 2017, 177, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.J.; Mihm, M.C., Jr. Melanoma. N. Engl. J. Med. 2006, 355, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Statescu, L.; Trandafir, L.M.; Tarca, E.; Moscalu, M.; Leon Constantin, M.M.; Butnariu, L.I.; Trandafirescu, M.F.; Tirnovanu, M.C.; Heredea, R.; Patrascu, A.V.; et al. Advancing Cancer Research: Current Knowledge on Cutaneous Neoplasia. Int. J. Mol. Sci. 2023, 24, 11176. [Google Scholar] [CrossRef]
- Que, S.K.T.; Zwald, F.O.; Schmults, C.D. Cutaneous Squamous Cell Carcinoma: Incidence, Risk Factors, Diagnosis, and Staging. J. Am. Acad. Dermatol. 2018, 78, 237–247. [Google Scholar] [CrossRef]
- Zouboulis, C.C.; Coenye, T.; He, L.; Kabashima, K.; Kobayashi, T.; Niemann, C.; Nomura, T.; Oláh, A.; Picardo, M.; Quist, S.R.; et al. Sebaceous Immunobiology—Skin Homeostasis, Pathophysiology, Coordination of Innate Immunity and Inflammatory Response and Disease Associations. Front. Immunol. 2022, 13, 1029818. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular Senescence: From Physiology to Pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Kalluri, R. The Biology and Function of Fibroblasts in Cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. A Peek into Cancer-Associated Fibroblasts: Origins, Functions and Translational Impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef]
- Chhabra, Y.; Weeraratna, A.T. Fibroblasts in Cancer: Unity in Heterogeneity. Cell 2023, 186, 1580–1609. [Google Scholar] [CrossRef]
- De Veirman, K.; Rao, L.; De Bruyne, E.; Menu, E.; Van Valckenborgh, E.; Van Riet, I.; Frassanito, M.A.; Di Marzo, L.; Vacca, A.; Vanderkerken, K. Cancer Associated Fibroblasts and Tumor Growth: Focus on Multiple Myeloma. Cancers 2014, 6, 1363–1381. [Google Scholar] [CrossRef] [PubMed]
- Bellei, B.; Picardo, M. Premature Cell Senescence in Human Skin: Dual Face in Chronic Acquired Pigmentary Disorders. Ageing Res. Rev. 2020, 57, 100981. [Google Scholar] [CrossRef] [PubMed]
- Chatsirisupachai, K.; Lagger, C.; de Magalhaes, J.P. Age-Associated Differences in the Cancer Molecular Landscape. Trends Cancer 2022, 8, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Paccosi, E.; Balajee, A.S.; Proietti-De-Santis, L. A Matter of Delicate Balance: Loss and Gain of Cockayne Syndrome Proteins in Premature Aging and Cancer. Front. Aging 2022, 3, 960662. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, E.; Golemis, E.A.; Arora, S. POLD1: Central Mediator of DNA Replication and Repair, and Implication in Cancer and Other Pathologies. Gene 2016, 590, 128–141. [Google Scholar] [CrossRef]
- Scharffetter-Kochanek, K.; Wang, Y.; Makrantonaki, E.; Crisan, D.; Wlaschek, M.; Geiger, H.; Maity, P. Skin aging-cellular senescence: What is the future? Dermatologie. 2023, 74, 645. [Google Scholar] [CrossRef]
- Lyu, Y.; Ge, Y. Toward Elucidating Epigenetic and Metabolic Regulation of Stem Cell Lineage Plasticity in Skin Aging. Front. Cell. Dev. Biol. 2022, 10, 903904. [Google Scholar] [CrossRef]
- Leonardi, G.C.; Accardi, G.; Monastero, R.; Nicoletti, F.; Libra, M. Ageing: From Inflammation to Cancer. Immun. Ageing 2018, 15, 1. [Google Scholar] [CrossRef]
- Schmitt, R. Senotherapy: Growing Old and Staying Young? Pflug. Arch. 2017, 469, 1051–1059. [Google Scholar] [CrossRef]
- Laconi, E.; Cheri, S.; Fanti, M.; Marongiu, F. Aging and Cancer: The Waning of Community Bonds. Cells 2021, 10, 2269. [Google Scholar] [CrossRef]
- Hinds, P.; Pietruska, J. Senescence and Tumor Suppression. F1000Research 2017, 6, 2121. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, T.; Kitagawa, M.; Kasuga, T. Proliferative Activity of Primary Cutaneous Melanocytic Tumours. Virchows Arch. A Pathol. Anat. Histopathol. 1993, 423, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, J.L.; Timmerman, L.; Fridlyand, J.; Bastian, B.C. Mechanisms of Cell-Cycle Arrest in Spitz Nevi with Constitutive Activation of the MAP-Kinase Pathway. Am. J. Pathol. 2004, 164, 1783–1787. [Google Scholar] [CrossRef] [PubMed]
- Dankort, D.; Curley, D.P.; Cartlidge, R.A.; Nelson, B.; Karnezis, A.N.; Damsky, W.E.J.; You, M.J.; DePinho, R.A.; McMahon, M.; Bosenberg, M. Braf(V600E) Cooperates with Pten Loss to Induce Metastatic Melanoma. Nat. Genet. 2009, 41, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Dhomen, N.; Marais, R. New Insight into BRAF Mutations in Cancer. Curr. Opin. Genet. Dev. 2007, 17, 31–39. [Google Scholar] [CrossRef]
- Williams, M.L.; Sagebiel, R.W. Melanoma Risk Factors and Atypical Moles. West. J. Med. 1994, 160, 343–350. [Google Scholar]
- Zhang, G.; Herlyn, M. Human Nevi: No Longer Precursors of Melanomas? J. Investig. Dermatol. 2012, 132, 2133–2134. [Google Scholar] [CrossRef]
- Tsao, H.; Bevona, C.; Goggins, W.; Quinn, T. The Transformation Rate of Moles (Melanocytic Nevi) into Cutaneous Melanoma: A Population-Based Estimate. Arch. Dermatol. 2003, 139, 282–288. [Google Scholar] [CrossRef]
- Bataille, V.; Kato, B.S.; Falchi, M.; Gardner, J.; Kimura, M.; Lens, M.; Perks, U.; Valdes, A.M.; Bennett, D.C.; Aviv, A.; et al. Nevus Size and Number are Associated with Telomere Length and Represent Potential Markers of a Decreased Senescence in Vivo. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1499–1502. [Google Scholar] [CrossRef]
- Tsatsakis, A.; Oikonomopoulou, T.; Nikolouzakis, T.K.; Vakonaki, E.; Tzatzarakis, M.; Flamourakis, M.; Renieri, E.; Fragkiadaki, P.; Iliaki, E.; Bachlitzanaki, M.; et al. Role of Telomere Length in Human Carcinogenesis (Review). Int. J. Oncol. 2023, 63, 78. [Google Scholar] [CrossRef]
- Ramirez, R.D.; D’Atri, S.; Pagani, E.; Faraggiana, T.; Lacal, P.M.; Taylor, R.S.; Shay, J.W. Progressive Increase in Telomerase Activity from Benign Melanocytic Conditions to Malignant Melanoma. Neoplasia 1999, 1, 42–49. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Glaessl, A.; Bosserhoff, A.K.; Buettner, R.; Hohenleutner, U.; Landthaler, M.; Stolz, W. Increase in Telomerase Activity during Progression of Melanocytic Cells from Melanocytic Naevi to Malignant Melanomas. Arch. Dermatol. Res. 1999, 291, 81–87. [Google Scholar] [CrossRef] [PubMed]
- De Vitis, M.; Berardinelli, F.; Sgura, A. Telomere Length Maintenance in Cancer: At the Crossroad between Telomerase and Alternative Lengthening of Telomeres (ALT). Int. J. Mol. Sci. 2018, 19, 606. [Google Scholar] [CrossRef]
- Son, N.; Cui, Y.; Xi, W. Association between Telomere Length and Skin Cancer and Aging: A Mendelian Randomization Analysis. Front. Genet. 2022, 13, 931785. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, W. Insights into Risk Factors for Basal Cell Carcinoma: A Mendelian Randomization Study. Clin. Exp. Dermatol. 2023, 48, 676–680. [Google Scholar] [CrossRef]
- Ismail, H.; Helby, J.; Holmich, L.R.; Chakera, A.H.; Bastholt, L.; Klyver, H.; Sjogren, P.; Schmidt, H.; Schollhammer, L.; Nordestgaard, B.G.; et al. Genetic Predisposition to Long Telomeres is Associated with Increased Mortality After Melanoma: A Study of 2101 Melanoma Patients from Hospital Clinics and the General Population. Pigment Cell Melanoma Res. 2021, 34, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Ashford, B.G.; Clark, J.; Gupta, R.; Iyer, N.G.; Yu, B.; Ranson, M. Reviewing the Genetic Alterations in High-Risk Cutaneous Squamous Cell Carcinoma: A Search for Prognostic Markers and Therapeutic Targets. Head Neck 2017, 39, 1462–1469. [Google Scholar] [CrossRef]
- Tinaburri, L.; Valente, C.; Teson, M.; Minafo, Y.A.; Cordisco, S.; Guerra, L.; Dellambra, E. The Secretome of Aged Fibroblasts Promotes EMT-Like Phenotype in Primary Keratinocytes from Elderly Donors through BDNF-TrkB Axis. J. Investig. Dermatol. 2021, 141, 1052–1062.e12. [Google Scholar] [CrossRef]
- Brash, D.E. Cancer. Preprocancer Sci. 2015, 348, 867–868. [Google Scholar]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor Evolution. High Burden and Pervasive Positive Selection of Somatic Mutations in Normal Human Skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef]
- Gosselin, K.; Deruy, E.; Martien, S.; Vercamer, C.; Bouali, F.; Dujardin, T.; Slomianny, C.; Houel-Renault, L.; Chelli, F.; De Launoit, Y.; et al. Senescent Keratinocytes Die by Autophagic Programmed Cell Death. Am. J. Pathol. 2009, 174, 423–435. [Google Scholar] [CrossRef]
- Kakiuchi, N.; Ogawa, S. Clonal Expansion in Non-Cancer Tissues. Nat. Rev. Cancer 2021, 21, 239–256. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.J.; Abascal, F.; Hall, M.W.J.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M.R.; et al. Somatic Mutant Clones Colonize the Human Esophagus with Age. Science 2018, 362, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Dotto, G.P. Multifocal Epithelial Tumors and Field Cancerization: Stroma as a Primary Determinant. J. Clin. Investig. 2014, 124, 1446–1453. [Google Scholar] [CrossRef] [PubMed]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking Senescence: Context-Dependent Effects of SASP in Cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Abbadie, C.; Pluquet, O.; Pourtier, A. Epithelial Cell Senescence: An Adaptive Response to Pre-Carcinogenic Stresses? Cell Mol. Life Sci. 2017, 74, 4471–4509. [Google Scholar] [CrossRef]
- Posch, C. Ageing Research: Rethinking Primary Prevention of Skin Cancer. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 2216–2218. [Google Scholar] [CrossRef]
- Yokoyama, N.N.; Denmon, A.; Uchio, E.M.; Jordan, M.; Mercola, D.; Zi, X. When Anti-Aging Studies Meet Cancer Chemoprevention: Can Anti-Aging Agent Kill Two Birds with One Blow? Curr. Pharmacol. Rep. 2015, 1, 420–433. [Google Scholar] [CrossRef]
- Law, M.H.; Medland, S.E.; Zhu, G.; Yazar, S.; Viñuela, A.; Wallace, L.; Shekar, S.N.; Duffy, D.L.; Bataille, V.; Glass, D.; et al. Genome-Wide Association shows that Pigmentation Genes Play a Role in Skin Aging. J. Investig. Dermatol. 2017, 137, 1887–1894. [Google Scholar] [CrossRef]
- Guida, S.; Ciardo, S.; De Pace, B.; De Carvalho, N.; Farnetani, F.; Pezzini, C.; Chester, J.; Shaniko, K.; Manganelli, M.; Guida, G.; et al. Atrophic and Hypertrophic Skin Photoaging and Melanocortin-1 Receptor (MC1R): The Missing Link. J. Am. Acad. Dermatol. 2021, 84, 187–190. [Google Scholar] [CrossRef]
- Latreille, J.; Ezzedine, K.; Elfakir, A.; Ambroisine, L.; Jdid, R.; Galan, P.; Hercberg, S.; Gruber, F.; Malvy, D.; Tschachler, E.; et al. MC1R Polymorphisms and Facial Photoaging. Ann. Dermatol. Venereol. 2011, 138, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Elfakir, A.; Ezzedine, K.; Latreille, J.; Ambroisine, L.; Jdid, R.; Galan, P.; Hercberg, S.; Gruber, F.; Malvy, D.; Tschachler, E.; et al. Functional MC1R-Gene Variants are Associated with Increased Risk for Severe Photoaging of Facial Skin. J. Investig. Dermatol. 2010, 130, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Böhm, M.; Luger, T.A. Melanocortins in Fibroblast Biology—Current Update and Future Perspective for Dermatology. Exp. Dermatol. 2004, 13 (Suppl. S4), 16–21. [Google Scholar] [CrossRef] [PubMed]
- Böhm, M.; Schiller, M.; Luger, T.A. Non-Pigmentary Actions of Alpha-Melanocyte-Stimulating Hormone—Lessons from the Cutaneous Melanocortin System. Cell. Mol. Biol. 2006, 52, 61–68. [Google Scholar]
- Naval, J.; Alonso, V.; Herranz, M.A. Genetic Polymorphisms and Skin Aging: The Identification of Population Genotypic Groups Holds Potential for Personalized Treatments. Clin. Cosmet. Investig. Dermatol. 2014, 7, 207–214. [Google Scholar] [CrossRef]
- Martic, I.; Jansen-Durr, P.; Cavinato, M. Effects of Air Pollution on Cellular Senescence and Skin Aging. Cells 2022, 11, 2220. [Google Scholar] [CrossRef]
- Wigmann, C.; Huls, A.; Krutmann, J.; Schikowski, T. Estimating the Relative Contribution of Environmental and Genetic Risk Factors to Different Aging Traits by Combining Correlated Variables into Weighted Risk Scores. Int. J. Environ. Res. Public Health 2022, 19, 16746. [Google Scholar] [CrossRef]
- Geng, R.; Kang, S.; Huang, K.; Tong, T. Boosting the Photoaged Skin: The Potential Role of Dietary Components. Nutrients 2021, 13, 1691. [Google Scholar] [CrossRef]
- Welti, M.; Ramelyte, E.; Dummer, R.; Imhof, L. Evaluation of the Minimal Erythema Dose for UVB and UVA in Context of Skin Phototype and Nature of Photodermatosis. Photodermatol. Photoimmunol. Photomed. 2020, 36, 200–207. [Google Scholar] [CrossRef]
- Del Bino, S.; Duval, C.; Bernerd, F. Clinical and Biological Characterization of Skin Pigmentation Diversity and its Consequences on UV Impact. Int. J. Mol. Sci. 2018, 19, 2668. [Google Scholar] [CrossRef]
- Moutraji, R.; Taylor, S.C. Skin Aging Exposome in Skin of Color Populations: Review of the Literature. Dermatol. Surg. 2023, 49, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Gilchrest, B.A. Photoaging. J. Investig. Dermatol. 2013, 133, 2. [Google Scholar] [CrossRef] [PubMed]
- Friedman, O. Changes Associated with the Aging Face. Facial Plast. Surg. Clin. N. Am. 2005, 13, 371–380. [Google Scholar] [CrossRef]
- Zouboulis, C.C.; Makrantonaki, E. Clinical Aspects and Molecular Diagnostics of Skin Aging. Clin. Dermatol. 2011, 29, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Varga, R.; Gross, J. Oxidative Stress Status and its Relationship to Skin Aging. Plast. Aesthet. Nurs. 2023, 43, 141–148. [Google Scholar] [CrossRef]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide Metabolism in Mammalian Organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef]
- Leiter, U.; Eigentler, T.; Garbe, C. Epidemiology of Skin Cancer. Adv. Exp. Med. Biol. 2014, 810, 120–140. [Google Scholar]
- Cirri, P.; Chiarugi, P. Cancer Associated Fibroblasts: The Dark Side of the Coin. Am. J. Cancer Res. 2011, 1, 482–497. [Google Scholar]
- Waldera-Lupa, D.M.; Kalfalah, F.; Florea, A.; Sass, S.; Kruse, F.; Rieder, V.; Tigges, J.; Fritsche, E.; Krutmann, J.; Busch, H.; et al. Proteome-Wide Analysis Reveals an Age-Associated Cellular Phenotype of in Situ Aged Human Fibroblasts. Aging 2014, 6, 856–878. [Google Scholar] [CrossRef]
- Knops, A.M.; South, A.; Rodeck, U.; Martinez-Outschoorn, U.; Harshyne, L.A.; Johnson, J.; Luginbuhl, A.J.; Curry, J.M. Cancer-Associated Fibroblast Density, Prognostic Characteristics, and Recurrence in Head and Neck Squamous Cell Carcinoma: A Meta-Analysis. Front. Oncol. 2020, 10, 565306. [Google Scholar] [CrossRef]
- Quan, T.; Fisher, G.J. Role of Age-Associated Alterations of the Dermal Extracellular Matrix Microenvironment in Human Skin Aging: A Mini-Review. Gerontology 2015, 61, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Waaijer, M.E.; Parish, W.E.; Strongitharm, B.H.; van Heemst, D.; Slagboom, P.E.; de Craen, A.J.; Sedivy, J.M.; Westendorp, R.G.; Gunn, D.A.; Maier, A.B. The Number of p16INK4a Positive Cells in Human Skin Reflects Biological Age. Aging Cell 2012, 11, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between Cancer-Associated Fibroblasts and Immune Cells in the Tumor Microenvironment: New Findings and Future Perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef]
- Arandkar, S.; Furth, N.; Elisha, Y.; Nataraj, N.B.; van der Kuip, H.; Yarden, Y.; Aulitzky, W.; Ulitsky, I.; Geiger, B.; Oren, M. Altered p53 Functionality in Cancer-Associated Fibroblasts Contributes to their Cancer-Supporting Features. Proc. Natl. Acad. Sci. USA 2018, 115, 6410–6415. [Google Scholar] [CrossRef] [PubMed]
- Procopio, M.; Laszlo, C.; Dotto, G.P. CSL-p53: From Senescence to CAF Activation. Cell Cycle 2016, 15, 485–486. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Procopio, M.; Laszlo, C.; Al Labban, D.; Kim, D.E.; Bordignon, P.; Jo, S.; Goruppi, S.; Menietti, E.; Ostano, P.; Ala, U.; et al. Combined CSL and p53 Downregulation Promotes Cancer-Associated Fibroblast Activation. Nat. Cell Biol. 2015, 17, 1193–1204. [Google Scholar] [CrossRef]
- Schmid, J.O.; Dong, M.; Haubeiss, S.; Friedel, G.; Bode, S.; Grabner, A.; Ott, G.; Mürdter, T.E.; Oren, M.; Aulitzky, W.E.; et al. Cancer Cells Cue the p53 Response of Cancer-Associated Fibroblasts to Cisplatin. Cancer Res. 2012, 72, 5824–5832. [Google Scholar] [CrossRef]
- Krylova, S.V.; Feng, D. The Machinery of Exosomes: Biogenesis, Release, and Uptake. Int. J. Mol. Sci. 2023, 24, 1337. [Google Scholar] [CrossRef]
- Ghosh, K.; Capell, B.C. The Senescence-Associated Secretory Phenotype: Critical Effector in Skin Cancer and Aging. J. Investig. Dermatol. 2016, 136, 2133–2139. [Google Scholar] [CrossRef]
- Laberge, R.; Awad, P.; Campisi, J.; Desprez, P. Epithelial-Mesenchymal Transition Induced by Senescent Fibroblasts. Cancer Microenviron. 2012, 5, 39–44. [Google Scholar] [CrossRef]
- Deruy, E.; Nassour, J.; Martin, N.; Vercamer, C.; Malaquin, N.; Bertout, J.; Chelli, F.; Pourtier, A.; Pluquet, O.; Abbadie, C. Level of Macroautophagy Drives Senescent Keratinocytes into Cell Death Or Neoplastic Evasion. Cell Death Dis. 2014, 5, e1577. [Google Scholar] [CrossRef] [PubMed]
- Victorelli, S.; Lagnado, A.; Halim, J.; Moore, W.; Talbot, D.; Barrett, K.; Chapman, J.; Birch, J.; Ogrodnik, M.; Meves, A.; et al. Senescent Human Melanocytes Drive Skin Ageing Via Paracrine Telomere Dysfunction. EMBO J. 2019, 38, e101982. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Hetz, C. Mastering Organismal Aging through the Endoplasmic Reticulum Proteostasis Network. Aging Cell 2020, 19, e13265. [Google Scholar] [CrossRef]
- Pieraggi, M.T.; Bouissou, H.; Angelier, C.; Uhart, D.; Magnol, J.P.; Kokolo, J. The Fibroblast. Ann. Pathol. 1985, 5, 65–76. [Google Scholar] [PubMed]
- Fenske, N.A.; Lober, C.W. Structural and Functional Changes of Normal Aging Skin. J. Am. Acad. Dermatol. 1986, 15, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Pochi, P.E.; Strauss, J.S.; Downing, D.T. Age-Related Changes in Sebaceous Gland Activity. J. Investig. Dermatol. 1979, 73, 108–111. [Google Scholar] [CrossRef]
- Makrantonaki, E.; Brink, T.C.; Zampeli, V.; Elewa, R.M.; Mlody, B.; Hossini, A.M.; Hermes, B.; Krause, U.; Knolle, J.; Abdallah, M.; et al. Identification of Biomarkers of Human Skin Ageing in both Genders. Wnt Signalling—A Label of Skin Ageing? PLoS ONE 2012, 7, e50393. [Google Scholar] [CrossRef]
- Zouboulis, C.C.; Picardo, M.; Ju, Q.; Kurokawa, I.; Törőcsik, D.; Bíró, T.; Schneider, M.R. Beyond Acne: Current Aspects of Sebaceous Gland Biology and Function. Rev. Endocr. Metab. Disord. 2016, 17, 319–334. [Google Scholar] [CrossRef]
- Leyden, J. What is Photoaged Skin? Eur. J. Dermatol. 2001, 11, 165–167. [Google Scholar]
- Hou, X.; Wei, Z.; Zouboulis, C.C.; Ju, Q. Aging in the Sebaceous Gland. Front. Cell Dev. Biol. 2022, 10, 909694. [Google Scholar] [CrossRef]
- Lee, W.J.; Park, K.H.; Sohn, M.Y.; Lee, W.C.; Lee, S.; Kim, D.W. Ultraviolet B Irradiation Increases the Expression of Inflammatory Cytokines in Cultured Sebocytes. J. Dermatol. 2013, 40, 993–997. [Google Scholar] [CrossRef]
- Bocheva, G.; Slominski, R.M.; Slominski, A.T. Neuroendocrine Aspects of Skin Aging. Int. J. Mol. Sci. 2019, 20, 2798. [Google Scholar] [CrossRef]
- Lupu, M.; Caruntu, A.; Caruntu, C.; Papagheorghe, L.M.L.; Ilie, M.A.; Voiculescu, V.; Boda, D.; Constantin, C.; Tanase, C.; Sifaki, M.; et al. Neuroendocrine Factors: The Missing Link in Non-melanoma Skin Cancer (Review). Oncol. Rep. 2017, 38, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Seiffert, K.; Granstein, R.D. Neuropeptides and Neuroendocrine Hormones in Ultraviolet Radiation-Induced Immunosuppression. Methods 2002, 28, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, J.V.; Blum, A.; Walder, J.; Walder, R. Eosinophils from Granulomas in Murine Schistosomiasis Mansoni Produce Substance P. J. Immunol. 1988, 141, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chawla, M.K.; Rios-Monterrosa, J.L.; Wang, L.; Zempare, M.A.; Hruby, V.J.; Barnes, C.A.; Cai, M. Aged Brains Express Less Melanocortin Receptors, which Correlates with Age-Related Decline of Cognitive Functions. Molecules 2021, 26, 6266. [Google Scholar] [CrossRef] [PubMed]
- Waldera Lupa, D.M.; Kalfalah, F.; Safferling, K.; Boukamp, P.; Poschmann, G.; Volpi, E.; Götz-Rösch, C.; Bernerd, F.; Haag, L.; Huebenthal, U.; et al. Characterization of Skin Aging-Associated Secreted Proteins (SAASP) Produced by Dermal Fibroblasts Isolated from Intrinsically Aged Human Skin. J. Investig. Dermatol. 2015, 135, 1954–1968. [Google Scholar] [CrossRef]
- Rho, O.; Kim, D.J.; Kiguchi, K.; Digiovanni, J. Growth Factor Signaling Pathways as Targets for Prevention of Epithelial Carcinogenesis. Mol. Carcinog. 2011, 50, 264–279. [Google Scholar] [CrossRef]
- Kasprzak, A.; Kwasniewski, W.; Adamek, A.; Gozdzicka-Jozefiak, A. Insulin-like Growth Factor (IGF) Axis in Cancerogenesis. Mutat. Res. Rev. Mutat. Res. 2017, 772, 78–104. [Google Scholar] [CrossRef]
- Cardoso, A.L.; Fernandes, A.; Aguilar-Pimentel, J.A.; de Angelis, M.H.; Guedes, J.R.; Brito, M.A.; Ortolano, S.; Pani, G.; Athanasopoulou, S.; Gonos, E.S.; et al. Towards Frailty Biomarkers: Candidates from Genes and Pathways Regulated in Aging and Age-Related Diseases. Ageing Res. Rev. 2018, 47, 214–277. [Google Scholar] [CrossRef]
- Sepúlveda, M.; Arauna, D.; García, F.; Albala, C.; Palomo, I.; Fuentes, E. Frailty in Aging and the Search for the Optimal Biomarker: A Review. Biomedicines 2022, 10, 1426. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Huang, Y.; Lyu, Y.; Dai, W.; Tong, Y.; Li, Y. GDF15 as a Biomarker of Ageing. Exp. Gerontol. 2021, 146, 111228. [Google Scholar] [CrossRef] [PubMed]
- Joo, M.; Kim, D.; Lee, M.; Lee, H.J.; Kim, J. GDF15 Promotes Cell Growth, Migration, and Invasion in Gastric Cancer by Inducing STAT3 Activation. Int. J. Mol. Sci. 2023, 24, 2925. [Google Scholar] [CrossRef] [PubMed]
- Wedel, S.; Martic, I.; Guerrero Navarro, L.; Ploner, C.; Pierer, G.; Jansen-Durr, P.; Cavinato, M. Depletion of Growth Differentiation Factor 15 (GDF15) Leads to Mitochondrial Dysfunction and Premature Senescence in Human Dermal Fibroblasts. Aging Cell 2023, 22, e13752. [Google Scholar] [CrossRef]
- Kim, Y.; Kang, B.; Kim, J.C.; Park, T.J.; Kang, H.Y. Senescent Fibroblast-Derived GDF15 Induces Skin Pigmentation. J. Investig. Dermatol. 2020, 140, 2478–2486.e4. [Google Scholar] [CrossRef]
- Yang, G.; Zhang, G.; Pittelkow, M.R.; Ramoni, M.; Tsao, H. Expression Profiling of UVB Response in Melanocytes Identifies a Set of p53-Target Genes. J. Investig. Dermatol. 2006, 126, 2490–2506. [Google Scholar] [CrossRef]
- Zhang, J.; He, L.; Wang, Z.; Shao, S.; Qiao, P.; Zhang, J.; Zhang, K.; Li, C.; Zhang, Y.; Wang, G.; et al. Decreasing GDF15 Promotes Inflammatory Signals and Neutrophil Infiltration in Psoriasis Models. J. Investig. Dermatol. 2023, 143, 419–430.e8. [Google Scholar] [CrossRef]
- Boyle, G.M.; Pedley, J.; Martyn, A.C.; Banducci, K.J.; Strutton, G.M.; Brown, D.A.; Breit, S.N.; Parsons, P.G. Macrophage Inhibitory Cytokine-1 is Overexpressed in Malignant Melanoma and is Associated with Tumorigenicity. J. Investig. Dermatol. 2009, 129, 383–391. [Google Scholar] [CrossRef]
- Weide, B.; Schafer, T.; Martens, A.; Kuzkina, A.; Uder, L.; Noor, S.; Garbe, C.; Harter, P.N.; Mittelbronn, M.; Wischhusen, J. High GDF-15 Serum Levels Independently Correlate with Poorer overall Survival of Patients with Tumor-Free Stage III and Unresectable Stage IV Melanoma. J. Investig. Dermatol. 2016, 136, 2444–2452. [Google Scholar] [CrossRef]
- Lewis, D.A.; Travers, J.B.; Somani, A.K.; Spandau, D.F. The IGF-1/IGF-1R Signaling Axis in the Skin: A New Role for the Dermis in Aging-Associated Skin Cancer. Oncogene 2010, 29, 1475–1485. [Google Scholar] [CrossRef]
- Mainzer, C.; Remoué, N.; Molinari, J.; Rousselle, P.; Barricchello, C.; Lago, J.C.; Sommer, P.; Sigaudo-Roussel, D.; Debret, R. In Vitro Epidermis Model Mimicking IGF-1-Specific Age-Related Decline. Exp. Dermatol. 2018, 27, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Rivas, S.; Marin, A.; Samtani, S.; Gonzalez-Feliu, E.; Armisen, R. MET Signaling Pathways, Resistance Mechanisms, and Opportunities for Target Therapies. Int. J. Mol. Sci. 2022, 23, 13898. [Google Scholar] [CrossRef] [PubMed]
- LeRoith, D.; Holly, J.M.P.; Forbes, B.E. Insulin-like Growth Factors: Ligands, Binding Proteins, and Receptors. Mol. Metab. 2021, 52, 101245. [Google Scholar] [CrossRef]
- Bach, L.A. IGF-Binding Proteins. J. Mol. Endocrinol. 2018, 61, T11–T28. [Google Scholar] [CrossRef] [PubMed]
- Wajapeyee, N.; Kapoor, V.; Mahalingam, M.; Green, M.R. Efficacy of IGFBP7 for Treatment of Metastatic Melanoma and Other Cancers in Mouse Models and Human Cell Lines. Mol. Cancer Ther. 2009, 8, 3009–3014. [Google Scholar] [CrossRef] [PubMed]
- Fane, M.; Weeraratna, A.T. Normal Aging and its Role in Cancer Metastasis. Cold Spring Harb Perspect. Med. 2019, 10, a037341. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, E. Turning Foes to Friends: Targeting Cancer-Associated Fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- Bellei, B.; Pitisci, A.; Ottaviani, M.; Ludovici, M.; Cota, C.; Luzi, F.; Dell’Anna, M.L.; Picardo, M. Vitiligo: A Possible Model of Degenerative Diseases. PLoS ONE 2013, 8, e59782. [Google Scholar] [CrossRef]
- Kovacs, D.; Cardinali, G.; Aspite, N.; Cota, C.; Luzi, F.; Bellei, B.; Briganti, S.; Amantea, A.; Torrisi, M.R.; Picardo, M. Role of Fibroblast-Derived Growth Factors in Regulating Hyperpigmentation of Solar Lentigo. Br. J. Dermatol. 2010, 163, 1020–1027. [Google Scholar] [CrossRef]
- Kemp, M.G.; Spandau, D.F.; Travers, J.B. Impact of Age and Insulin-Like Growth Factor-1 on DNA Damage Responses in UV-Irradiated Human Skin. Molecules. 2017, 22, 356. [Google Scholar] [CrossRef]
- Tyrrell, R.M. The Molecular and Cellular Pathology of Solar Ultraviolet Radiation. Mol. Asp. Med. 1994, 15, 1–77. [Google Scholar]
- Lewis, D.A.; Spandau, D.F. UVB-Induced Activation of NF-kappaB is Regulated by the IGF-1R and Dependent on p38 MAPK. J. Investig. Dermatol. 2008, 128, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Frommeyer, T.C.; Rohan, C.A.; Spandau, D.F.; Kemp, M.G.; Wanner, M.A.; Tanzi, E.; Travers, J.B. Wounding Therapies for Prevention of Photocarcinogenesis. Front. Oncol. 2022, 11, 813132. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.; Lewis, D.A.; Spandau, D.F. Decreased Incidence of Nonmelanoma Skin Cancer in Patients with Type 2 Diabetes Mellitus using Insulin: A Pilot Study. Br. J. Dermatol. 2005, 153, 552–557. [Google Scholar] [CrossRef] [PubMed]
- de Araújo, R.; Lôbo, M.; Trindade, K.; Silva, D.F.; Pereira, N. Fibroblast Growth Factors: A Controlling Mechanism of Skin Aging. Skin Pharmacol. Physiol. 2019, 32, 275–282. [Google Scholar] [CrossRef]
- Lee, H.; Hong, Y.; Kim, M. Structural and Functional Changes and Possible Molecular Mechanisms in Aged Skin. Int. J. Mol. Sci. 2021, 22, 12489. [Google Scholar] [CrossRef]
- Prudovsky, I. Cellular Mechanisms of FGF-Stimulated Tissue Repair. Cells 2021, 10, 1830. [Google Scholar] [CrossRef]
- Kinn, P.M.; Holdren, G.O.; Westermeyer, B.A.; Abuissa, M.; Fischer, C.L.; Fairley, J.A.; Brogden, K.A.; Brogden, N.K. Age-Dependent Variation in Cytokines, Chemokines, and Biologic Analytes Rinsed from the Surface of Healthy Human Skin. Sci. Rep. 2015, 5, 10472. [Google Scholar] [CrossRef]
- Okazaki, M.; Yoshimura, K.; Uchida, G.; Harii, K. Correlation between Age and the Secretions of Melanocyte-Stimulating Cytokines in Cultured Keratinocytes and Fibroblasts. Br. J. Dermatol. 2005, 153 (Suppl. S2), 23–29. [Google Scholar] [CrossRef]
- Papaccio, F.; Kovacs, D.; Bellei, B.; Caputo, S.; Migliano, E.; Cota, C.; Picardo, M. Profiling Cancer-Associated Fibroblasts in Melanoma. Int. J. Mol. Sci. 2021, 22, 7255. [Google Scholar] [CrossRef]
- Tsang, M.; Quesnel, K.; Vincent, K.; Hutchenreuther, J.; Postovit, L.M.; Leask, A. Insight into Fibroblast Plasticity: Celular Communication Network 2 Is Required for Activation of Cancer-Associated Fibroblasts in a Murine Model of Melanoma. Am. J. Pathol. 2020, 1, 206–221. [Google Scholar] [CrossRef]
- Pradip, D.; Jennifer, A.; Nandini, D. Cancer-Associated Fibroblasts in Conversation with Tumor Cells in Endometrial Cancers: A Partner in Crime. Int. J. Mol. Sci. 2021, 22, 9121. [Google Scholar] [CrossRef] [PubMed]
- Brenner, M.; Degitz, K.; Besch, R.; Berking, C. Differential Expression of Melanoma-Associated Growth Factors in Keratinocytes and Fibroblasts by Ultraviolet A and Ultraviolet B Radiation. Br. J. Dermatol. 2005, 153, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Briganti, S.; Wlaschek, M.; Hinrichs, C.; Bellei, B.; Flori, E.; Treiber, N.; Iben, S.; Picardo, M.; Scharffetter-Kochanek, K. Small Molecular Antioxidants Effectively Protect from PUVA-Induced Oxidative Stress Responses Underlying Fibroblast Senescence and Photoaging. Free Radic. Biol. Med. 2008, 45, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Kim, J.; Kim, E.K. Repeated Exposure of Human Fibroblasts to UVR Induces Secretion of Stem Cell Factor and Senescence. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 1577–1580. [Google Scholar] [CrossRef]
- Unver, N.; Freyschmidt-Paul, P.; Horster, S.; Wenck, H.; Stab, F.; Blatt, T.; Elsasser, H. Alterations in the Epidermal-Dermal Melanin Axis and Factor XIIIa Melanophages in Senile Lentigo and Ageing Skin. Br. J. Dermatol. 2006, 155, 119–128. [Google Scholar] [CrossRef]
- Salducci, M.; André, N.; Guéré, C.; Martin, M.; Fitoussi, R.; Vié, K.; Cario-André, M. Factors Secreted by Irradiated Aged Fibroblasts Induce Solar Lentigo in Pigmented Reconstructed Epidermis. Pigment Cell Melanoma Res. 2014, 27, 502–504. [Google Scholar] [CrossRef]
- Goorochurn, R.; Viennet, C.; Tissot, M.; Locatelli, F.; Granger, C.; Varin-Blank, N.; Humbert, P.; Le Roy, C. Differential Morphological and Functional Features of Fibroblasts Explanted from Solar Lentigo. Br. J. Dermatol. 2017, 177, e109–e111. [Google Scholar] [CrossRef]
- Czyz, M. HGF/C-MET Signaling in Melanocytes and Melanoma. Int. J. Mol. Sci. 2018, 19, 3844. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, D.; Lee, S.; Kim, D.; Nam, H.; Cho, M.K. Expression of the C-Met Proteins in Malignant Skin Cancers. Ann. Dermatol. 2011, 23, 33–38. [Google Scholar] [CrossRef]
- Welsh, S.J.; Rizos, H.; Scolyer, R.A.; Long, G.V. Resistance to Combination BRAF and MEK Inhibition in Metastatic Melanoma: Where to Next? Eur. J. Cancer 2016, 62, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Montone, K.T.; van Belle, P.; Elenitsas, R.; Elder, D.E. Proto-Oncogene C-Kit Expression in Malignant Melanoma: Protein Loss with Tumor Progression. Mod. Pathol. 1997, 10, 939–944. [Google Scholar] [PubMed]
- Prignano, F.; Gerlini, G.; Salvatori, B.; Orlando, C.; Mazzoli, S.; Pimpinelli, N.; Moretti, S. Stem Cell Factor Affects Tumour Progression Markers in Metastatic Melanoma Cells. Clin. Exp. Metastasis 2006, 23, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Räsänen, K.; Vaheri, A. Activation of Fibroblasts in Cancer Stroma. Exp. Cell Res. 2010, 316, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Ben Baruch, B.; Mantsur, E.; Franco-Barraza, J.; Blacher, E.; Cukierman, E.; Stein, R. CD38 in Cancer-Associated Fibroblasts Promotes Pro-Tumoral Activity. Lab. Investig. 2020, 100, 1517–1531. [Google Scholar] [CrossRef]
- Ke, Y.; Wang, X. TGFβ Signaling in Photoaging and UV-Induced Skin Cancer. J. Investig. Dermatol. 2021, 141, 1104–1110. [Google Scholar] [CrossRef]
- Hasegawa, T.; Yashiro, M.; Nishii, T.; Matsuoka, J.; Fuyuhiro, Y.; Morisaki, T.; Fukuoka, T.; Shimizu, K.; Shimizu, T.; Miwa, A.; et al. Cancer-Associated Fibroblasts might Sustain the Stemness of Scirrhous Gastric Cancer Cells Via Transforming Growth Factor-Β Signaling. Int. J. Cancer 2014, 134, 1785–1795. [Google Scholar] [CrossRef]
- Elston, R.; Inman, G.J. Crosstalk between p53 and TGF-Beta Signalling. J. Signal. Transduct. 2012, 2012, 294097. [Google Scholar] [CrossRef]
- Hasegawa, T.; Sakamoto, A.; Wada, A.; Fukai, T.; Iida, H.; Ikeda, S. Keratinocyte Progenitor Cells Reside in Human Subcutaneous Adipose Tissue. PLoS ONE 2015, 10, e0118402. [Google Scholar] [CrossRef]
- Berking, C.; Takemoto, R.; Schaider, H.; Showe, L.; Satyamoorthy, K.; Robbins, P.; Herlyn, M. Transforming Growth Factor-Beta1 Increases Survival of Human Melanoma through Stroma Remodeling. Cancer Res. 2001, 61, 8306–8316. [Google Scholar]
- Nestle, F.O.; Di Meglio, P.; Qin, J.; Nickoloff, B.J. Skin Immune Sentinels in Health and Disease. Nat. Rev. Immunol. 2009, 9, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.V.; Soulika, A.M. The Dynamics of the Skin’s Immune System. Int. J. Mol. Sci. 2019, 20, 1811. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, F.; Mijouin, L.; Pichon, C. Skin Immune Landscape: Inside and Outside the Organism. Mediat. Inflamm. 2017, 2017, 5095293. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.S.; Vukmanovic-Stejic, M. Skin Barrier Immunity and Ageing. Immunology 2020, 160, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Photoaging: UV Radiation-Induced Inflammation and Immunosuppression Accelerate the Aging Process in the Skin. Inflamm. Res. 2022, 71, 817–831. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Vitale, G.; Capri, M.; Salvioli, S. Inflammaging and ‘Garb-Aging’. Trends Endocrinol. Metab. 2017, 28, 199–212. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.; Lasitschka, F.; Andrulis, M.; et al. A Complex Secretory Program Orchestrated by the Inflammasome Controls Paracrine Senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Giunta, S.; Wei, Y.; Xu, K.; Xia, S. Cold-Inflammaging: When a State of Homeostatic-Imbalance Associated with Aging Precedes the Low-Grade Pro-Inflammatory-State (Inflammaging): Meaning, Evolution, Inflammaging Phenotypes. Clin. Exp. Pharmacol. Physiol. 2022, 49, 925–934. [Google Scholar] [CrossRef]
- Pajak, J.; Nowicka, D.; Szepietowski, J.C. Inflammaging and Immunosenescence as Part of Skin Aging—A Narrative Review. Int. J. Mol. Sci. 2023, 24, 7784. [Google Scholar] [CrossRef]
- Vukmanovic-Stejic, M.; Rustin, M.H.A.; Nikolich-Zugich, J.; Akbar, A.N. Immune Responses in the Skin in Old Age. Curr. Opin. Immunol. 2011, 23, 525–531. [Google Scholar] [CrossRef]
- Jarrold, B.B.; Tan, C.Y.R.; Ho, C.Y.; Soon, A.L.; Lam, T.T.; Yang, X.; Nguyen, C.; Guo, W.; Chew, Y.C.; DeAngelis, Y.M.; et al. Early Onset of Senescence and Imbalanced Epidermal Homeostasis Across the Decades in Photoexposed Human Skin: Fingerprints of Inflammaging. Exp. Dermatol. 2022, 31, 1748–1760. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Xiang, X.; Wang, L.; Zhu, B.; Huang, S.; Tang, X.; Chen, J.; Qiu, L. Percutaneous Contrast-Enhanced Ultrasound for Localization and Qualitative Diagnosis of Sentinel Lymph Nodes in Cutaneous Malignant Melanoma of Lower Extremities: A Preliminary Study. Quant. Imaging Med. Surg. 2022, 12, 366–375. [Google Scholar] [CrossRef]
- Pilkington, S.M.; Bulfone-Paus, S.; Griffiths, C.E.M.; Watson, R.E.B. Inflammaging and the Skin. J. Investig. Dermatol. 2021, 141, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Ruhland, M.K.; Loza, A.J.; Capietto, A.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal Senescence Establishes an Immunosuppressive Microenvironment that Drives Tumorigenesis. Nat. Commun. 2016, 7, 11762. [Google Scholar] [CrossRef]
- Agius, E.; Lacy, K.E.; Vukmanovic-Stejic, M.; Jagger, A.L.; Papageorgiou, A.; Hall, S.; Reed, J.R.; Curnow, S.J.; Fuentes-Duculan, J.; Buckley, C.D.; et al. Decreased TNF-Alpha Synthesis by Macrophages Restricts Cutaneous Immunosurveillance by Memory CD4+ T Cells during Aging. J. Exp. Med. 2009, 206, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.P.; Newson, J.; Kwong, S.; Richard-Loendt, A.; Colas, R.; Dalli, J.; Gilroy, D.W. Prolonged Immune Alteration Following Resolution of Acute Inflammation in Humans. PLoS ONE 2017, 12, e0186964. [Google Scholar] [CrossRef] [PubMed]
- Newson, J.; Motwani, M.P.; Kendall, A.C.; Nicolaou, A.; Muccioli, G.G.; Alhouayek, M.; Bennett, M.; Van De Merwe, R.; James, S.; De Maeyer, R.P.H.; et al. Inflammatory Resolution Triggers a Prolonged Phase of Immune Suppression through COX-1/mPGES-1-Derived Prostaglandin E(2). Cell. Rep. 2017, 20, 3162–3175. [Google Scholar] [CrossRef]
- Enk, C.D.; Sredni, D.; Blauvelt, A.; Katz, S.I. Induction of IL-10 Gene Expression in Human Keratinocytes by UVB Exposure in Vivo and in Vitro. J. Immunol. 1995, 154, 4851–4856. [Google Scholar] [CrossRef]
- Kang, K.; Hammerberg, C.; Meunier, L.; Cooper, K.D. CD11b+ Macrophages that Infiltrate Human Epidermis After in Vivo Ultraviolet Exposure Potently Produce IL-10 and Represent the Major Secretory Source of Epidermal IL-10 Protein. J. Immunol. 1994, 153, 5256–5264. [Google Scholar] [CrossRef]
- Piskin, G.; Bos, J.D.; Teunissen, M.B.M. Neutrophils Infiltrating Ultraviolet B-Irradiated Normal Human Skin Display High IL-10 Expression. Arch. Dermatol. Res. 2005, 296, 339–342. [Google Scholar] [CrossRef]
- Bos, P.D. T(REG) Cells in Cancer: Beyond Classical Immunological Control. Immunol. Investig. 2016, 45, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Kaporis, H.G.; Guttman-Yassky, E.; Lowes, M.A.; Haider, A.S.; Fuentes-Duculan, J.; Darabi, K.; Whynot-Ertelt, J.; Khatcherian, A.; Cardinale, I.; Novitskaya, I.; et al. Human Basal Cell Carcinoma is Associated with Foxp3+ T Cells in a Th2 Dominant Microenvironment. J. Investig. Dermatol. 2007, 127, 2391–2398. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Felipe-Silva, A.; Heemskerk, B.; Powell, D.J.J.; Wunderlich, J.R.; Merino, M.J.; Rosenberg, S.A. FOXP3 Expression Accurately Defines the Population of Intratumoral Regulatory T Cells that Selectively Accumulate in Metastatic Melanoma Lesions. Blood 2008, 112, 4953–4960. [Google Scholar] [CrossRef] [PubMed]
- Salmi, S.; Lin, A.; Hirschovits-Gerz, B.; Valkonen, M.; Aaltonen, N.; Sironen, R.; Siiskonen, H.; Pasonen-Seppanen, S. The Role of FoxP3+ Regulatory T Cells and IDO+ Immune and Tumor Cells in Malignant Melanoma—An Immunohistochemical Study. BMC Cancer 2021, 21, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Ferronika, P.; Dhiyani, S.A.; Budiarti, T.; Widodo, I.; Rinonce, H.T.; Anwar, S.L. Regulatory T Cells but Not Tumour-Infiltrating Lymphocytes Correlate with Tumour Invasion Depth in Basal Cell Carcinoma. Diagnostics 2022, 12, 2987. [Google Scholar] [CrossRef]
- Dunne, P.J.; Fletcher, J.M. Recent Advances in Regulatory T Cell Therapy of Autoimmunity, Graft Rejection and Cancer. Recent Pat. Inflamm. Allergy Drug Discov. 2010, 4, 231–243. [Google Scholar] [CrossRef]
- Li, X.; Kostareli, E.; Suffner, J.; Garbi, N.; Hammerling, G.J. Efficient Treg Depletion Induces T-Cell Infiltration and Rejection of Large Tumors. Eur. J. Immunol. 2010, 40, 3325–3335. [Google Scholar] [CrossRef]
- Zhang, B.; Pan, C.; Feng, C.; Yan, C.; Yu, Y.; Chen, Z.; Guo, C.; Wang, X. Role of Mitochondrial Reactive Oxygen Species in Homeostasis Regulation. Redox Rep. 2022, 27, 45–52. [Google Scholar] [CrossRef]
- Conley, K.E.; Jubrias, S.A.; Esselman, P.C. Oxidative Capacity and Ageing in Human Muscle. J. Physiol. 2000, 526 Pt 1, 203–210. [Google Scholar] [CrossRef]
- Stocco, D.M.; Cascarano, J.; Wilson, M.A. Quantitation of Mitochondrial DNA, RNA, and Protein in Starved and Starved-Refed Rat Liver. J. Cell. Physiol. 1977, 90, 295–306. [Google Scholar] [CrossRef]
- Lee, S.; Jeong, S.; Lim, W.; Kim, S.; Park, Y.; Sun, X.; Youle, R.J.; Cho, H. Mitochondrial Fission and Fusion Mediators, hFis1 and OPA1, Modulate Cellular Senescence. J. Biol. Chem. 2007, 282, 22977–22983. [Google Scholar] [CrossRef]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in Skeletal Muscle Mitochondrial Function with Aging in Humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef]
- Chen, Q.; Young, L.; Barsotti, R. Mitochondria in Cell Senescence: A Friend Or Foe? Adv. Protein Chem. Struct. Biol. 2023, 136, 35–91. [Google Scholar] [PubMed]
- Akbari, M.; Nilsen, H.L.; Montaldo, N.P. Dynamic Features of Human Mitochondrial DNA Maintenance and Transcription. Front. Cell Dev. Biol. 2022, 10, 984245. [Google Scholar] [CrossRef]
- Fang, W.; Chen, S.; Jin, X.; Liu, S.; Cao, X.; Liu, B. Metabolomics in Aging Research: Aging Markers from Organs. Front. Cell Dev. Biol. 2023, 11, 1198794. [Google Scholar] [CrossRef] [PubMed]
- Kuehne, A.; Hildebrand, J.; Soehle, J.; Wenck, H.; Terstegen, L.; Gallinat, S.; Knott, A.; Winnefeld, M.; Zamboni, N. An Integrative Metabolomics and Transcriptomics Study to Identify Metabolic Alterations in Aged Skin of Humans in Vivo. BMC Genom. 2017, 18, 169. [Google Scholar] [CrossRef] [PubMed]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef]
- Sreedhar, A.; Aguilera-Aguirre, L.; Singh, K.K. Mitochondria in Skin Health, Aging, and Disease. Cell Death Dis. 2020, 11, 444. [Google Scholar] [CrossRef]
- Berneburg, M.; Gattermann, N.; Stege, H.; Grewe, M.; Vogelsang, K.; Ruzicka, T.; Krutmann, J. Chronically Ultraviolet-Exposed Human Skin shows a Higher Mutation Frequency of Mitochondrial DNA as Compared to Unexposed Skin and the Hematopoietic System. Photochem. Photobiol. 1997, 66, 271–275. [Google Scholar] [CrossRef]
- Birch-Machin, M.A.; Tindall, M.; Turner, R.; Haldane, F.; Rees, J.L. Mitochondrial DNA Deletions in Human Skin Reflect Photo- rather than Chronologic Aging. J. Investig. Dermatol. 1998, 110, 149–152. [Google Scholar] [CrossRef]
- Krishnan, K.J.; Birch-Machin, M.A. The Incidence of both Tandem Duplications and the Common Deletion in mtDNA from Three Distinct Categories of Sun-Exposed Human Skin and in Prolonged Culture of Fibroblasts. J. Investig. Dermatol. 2006, 126, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.J.; Turner, R.; Nikaido, O.; Rees, J.L.; Birch-Machin, M.A. The Spectrum of Mitochondrial DNA Deletions is a Ubiquitous Marker of Ultraviolet Radiation Exposure in Human Skin. J. Investig. Dermatol. 2000, 115, 674–679. [Google Scholar] [CrossRef]
- Powers, J.M.; Murphy, G.; Ralph, N.; O’Gorman, S.M.; Murphy, J.E.J. Mitochondrial DNA Deletion Percentage in Sun Exposed and Non Sun Exposed Skin. J. Photochem. Photobiol. B 2016, 165, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Schlieben, L.D.; Prokisch, H. Genetics of Mitochondrial Diseases: Current Approaches for the Molecular Diagnosis. Handb. Clin. Neurol. 2023, 194, 141–165. [Google Scholar] [PubMed]
- Yang, J.; Lee, H.; Chung, J.; Wei, Y. Mitochondrial DNA Mutations in Light-Associated Skin Tumors. Anticancer Res. 2004, 24, 1753–1758. [Google Scholar] [PubMed]
- Liu, C.; Li, J. O-GlcNAc: A Sweetheart of the Cell Cycle and DNA Damage Response. Front. Endocrinol. 2018, 9, 415. [Google Scholar] [CrossRef]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. Activated p53 Induces NF-kappaB DNA Binding but Suppresses its Transcriptional Activation. Biochem. Biophys. Res. Commun. 2008, 372, 137–141. [Google Scholar] [CrossRef]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. Loss of p53 Enhances Catalytic Activity of IKKbeta through O-Linked Beta-N-Acetyl Glucosamine Modification. Proc. Natl. Acad. Sci. USA 2009, 106, 3431–3436. [Google Scholar] [CrossRef]
- Taylor, D.J.; Faragher, E.B.; Evanson, J.M. Inflammatory Cytokines Stimulate Glucose Uptake and Glycolysis but Reduce Glucose Oxidation in Human Dermal Fibroblasts in Vitro. Circ. Shock 1992, 37, 105–110. [Google Scholar]
- Lee, J.B.; Pyo, K.; Kim, H.R. Role and Function of O-GlcNAcylation in Cancer. Cancers 2021, 13, 5365. [Google Scholar] [CrossRef]
- Ouyang, M.; Yu, C.; Deng, X.; Zhang, Y.; Zhang, X.; Duan, F. O-GlcNAcylation and its Role in Cancer-Associated Inflammation. Front. Immunol. 2022, 13, 861559. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Jin, J.; Qiu, Z.; Liu, D.; Luo, H. Functional Analysis of O-GlcNAcylation in Cancer Metastasis. Front. Oncol. 2020, 10, 585288. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Hooper, R.; Tomar, D.; Armstead, A.P.; Shanas, N.; Mallu, P.; Joshi, H.; Ray, S.; Chong, P.L.; Astsaturov, I.; et al. Suppression of Ca(2+) Signaling Enhances Melanoma Progression. EMBO J. 2022, 41, e110046. [Google Scholar] [CrossRef] [PubMed]
- Noordam, R.; Gunn, D.A.; Tomlin, C.C.; Maier, A.B.; Griffiths, T.; Catt, S.D.; Ogden, S.; Slagboom, P.E.; Westendorp, R.G.; Griffiths, C.E.; et al. Serum Insulin-Like Growth Factor 1 and Facial Ageing: High Levels Associate with Reduced Skin Wrinkling in a Cross-Sectional Study. Br. J. Dermatol. 2013, 168, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Dekker, P.; Maier, A.B.; van Heemst, D.; de Koning-Treurniet, C.; Blom, J.; Dirks, R.W.; Tanke, H.J.; Westendorp, R.G.J. Stress-Induced Responses of Human Skin Fibroblasts in Vitro Reflect Human Longevity. Aging Cell. 2009, 8, 595–603. [Google Scholar] [CrossRef]
- Blazer, S.; Khankin, E.; Segev, Y.; Ofir, R.; Yalon-Hacohen, M.; Kra-Oz, Z.; Gottfried, Y.; Larisch, S.; Skorecki, K.L. High Glucose-Induced Replicative Senescence: Point of no Return and Effect of Telomerase. Biochem. Biophys. Res. Commun. 2002, 296, 93–101. [Google Scholar] [CrossRef]
- Stabenow, L.K.; Zibrova, D.; Ender, C.; Helbing, D.L.; Spengler, K.; Marx, C.; Wang, Z.; Heller, R. Oxidative Glucose Metabolism Promotes Senescence in Vascular Endothelial Cells. Cells 2022, 11, 2213. [Google Scholar] [CrossRef]
- Mellem, D.; Sattler, M.; Pagel-Wolff, S.; Jaspers, S.; Wenck, H.; Rübhausen, M.A.; Fischer, F. Fragmentation of the Mitochondrial Network in Skin in Vivo. PLoS ONE 2017, 12, e0174469. [Google Scholar] [CrossRef]
- Lu, J.; Tan, M.; Cai, Q. The Warburg Effect in Tumor Progression: Mitochondrial Oxidative Metabolism as an Anti-Metastasis Mechanism. Cancer Lett. 2015, 356, 156–164. [Google Scholar] [CrossRef]
- Sergentanis, T.N.; Antoniadis, A.G.; Gogas, H.J.; Antonopoulos, C.N.; Adami, H.; Ekbom, A.; Petridou, E.T. Obesity and Risk of Malignant Melanoma: A Meta-Analysis of Cohort and Case-Control Studies. Eur. J. Cancer 2013, 49, 642–657. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, K.K.L.; Jiang, X.; Xu, A.; Cheng, K.K.Y. The Role of Adipose Tissue Senescence in Obesity- and Ageing-Related Metabolic Disorders. Clin. Sci. 2020, 134, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Danby, F.W. Nutrition and Aging Skin: Sugar and Glycation. Clin. Dermatol. 2010, 28, 409–411. [Google Scholar] [CrossRef] [PubMed]
- Swislocki, A.L.M. Glucose Trajectory: More than Changing Glucose Tolerance with Age? Metab. Syndr. Relat. Disord. 2022, 20, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, J.; Duan, Y.; Li, Y.; Sun, Y.; Sun, H.; Yu, X.; Gao, X.; Zhang, C.; Zhang, H.; et al. Metabolic Regulation: A Potential Strategy for Rescuing Stem Cell Senescence. Stem Cell Rev. Rep. 2022, 18, 1728–1742. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Qi, X.; Xiong, H.; Liu, Q.; Li, J.; Zhang, Y.; Ma, X.; Wu, N.; Liu, Q.; Feng, L. Type 2 Diabetes Mellitus and Risk of Malignant Melanoma: A Systematic Review and Meta-Analysis of Cohort Studies. Iran. J. Public Health 2014, 43, 857–866. [Google Scholar]
- Saewai, C.; Fumaneeshoat, O.; Thongsuksai, P.; Ingviya, T. Diabetes Mellitus as Cancer Risk: A 14-Year, Cross-Sectional Analysis. Nutr. Cancer 2023, 75, 1454–1463. [Google Scholar] [CrossRef]
- Kennedy, O.J.; Kicinski, M.; Valpione, S.; Gandini, S.; Suciu, S.; Blank, C.U.; Long, G.V.; Atkinson, V.G.; Dalle, S.; Haydon, A.M.; et al. Prognostic and Predictive Value of Metformin in the European Organisation for Research and Treatment of Cancer 1325/KEYNOTE-054 Phase III Trial of Pembrolizumab Versus Placebo in Resected High-Risk Stage III Melanoma. Eur. J. Cancer 2023, 189, 112900. [Google Scholar] [CrossRef]
- Krakowski, I.; Habel, H.; Nielsen, K.; Ingvar, C.; Andersson, T.M.L.; Girnita, A.; Smedby, K.E.; Eriksson, H. Association of Metformin use and Survival in Patients with Cutaneous Melanoma and Diabetes. Br. J. Dermatol. 2023, 188, 32–40. [Google Scholar] [CrossRef]
- Ferguson, J.; Smith, M.; Zudaire, I.; Wellbrock, C.; Arozarena, I. Glucose Availability Controls ATF4-Mediated MITF Suppression to Drive Melanoma Cell Growth. Oncotarget 2017, 8, 32946–32959. [Google Scholar] [CrossRef]
- Scott, D.A.; Richardson, A.D.; Filipp, F.V.; Knutzen, C.A.; Chiang, G.G.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Comparative Metabolic Flux Profiling of Melanoma Cell Lines: Beyond the Warburg Effect. J. Biol. Chem. 2011, 286, 42626–42634. [Google Scholar] [CrossRef]
- Filipp, F.V.; Ratnikov, B.; De Ingeniis, J.; Smith, J.W.; Osterman, A.L.; Scott, D.A. Glutamine-Fueled Mitochondrial Metabolism is Decoupled from Glycolysis in Melanoma. Pigment Cell Melanoma Res. 2012, 25, 732–739. [Google Scholar] [CrossRef]
- Corazao-Rozas, P.; Guerreschi, P.; Jendoubi, M.; Andre, F.; Jonneaux, A.; Scalbert, C.; Garcon, G.; Malet-Martino, M.; Balayssac, S.; Rocchi, S.; et al. Mitochondrial Oxidative Stress is the Achille’s Heel of Melanoma Cells Resistant to Braf-Mutant Inhibitor. Oncotarget 2013, 4, 1986–1998. [Google Scholar] [CrossRef] [PubMed]
- Brummer, C.; Faerber, S.; Bruss, C.; Blank, C.; Lacroix, R.; Haferkamp, S.; Herr, W.; Kreutz, M.; Renner, K. Metabolic Targeting Synergizes with MAPK Inhibition and Delays Drug Resistance in Melanoma. Cancer Lett. 2019, 442, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Li, H.; Go, Y.; Chan, X.H.F.; Huang, Q.; Wu, J. Research Advances on the Damage Mechanism of Skin Glycation and Related Inhibitors. Nutrients 2022, 14, 4588. [Google Scholar] [CrossRef]
- Rinnerthaler, M.; Bischof, J.; Streubel, M.K.; Trost, A.; Richter, K. Oxidative Stress in Aging Human Skin. Biomolecules 2015, 5, 545–589. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhang, J.; Li, L.; Guo, M.; He, Y.; Dong, Y.; Meng, H.; Yi, F. Advanced Glycation End Products in the Skin: Molecular Mechanisms, Methods of Measurement, and Inhibitory Pathways. Front. Med. 2022, 9, 837222. [Google Scholar] [CrossRef]
- Palanissami, G.; Paul, S.F.D. RAGE and its Ligands: Molecular Interplay between Glycation, Inflammation, and Hallmarks of Cancer–A Review. Horm. Cancer 2018, 9, 295–325. [Google Scholar] [CrossRef]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-Activated Protein Kinase Induces a p53-Dependent Metabolic Checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef]
- Donehower, L.A. Does p53 Affect Organismal Aging? J. Cell. Physiol. 2002, 192, 23–33. [Google Scholar] [CrossRef]
- Tyner, S.D.; Venkatachalam, S.; Choi, J.; Jones, S.; Ghebranious, N.; Igelmann, H.; Lu, X.; Soron, G.; Cooper, B.; Brayton, C.; et al. P53 Mutant Mice that Display Early Ageing-Associated Phenotypes. Nature 2002, 415, 45–53. [Google Scholar] [CrossRef]
- Bonafe, M.; Olivieri, F.; Mari, D.; Baggio, G.; Mattace, R.; Berardelli, M.; De Benedictis, G.; De Luca, M.; Marchegiani, F.; Cavallone, L.; et al. P53 Codon 72 Polymorphism and Longevity: Additional Data on Centenarians from Continental Italy and Sardinia. Am. J. Hum. Genet. 1999, 65, 1782–1785. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Papaccio, F.; Arino, A.D.; Caputo, S.; Bellei, B. Focus on the Contribution of Oxidative Stress in Skin Aging. Antioxidants. 2022, 11, 1121. [Google Scholar] [CrossRef] [PubMed]
- Hekimi, S. How Genetic Analysis Tests Theories of Animal Aging. Nat. Genet. 2006, 38, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Hellemans, L.; Corstjens, H.; Neven, A.; Declercq, L.; Maes, D. Antioxidant Enzyme Activity in Human Stratum Corneum shows Seasonal Variation with an Age-Dependent Recovery. J. Investig. Dermatol. 2003, 120, 434–439. [Google Scholar] [CrossRef]
- Bielli, A.; Scioli, M.G.; D’Amico, F.; Tarquini, C.; Agostinelli, S.; Costanza, G.; Doldo, E.; Campione, E.; Passeri, D.; Coniglione, F.; et al. Cellular Retinoic Acid Binding Protein-II Expression and its Potential Role in Skin Aging. Aging 2019, 11, 1619–1632. [Google Scholar] [CrossRef]
- Prahl, S.; Kueper, T.; Biernoth, T.; Wohrmann, Y.; Munster, A.; Furstenau, M.; Schmidt, M.; Schulze, C.; Wittern, K.; Wenck, H.; et al. Aging Skin is Functionally Anaerobic: Importance of Coenzyme Q10 for Anti Aging Skin Care. Biofactors 2008, 32, 245–255. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Mel’nikova, T.I.; Porozov, Y.B.; Terentiev, A.A. Oxidative Stress and Advanced Lipoxidation and Glycation End Products (ALEs and AGEs) in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 3085756. [Google Scholar] [CrossRef]
- Larroque-Cardoso, P.; Camare, C.; Nadal-Wollbold, F.; Grazide, M.; Pucelle, M.; Garoby-Salom, S.; Bogdanowicz, P.; Josse, G.; Schmitt, A.; Uchida, K.; et al. Elastin Modification by 4-Hydroxynonenal in Hairless Mice Exposed to UV-A. Role in Photoaging and Actinic Elastosis. J. Investig. Dermatol. 2015, 135, 1873–1881. [Google Scholar] [CrossRef]
- Williams, J.D.; Bermudez, Y.; Park, S.L.; Stratton, S.P.; Uchida, K.; Hurst, C.A.; Wondrak, G.T. Malondialdehyde-Derived Epitopes in Human Skin Result from Acute Exposure to Solar UV and Occur in Nonmelanoma Skin Cancer Tissue. J. Photochem. Photobiol. B 2014, 132, 56–65. [Google Scholar] [CrossRef]
- Jove, M.; Naudi, A.; Gambini, J.; Borras, C.; Cabre, R.; Portero-Otin, M.; Vina, J.; Pamplona, R. A Stress-Resistant Lipidomic Signature Confers Extreme Longevity to Humans. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 30–37. [Google Scholar] [CrossRef]
- Borchman, D.; Stimmelmayr, R.; George, J.C. Whales, Lifespan, Phospholipids, and Cataracts. J. Lipid Res. 2017, 58, 2289–2298. [Google Scholar] [CrossRef] [PubMed]
- Jobson, R.W.; Nabholz, B.; Galtier, N. An Evolutionary Genome Scan for Longevity-Related Natural Selection in Mammals. Mol. Biol. Evol. 2010, 27, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Gruber, F.; Marchetti-Deschmann, M.; Kremslehner, C.; Schosserer, M. The Skin Epilipidome in Stress, Aging, and Inflammation. Front. Endocrinol. 2021, 11, 607076. [Google Scholar] [CrossRef] [PubMed]
- Gruber, F.; Bicker, W.; Oskolkova, O.V.; Tschachler, E.; Bochkov, V.N. A Simplified Procedure for Semi-Targeted Lipidomic Analysis of Oxidized Phosphatidylcholines Induced by UVA Irradiation. J. Lipid Res. 2012, 53, 1232–1242. [Google Scholar] [CrossRef]
- Schmitz, S.; Thomas, P.D.; Allen, T.M.; Poznansky, M.J.; Jimbow, K. Dual Role of Melanins and Melanin Precursors as Photoprotective and Phototoxic Agents: Inhibition of Ultraviolet Radiation-Induced Lipid Peroxidation. Photochem. Photobiol. 1995, 61, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Ramprecht, C.; Jaritz, H.; Streith, I.; Zenzmaier, E.; Kofeler, H.; Hofmann-Wellenhof, R.; Schaider, H.; Hermetter, A. Toxicity of Oxidized Phosphatidylcholines in Cultured Human Melanoma Cells. Chem. Phys. Lipids 2015, 189, 39–47. [Google Scholar] [CrossRef]
- Halczy-Kowalik, L.; Drozd, A.; Stachowska, E.; Drozd, R.; Zabski, T.; Domagala, W. Fatty Acids Distribution and Content in Oral Squamous Cell Carcinoma Tissue and its Adjacent Microenvironment. PLoS ONE 2019, 14, e0218246. [Google Scholar] [CrossRef]
- Heller, E.R.; Gor, A.; Wang, D.; Hu, Q.; Lucchese, A.; Kanduc, D.; Katdare, M.; Liu, S.; Sinha, A.A. Molecular Signatures of Basal Cell Carcinoma Susceptibility and Pathogenesis: A Genomic Approach. Int. J. Oncol. 2013, 42, 583–596. [Google Scholar] [CrossRef]
- Fisher, G.J.; Wang, B.; Cui, Y.; Shi, M.; Zhao, Y.; Quan, T.; Voorhees, J.J. Skin Aging from the Perspective of Dermal Fibroblasts: The Interplay between the Adaptation to the Extracellular Matrix Microenvironment and Cell Autonomous Processes. J. Cell Commun. Signal. 2023, 17, 523–529. [Google Scholar] [CrossRef]
- Wlaschek, M.; Tantcheva-Poor, I.; Naderi, L.; Ma, W.; Schneider, L.A.; Razi-Wolf, Z.; Schuller, J.; Scharffetter-Kochanek, K. Solar UV Irradiation and Dermal Photoaging. J. Photochem. Photobiol. B 2001, 63, 41–51. [Google Scholar] [CrossRef]
- Farage, M.A.; Miller, K.W.; Berardesca, E.; Maibach, H.I. Clinical Implications of Aging Skin: Cutaneous Disorders in the Elderly. Am. J. Clin. Dermatol. 2009, 10, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Le Varlet, B.; Chaudagne, C.; Saunois, A.; Barré, P.; Sauvage, C.; Berthouloux, B.; Meybeck, A.; Dumas, M.; Bonté, F. Age-Related Functional and Structural Changes in Human Dermo-Epidermal Junction Components. J. Investig. Dermatol. Symp. Proc. 1998, 3, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Langton, A.K.; Halai, P.; Griffiths, C.E.; Sherratt, M.J.; Watson, R.E. The Impact of Intrinsic Ageing on the Protein Composition of the Dermal-Epidermal Junction. Mech. Ageing Dev. 2016, 156, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.; Choi, K.; Kim, M.; Hong, S. Neural Stem Cells and the Secreted Proteins TIMPs Ameliorate UVB-Induced Skin Photodamage. Biochem. Biophys. Res. Commun. 2019, 518, 388–395. [Google Scholar] [CrossRef]
- Yokose, U.; Hachiya, A.; Sriwiriyanont, P.; Fujimura, T.; Visscher, M.O.; Kitzmiller, W.J.; Bello, A.; Tsuboi, R.; Kitahara, T.; Kobinger, G.P.; et al. The Endogenous Protease Inhibitor TIMP-1 Mediates Protection and Recovery from Cutaneous Photodamage. J. Investig. Dermatol. 2012, 132, 2800–2809. [Google Scholar] [CrossRef]
- Yasui, T.; Yonetsu, M.; Tanaka, R.; Tanaka, Y.; Fukushima, S.; Yamashita, T.; Ogura, Y.; Hirao, T.; Murota, H.; Araki, T. In Vivo Observation of Age-Related Structural Changes of Dermal Collagen in Human Facial Skin using Collagen-Sensitive Second Harmonic Generation Microscope Equipped with 1250-Nm Mode-Locked Cr:Forsterite Laser. J. Biomed. Opt. 2013, 18, 31108. [Google Scholar] [CrossRef]
- Pires, A.; Greenshields-Watson, A.; Jones, E.; Smart, K.; Lauder, S.N.; Somerville, M.; Milutinovic, S.; Kendrick, H.; Hindley, J.P.; French, R.; et al. Immune Remodeling of the Extracellular Matrix Drives Loss of Cancer Stem Cells and Tumor Rejection. Cancer Immunol. Res. 2020, 8, 1520–1531. [Google Scholar] [CrossRef]
- Shin, J.W.; Kwon, S.H.; Choi, J.Y.; Na, J.I.; Huh, C.H.; Choi, H.R.; Park, K.C. Molecular Mechanisms of Dermal Aging and Antiaging Approaches. Int. J. Mol. Sci. 2019, 20, 2126. [Google Scholar] [CrossRef]
- Worrede, A.; Douglass, S.M.; Weeraratna, A.T. The Dark Side of Daylight: Photoaging and the Tumor Microenvironment in Melanoma Progression. J. Clin. Investig. 2021, 131, e143763. [Google Scholar] [CrossRef]
- Tsukifuji, R.; Tagawa, K.; Hatamochi, A.; Shinkai, H. Expression of Matrix Metalloproteinase-1, -2 and -3 in Squamous Cell Carcinoma and Actinic Keratosis. Br. J. Cancer. 1999, 80, 1087–1091. [Google Scholar] [CrossRef]
- Kellow, N.J.; Coughlan, M.T.; Reid, C.M. Association between Habitual Dietary and Lifestyle Behaviours and Skin Autofluorescence (SAF), a Marker of Tissue Accumulation of Advanced Glycation Endproducts (AGEs), in Healthy Adults. Eur. J. Nutr. 2018, 57, 2209–2216. [Google Scholar] [CrossRef] [PubMed]
- Verzijl, N.; DeGroot, J.; Thorpe, S.R.; Bank, R.A.; Shaw, J.N.; Lyons, T.J.; Bijlsma, J.W.; Lafeber, F.P.; Baynes, J.W.; TeKoppele, J.M. Effect of Collagen Turnover on the Accumulation of Advanced Glycation End Products. J. Biol. Chem. 2000, 275, 39027–39031. [Google Scholar] [CrossRef] [PubMed]
- Avery, N.C.; Bailey, A.J. The Effects of the Maillard Reaction on the Physical Properties and Cell Interactions of Collagen. Pathol. Biol. 2006, 54, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Negre-Salvayre, A.; Salvayre, R. Post-Translational Modifications Evoke by Reactive Carbonyl Species in Ultraviolet-A-Exposed Skin: Implication in Fibroblast Senescence and Skin Photoaging. Antioxidants. 2022, 11, 2281. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, A.G.; Harper, J.M. Exploring the Role of Primary Fibroblast Cells in Comparative Physiology: A Historical and Contemporary Overview. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2023, 325, R45–R54. [Google Scholar] [CrossRef] [PubMed]
- Kueper, T.; Grune, T.; Prahl, S.; Lenz, H.; Welge, V.; Biernoth, T.; Vogt, Y.; Muhr, G.; Gaemlich, A.; Jung, T.; et al. Vimentin is the Specific Target in Skin Glycation. Structural Prerequisites, Functional Consequences, and Role in Skin Aging. J. Biol. Chem. 2007, 282, 23427–23436. [Google Scholar] [CrossRef]
- Sasaki, K.; Sugai, T.; Ishida, K.; Osakabe, M.; Amano, H.; Kimura, H.; Sakuraba, M.; Kashiwa, K.; Kobayashi, S. Analysis of Cancer-Associated Fibroblasts and the Epithelial-Mesenchymal Transition in Cutaneous Basal Cell Carcinoma, Squamous Cell Carcinoma, and Malignant Melanoma. Hum. Pathol. 2018, 79, 1–8. [Google Scholar] [CrossRef]
- Fujimura, T.; Kakizaki, A.; Furudate, S.; Kambayashi, Y.; Aiba, S. Tumor-Associated Macrophages in Skin: How to Treat their Heterogeneity and Plasticity. J. Dermatol. Sci. 2016, 83, 167–173. [Google Scholar] [CrossRef]
- Marcus, A.; Gowen, B.G.; Thompson, T.W.; Iannello, A.; Ardolino, M.; Deng, W.; Wang, L.; Shifrin, N.; Raulet, D.H. Recognition of Tumors by the Innate Immune System and Natural Killer Cells. Adv. Immunol. 2014, 122, 91–128. [Google Scholar]
- Muul, L.M.; Spiess, P.J.; Director, E.P.; Rosenberg, S.A. Identification of Specific Cytolytic Immune Responses Against Autologous Tumor in Humans Bearing Malignant Melanoma. J. Immunol. 1987, 138, 989–995. [Google Scholar] [CrossRef]
- Eze, K.C.; Ugochukwu, O.M.; Nzegwu, M.A. Death Patterns among Nigerian Leaders. J. Inj. Violence Res. 2010, 2, 61–65. [Google Scholar] [CrossRef][Green Version]
- Botti, G.; Cerrone, M.; Scognamiglio, G.; Anniciello, A.; Ascierto, P.A.; Cantile, M. Microenvironment and Tumor Progression of Melanoma: New Therapeutic Prospectives. J. Immunotoxicol. 2013, 10, 235–252. [Google Scholar] [CrossRef]
- Azimnasab-Sorkhabi, P.; Soltani-Asl, M.; Yoshinaga, T.T.; Zaidan Dagli, M.L.; Massoco, C.d.O.; Kfoury Junior, J.R. Indoleamine-2,3 Dioxygenase: A Fate-Changer of the Tumor Microenvironment. Mol. Biol. Rep. 2023, 50, 6133–6145. [Google Scholar] [CrossRef]
- Lacina, L.; Plzak, J.; Kodet, O.; Szabo, P.; Chovanec, M.; Dvorankova, B.; Smetana, K.J. Cancer Microenvironment: What can we Learn from the Stem Cell Niche. Int. J. Mol. Sci. 2015, 16, 24094–24110. [Google Scholar] [CrossRef]
- Wang, J.X.; Fukunaga-Kalabis, M.; Herlyn, M. Crosstalk in Skin: Melanocytes, Keratinocytes, Stem Cells, and Melanoma. J. Cell. Commun. Signal. 2016, 10, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Satyamoorthy, K.; Herlyn, M. N-Cadherin-Mediated Intercellular Interactions Promote Survival and Migration of Melanoma Cells. Cancer Res. 2001, 61, 3819–3825. [Google Scholar] [PubMed]
- Yang, K.; Wang, X.; Zhang, H.; Wang, Z.; Nan, G.; Li, Y.; Zhang, F.; Mohammed, M.K.; Haydon, R.C.; Luu, H.H.; et al. The Evolving Roles of Canonical WNT Signaling in Stem Cells and Tumorigenesis: Implications in Targeted Cancer Therapies. Lab. Investig. 2016, 96, 116–136. [Google Scholar] [CrossRef]
- Shields, B.D.; Koss, B.; Taylor, E.M.; Storey, A.J.; West, K.L.; Byrum, S.D.; Mackintosh, S.G.; Edmondson, R.; Mahmoud, F.; Shalin, S.C.; et al. Loss of E-Cadherin Inhibits CD103 Antitumor Activity and Reduces Checkpoint Blockade Responsiveness in Melanoma. Cancer Res. 2019, 79, 1113–1123. [Google Scholar] [CrossRef]
- Gambichler, T.; Rotterdam, S.; Tigges, C.; Altmeyer, P.; Bechara, F.G. Impact of Ultraviolet Radiation on the Expression of Marker Proteins of Gap and Adhesion Junctions in Human Epidermis. Photodermatol. Photoimmunol. Photomed. 2008, 24, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Arnette, C.R.; Roth-Carter, Q.R.; Koetsier, J.L.; Broussard, J.A.; Burks, H.E.; Cheng, K.; Amadi, C.; Gerami, P.; Johnson, J.L.; Green, K.J. Keratinocyte Cadherin Desmoglein 1 Controls Melanocyte Behavior through Paracrine Signaling. Pigment Cell Melanoma Res. 2020, 33, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Bellei, B.; Mastrofrancesco, A.; Briganti, S.; Aspite, N.; Ale-Agha, N.; Sies, H.; Picardo, M. Ultraviolet A Induced Modulation of Gap Junctional Intercellular Communication by P38 MAPK Activation in Human Keratinocytes. Exp. Dermatol. 2008, 17, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, G.R.; Divakar, S. Synthesis of Guaiacol-Alpha-D: -Glucoside and Curcumin-Bis-Alpha-D: -Glucoside by an Amyloglucosidase from Rhizopus. Biotechnol. Lett. 2005, 27, 1411–1415. [Google Scholar] [CrossRef] [PubMed]
- Panka, D.J.; Atkins, M.B.; Mier, J.W. Targeting the Mitogen-Activated Protein Kinase Pathway in the Treatment of Malignant Melanoma. Clin. Cancer Res. 2006, 12, 2371s–2375s. [Google Scholar] [CrossRef] [PubMed]
- Whipple, C.A.; Brinckerhoff, C.E. BRAF(V600E) Melanoma Cells Secrete Factors that Activate Stromal Fibroblasts and Enhance Tumourigenicity. Br. J. Cancer 2014, 111, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Khalili, J.S.; Liu, S.; Rodríguez-Cruz, T.G.; Whittington, M.; Wardell, S.; Liu, C.; Zhang, M.; Cooper, Z.A.; Frederick, D.T.; Li, Y.; et al. Oncogenic BRAF(V600E) Promotes Stromal Cell-Mediated Immunosuppression Via Induction of Interleukin-1 in Melanoma. Clin. Cancer Res. 2012, 18, 5329–5340. [Google Scholar] [CrossRef]
- Li, L.; Dragulev, B.; Zigrino, P.; Mauch, C.; Fox, J.W. The Invasive Potential of Human Melanoma Cell Lines Correlates with their Ability to Alter Fibroblast Gene Expression in Vitro and the Stromal Microenvironment in Vivo. Int. J. Cancer 2009, 125, 1796–1804. [Google Scholar] [CrossRef]
- Jobe, N.P.; Rösel, D.; Dvořánková, B.; Kodet, O.; Lacina, L.; Mateu, R.; Smetana, K.; Brábek, J. Simultaneous Blocking of IL-6 and IL-8 is Sufficient to Fully Inhibit CAF-Induced Human Melanoma Cell Invasiveness. Histochem. Cell Biol. 2016, 146, 205–217. [Google Scholar] [CrossRef]
- Kurgyis, Z.; Kemeny, L.V.; Buknicz, T.; Groma, G.; Olah, J.; Jakab, A.; Polyanka, H.; Zanker, K.; Dittmar, T.; Kemeny, L.; et al. Melanoma-Derived BRAF(V600E) Mutation in Peritumoral Stromal Cells: Implications for in Vivo Cell Fusion. Int. J. Mol. Sci. 2016, 17, 980. [Google Scholar] [CrossRef]
- Alicea, G.M.; Rebecca, V.W.; Goldman, A.R.; Fane, M.E.; Douglass, S.M.; Behera, R.; Webster, M.R.; Kugel, C.H., 3rd; Ecker, B.L.; Caino, M.C.; et al. Changes in Aged Fibroblast Lipid Metabolism Induce Age-Dependent Melanoma Cell Resistance to Targeted Therapy Via the Fatty Acid Transporter FATP2. Cancer Discov. 2020, 10, 1282–1295. [Google Scholar] [CrossRef]
- Kaur, A.; Webster, M.R.; Marchbank, K.; Behera, R.; Ndoye, A.; Kugel, C.H., 3rd; Dang, V.M.; Appleton, J.; O’Connell, M.P.; Cheng, P.; et al. sFRP2 in the Aged Microenvironment Drives Melanoma Metastasis and Therapy Resistance. Nature 2016, 532, 250–254. [Google Scholar] [CrossRef]
- Liang, X.; Lin, X.; Lin, Z.; Lin, W.; Peng, Z.; Wei, S. Genes Associated with Cellular Senescence Favor Melanoma Prognosis by Stimulating Immune Responses in Tumor Microenvironment. Comput. Biol. Med. 2023, 158, 106850. [Google Scholar] [CrossRef] [PubMed]
- Avagliano, A.; Arcucci, A. Insights into Melanoma Fibroblast Populations and Therapeutic Strategy Perspectives: Friends Or Foes? Curr. Med. Chem. 2022, 29, 6159–6168. [Google Scholar] [CrossRef] [PubMed]
- Romano, V.; Belviso, I.; Venuta, A.; Ruocco, M.R.; Masone, S.; Aliotta, F.; Fiume, G.; Montagnani, S.; Avagliano, A.; Arcucci, A. Influence of Tumor Microenvironment and Fibroblast Population Plasticity on Melanoma Growth, Therapy Resistance and Immunoescape. Int. J. Mol. Sci. 2021, 22, 5283. [Google Scholar] [CrossRef] [PubMed]
- Kavasi, R.; Neagu, M.; Constantin, C.; Munteanu, A.; Surcel, M.; Tsatsakis, A.; Tzanakakis, G.N.; Nikitovic, D. Matrix Effectors in the Pathogenesis of Keratinocyte-Derived Carcinomas. Front. Med. 2022, 9, 879500. [Google Scholar] [CrossRef]
- Thewes, M.; Worret, W.I.; Engst, R.; Ring, J. Stromelysin-3 (ST-3): Immunohistochemical Characterization of the Matrix Metalloproteinase (MMP)-11 in Benign and Malignant Skin Tumours and Other Skin Disorders. Clin. Exp. Dermatol. 1999, 24, 122–126. [Google Scholar] [CrossRef]
- Unden, A.B.; Sandstedt, B.; Bruce, K.; Hedblad, M.; Stahle-Backdahl, M. Stromelysin-3 mRNA Associated with Myofibroblasts is Overexpressed in Aggressive Basal Cell Carcinoma and in Dermatofibroma but Not in Dermatofibrosarcoma. J. Investig. Dermatol. 1996, 107, 147–153. [Google Scholar] [CrossRef]
- Majmudar, G.; Nelson, B.R.; Jensen, T.C.; Johnson, T.M. Increased Expression of Matrix Metalloproteinase-3 (Stromelysin-1) in Cultured Fibroblasts and Basal Cell Carcinomas of Nevoid Basal Cell Carcinoma Syndrome. Mol. Carcinog. 1994, 11, 29–33. [Google Scholar] [CrossRef]
- Pittayapruek, P.; Meephansan, J.; Prapapan, O.; Komine, M.; Ohtsuki, M. Role of Matrix Metalloproteinases in Photoaging and Photocarcinogenesis. Int. J. Mol. Sci. 2016, 17, 868. [Google Scholar] [CrossRef]
- Kadeh, H.; Saravani, S.; Heydari, F.; Shahraki, S. Differential Immunohistochemical Expression of Matrix Metalloproteinase-10 (MMP-10) in Non-Melanoma Skin Cancers of the Head and Neck. Pathol. Res. Pract. 2016, 212, 867–871. [Google Scholar] [CrossRef]
- Manola, I.; Mataic, A.; Drvar, D.L.; Pezelj, I.; Dzombeta, T.R.; Kruslin, B. Peritumoral Clefting and Expression of MMP-2 and MMP-9 in Basal Cell Carcinoma of the Skin. In Vivo 2020, 34, 1271–1275. [Google Scholar] [CrossRef]
- Tanpa, M.; Georgescu, S.R.; Mitran, M.I.; Mitran, C.I.; Caruntu, A.; Scheau, C.; Nicolae, A.; Matei, A.; Caruntu, C.; Costantin, C.; et al. Current Prospectives on the Role of Matrix Metalloproteinases in the Pathogenesis of Basal Cel Carcinoma. Biomolecules. 2021, 11, 903. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.E.; Dame, M.K.; Remick, D.R.; Elder, J.T.; Varani, J. Induction of Matrix Metalloproteinase-1 (MMP-1) during Epidermal Invasion of the Stroma in Human Skin Organ Culture: Keratinocyte Stimulation of Fibroblast MMP-1 Production. Br. J. Cancer 2001, 85, 1600–1605. [Google Scholar] [CrossRef] [PubMed]
- Uribe, P.; Gonzalez, S. Epidermal Growth Factor Receptor (EGFR) and Squamous Cell Carcinoma of the Skin: Molecular Bases for EGFR-Targeted Therapy. Pathol. Res. Pract. 2011, 207, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Pena-Duque, M.A.; Martinez-Rios, M.A.; Calderon, G.E.; Mejia, A.M.; Gomez, E.; Martinez-Sanchez, C.; Figueroa, J.; Gaspar, J.; Gonzalez, H.; Bialoztosky, D.; et al. Design and Implementation of the TRACIA: Intracoronary Autologous Transplant of Bone Marrow-Derived Stem Cells for Acute ST Elevation Myocardial Infarction. Arch. Cardiol. Mex. 2011, 81, 183–187. [Google Scholar] [PubMed]
- Tran, K.T.; Lamb, P.; Deng, J. Matrikines and Matricryptins: Implications for Cutaneous Cancers and Skin Repair. J. Dermatol. Sci. 2005, 40, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Hunzelmann, N.; Schonherr, E.; Bonnekoh, B.; Hartmann, C.; Kresse, H.; Krieg, T. Altered Immunohistochemical Expression of Small Proteoglycans in the Tumor Tissue and Stroma of Basal Cell Carcinoma. J. Investig. Dermatol. 1995, 104, 509–513. [Google Scholar] [CrossRef]
- Omland, S.H.; Wettergren, E.E.; Mollerup, S.; Asplund, M.; Mourier, T.; Hansen, A.J.; Gniadecki, R. Cancer Associated Fibroblasts (CAFs) are Activated in Cutaneous Basal Cell Carcinoma and in the Peritumoural Skin. BMC Cancer 2017, 17, 675. [Google Scholar] [CrossRef]
- Omland, S.H. Local immune response in cutaneous basal cell carcinoma. Dan. Med. J. 2017, 64, B5412. [Google Scholar]
- Micke, P.; Kappert, K.; Ohshima, M.; Sundquist, C.; Scheidl, S.; Lindahl, P.; Heldin, C.; Botling, J.; Ponten, F.; Ostman, A. In Situ Identification of Genes Regulated Specifically in Fibroblasts of Human Basal Cell Carcinoma. J. Investig. Dermatol. 2007, 127, 1516–1523. [Google Scholar] [CrossRef]
- Aycock, R.L.; Bradshaw, A.C.; Sage, E.H.; Starcher, B. Development of UV-Induced Squamous Cell Carcinomas is Suppressed in the Absence of SPARC. J. Investig. Dermatol. 2004, 123, 592–599. [Google Scholar] [CrossRef]
- Ledda, F.; Bravo, A.I.; Adris, S.; Bover, L.; Mordoh, J.; Podhajcer, O.L. The Expression of the Secreted Protein Acidic and Rich in Cysteine (SPARC) is Associated with the Neoplastic Progression of Human Melanoma. J. Investig. Dermatol. 1997, 108, 210–214. [Google Scholar] [CrossRef]
- Massi, D.; Franchi, A.; Borgognoni, L.; Reali, U.M.; Santucci, M. Osteonectin Expression Correlates with Clinical Outcome in Thin Cutaneous Malignant Melanomas. Hum. Pathol. 1999, 30, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Said, N.; Frierson, H.F.J.; Chernauskas, D.; Conaway, M.; Motamed, K.; Theodorescu, D. The Role of SPARC in the TRAMP Model of Prostate Carcinogenesis and Progression. Oncogene 2009, 28, 3487–3498. [Google Scholar] [CrossRef] [PubMed]
- Koblinski, J.E.; Kaplan-Singer, B.R.; VanOsdol, S.J.; Wu, M.; Engbring, J.A.; Wang, S.; Goldsmith, C.M.; Piper, J.T.; Vostal, J.G.; Harms, J.F.; et al. Endogenous Osteonectin/SPARC/BM-40 Expression Inhibits MDA-MB-231 Breast Cancer Cell Metastasis. Cancer Res. 2005, 65, 7370–7377. [Google Scholar] [CrossRef] [PubMed]
- Chlenski, A.; Liu, S.; Guerrero, L.J.; Yang, Q.; Tian, Y.; Salwen, H.R.; Zage, P.; Cohn, S.L. SPARC Expression is Associated with Impaired Tumor Growth, Inhibited Angiogenesis and Changes in the Extracellular Matrix. Int. J. Cancer 2006, 118, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Rousselle, P.; Beck, K. Laminin 332 Processing Impacts Cellular Behavior. Cell Adh. Migr. 2013, 7, 122–134. [Google Scholar] [CrossRef]
- Skyldberg, B.; Salo, S.; Eriksson, E.; Aspenblad, U.; Moberger, B.; Tryggvason, K.; Auer, G. Laminin-5 as a Marker of Invasiveness in Cervical Lesions. J. Natl. Cancer Inst. 1999, 91, 1882–1887. [Google Scholar] [CrossRef]
- Zhang, K.; Kramer, R.H. Laminin 5 Deposition Promotes Keratinocyte Motility. Exp. Cell Res. 1996, 227, 309–322. [Google Scholar] [CrossRef]
- Janes, S.M.; Watt, F.M. New Roles for Integrins in Squamous-Cell Carcinoma. Nat. Rev. Cancer 2006, 6, 175–183. [Google Scholar] [CrossRef]
- Farsam, V.; Basu, A.; Gatzka, M.; Treiber, N.; Schneider, L.A.; Mulaw, M.A.; Lucas, T.; Kochanek, S.; Dummer, R.; Levesque, M.P.; et al. Senescent Fibroblast-Derived Chemerin Promotes Squamous Cell Carcinoma Migration. Oncotarget 2016, 7, 83554–83569. [Google Scholar] [CrossRef]
- Aden, D.; Zaheer, S.; Ahluwalia, H.; Ranga, S. Cancer-Associated Fibroblasts: Is it a Key to an Intricate Lock of Tumorigenesis? Cell Biol. Int. 2023, 47, 859–893. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Meng, Q.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Zhao, Y.; Yu, X.; et al. Signaling Pathways in Cancer-Associated Fibroblasts: Recent Advances and Future Perspectives. Cancer Commun. 2023, 43, 3–41. [Google Scholar] [CrossRef] [PubMed]
- Eibenschutz, L.; Caputo, S.; Camera, E.; Carbone, A.; Silipo, V.; Migliano, E.; Aurizi, C.; Cota, C.; Frascione, P.; Bellei, B. Evaluation of Hedgehog Pathway Inhibition on Nevoid Basal Cell Carcinoma Syndrome Fibroblasts and Basal Cell Carcinoma-Associated Fibroblasts: Are Vismodegib and Sonidegib Useful to Target Cancer-Prone Fibroblasts? Cancers 2021, 13, 5858. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.; Potestio, L.; Fabbrocini, G.; Scalvezi, M. New Emerging Treatment Options for Advanced Basal Cell Cacinoma and Squamous Cell Carcinoma. Adv. Ther. 2022, 39, 1164–1178. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Arino, A.; Caputo, S.; Eibenschutz, L.; Piemonte, P.; Buccini, P.; Frascione, P.; Bellei, B. Skin Cancer Microenvironment: What We Can Learn from Skin Aging? Int. J. Mol. Sci. 2023, 24, 14043. https://doi.org/10.3390/ijms241814043
D’Arino A, Caputo S, Eibenschutz L, Piemonte P, Buccini P, Frascione P, Bellei B. Skin Cancer Microenvironment: What We Can Learn from Skin Aging? International Journal of Molecular Sciences. 2023; 24(18):14043. https://doi.org/10.3390/ijms241814043
Chicago/Turabian StyleD’Arino, Andrea, Silvia Caputo, Laura Eibenschutz, Paolo Piemonte, Pierluigi Buccini, Pasquale Frascione, and Barbara Bellei. 2023. "Skin Cancer Microenvironment: What We Can Learn from Skin Aging?" International Journal of Molecular Sciences 24, no. 18: 14043. https://doi.org/10.3390/ijms241814043
APA StyleD’Arino, A., Caputo, S., Eibenschutz, L., Piemonte, P., Buccini, P., Frascione, P., & Bellei, B. (2023). Skin Cancer Microenvironment: What We Can Learn from Skin Aging? International Journal of Molecular Sciences, 24(18), 14043. https://doi.org/10.3390/ijms241814043