

The Role of Acyl-CoA β-Oxidation in Brain Metabolism and Neurodegenerative Diseases

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Different Roles of Astrocytic βOX in Brain Metabolism
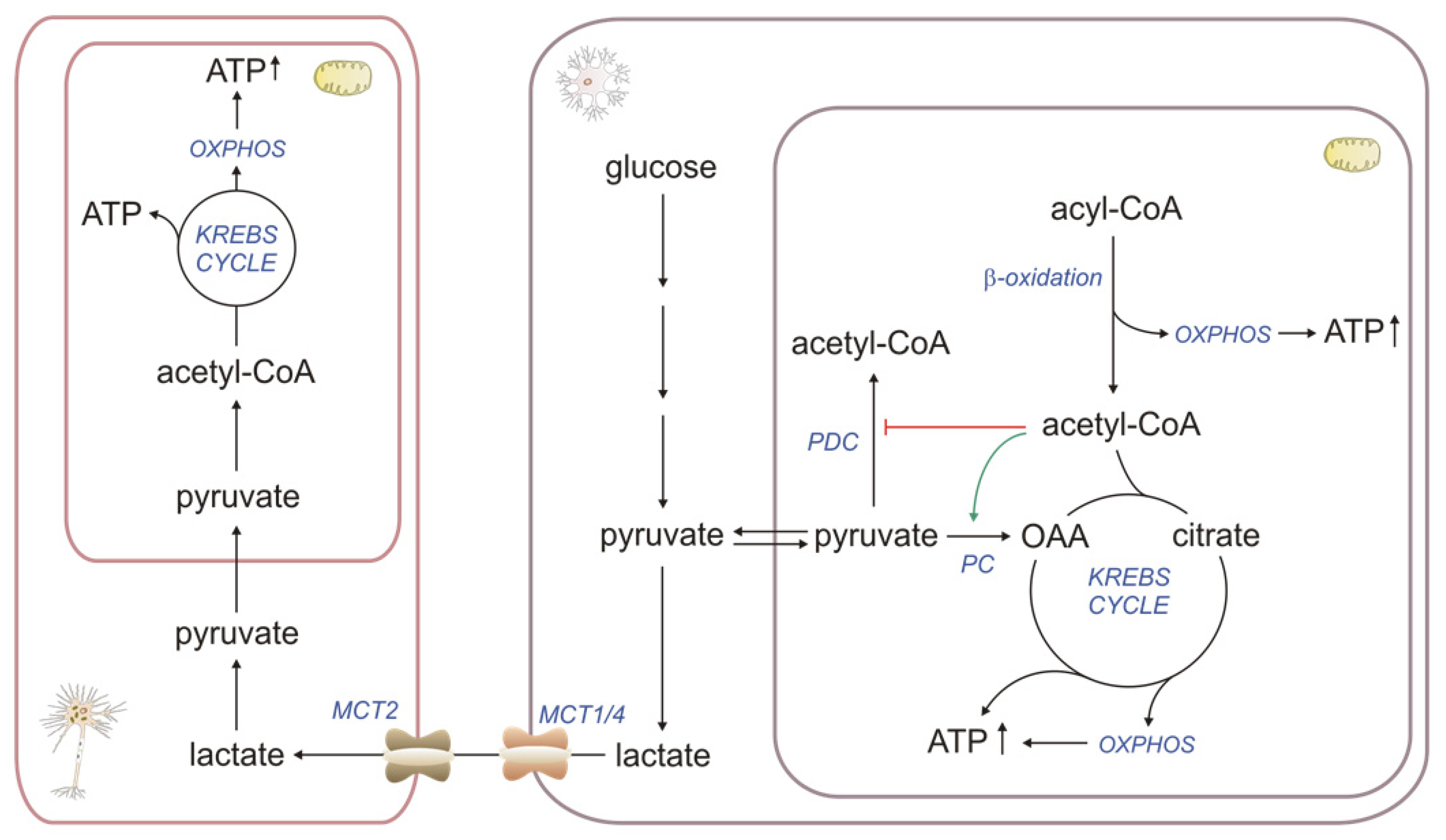
2.1. βOX as a Source of Energy
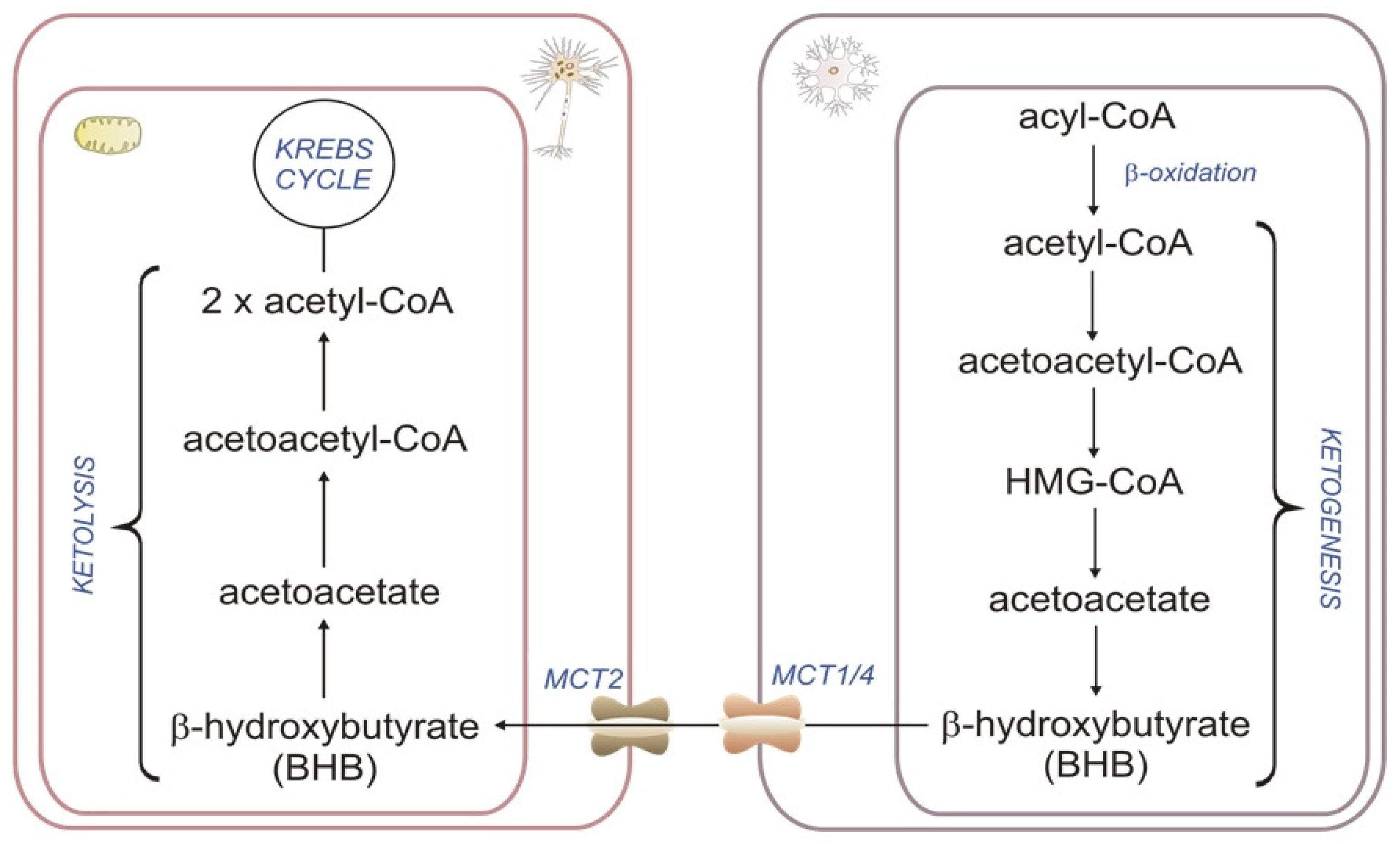
2.2. Astrocyte βOX as a Source of Ketone Bodies
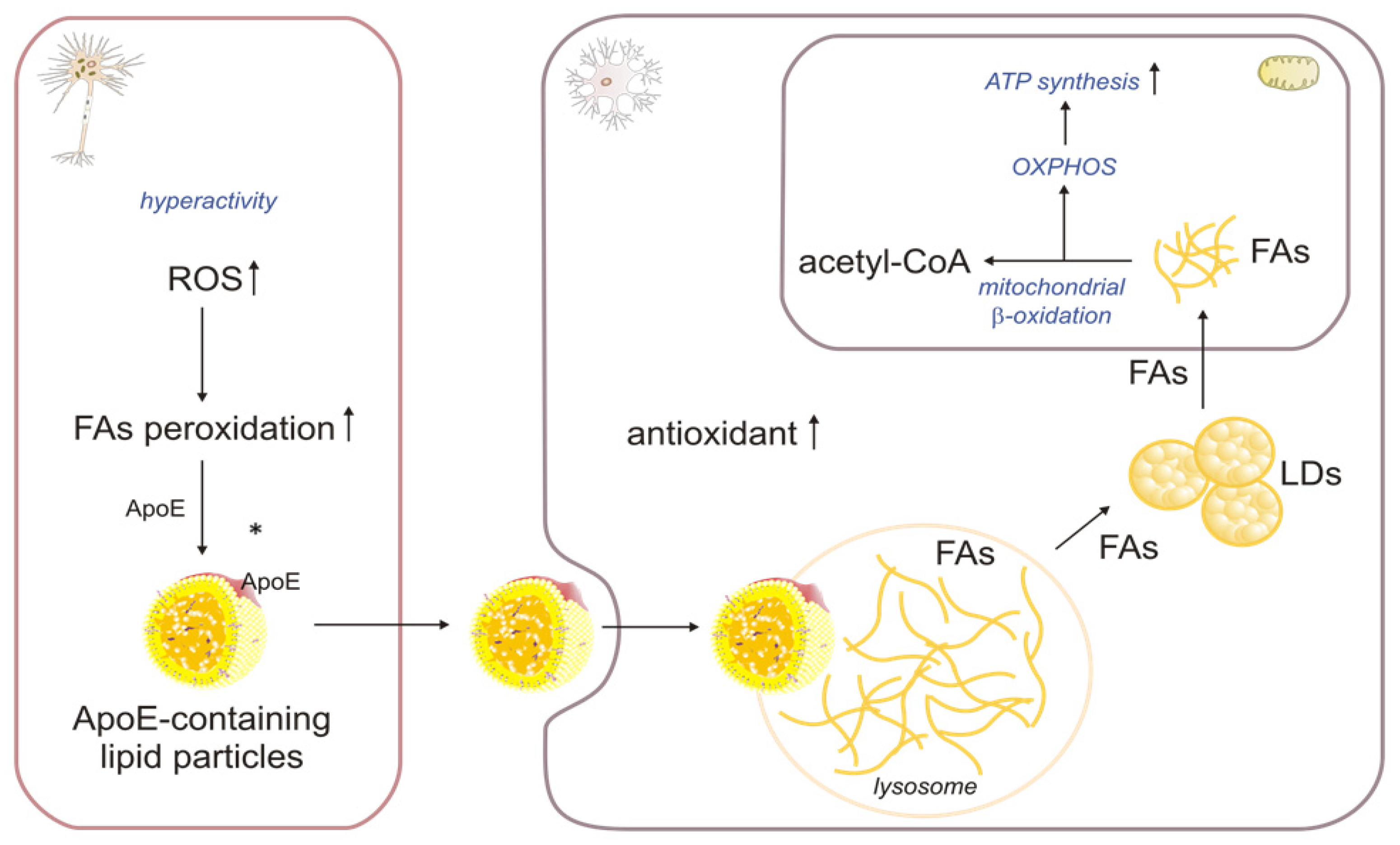
2.3. The Role of βOX in the Oxidation of Fatty Acids Transported from Neurons to Astrocytes
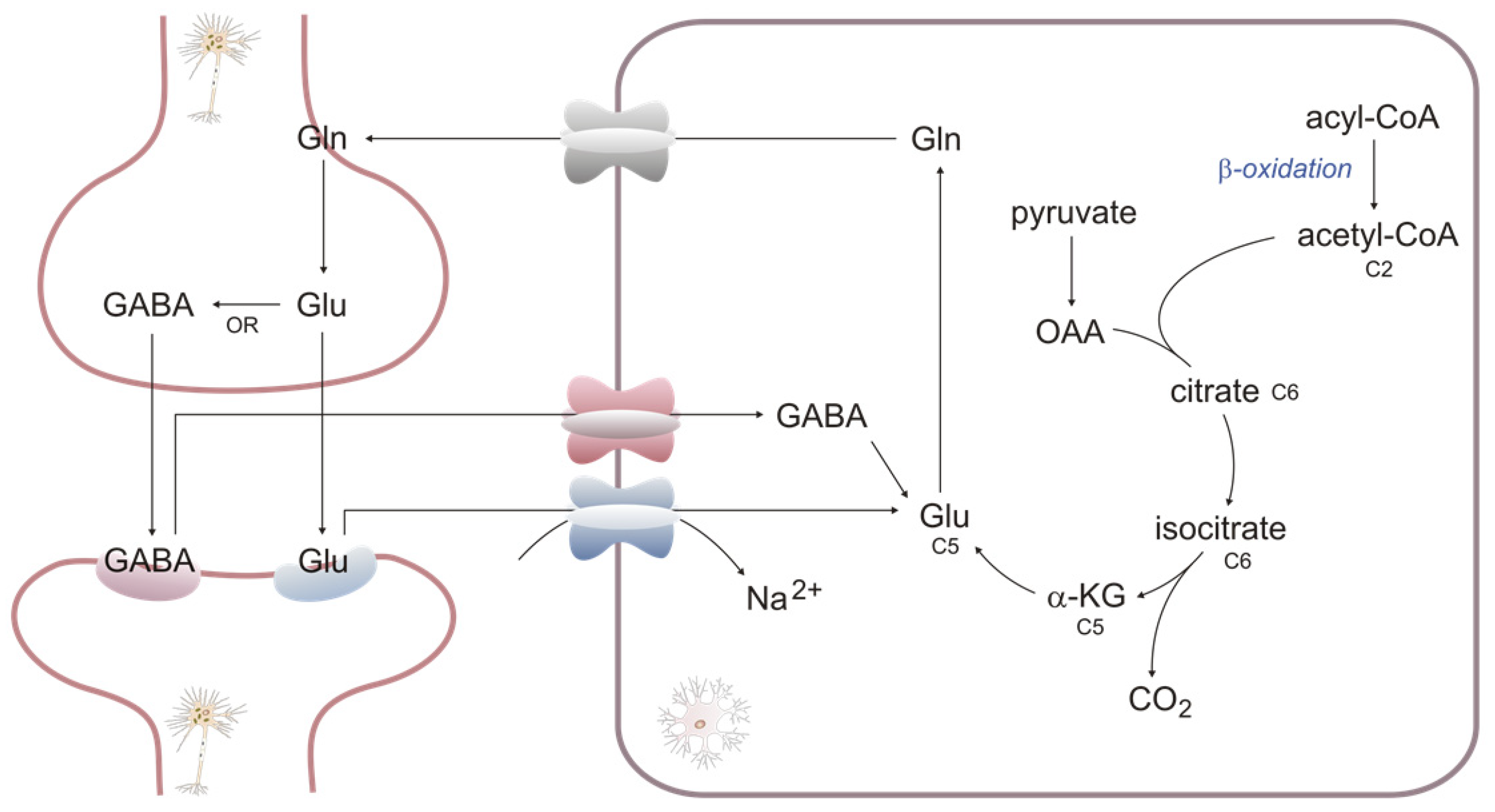
2.4. βOX as a Source of Carbons for Neurotransmitter Synthesis
2.5. βOX as a Source of Acetyl-CoA for Acetylation of Proteins
3. βOX in Neurodegenerative Diseases and Aging
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef] [PubMed]
- Tracey, T.J.; Steyn, F.J.; Wolvetang, E.J.; Ngo, S.T. Neuronal Lipid Metabolism: Multiple Pathways Driving Functional Outcomes in Health and Disease. Front. Mol. Neurosci. 2018, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.L. The Support of Energy Metabolism in the Central Nervous System with Substrates Other than Glucose. In Handbook of Neurochemistry and Molecular Neurobiology: Brain Energetics; Integration of Molecular and Cellular Processes; Springer: Boston, MA, USA, 2007. [Google Scholar]
- Mi, Y.; Qi, G.; Vitali, F.; Shang, Y.; Raikes, A.C.; Wang, T.; Jin, Y.; Brinton, R.D.; Gu, H.; Yin, F. Loss of Fatty Acid Degradation by Astrocytic Mitochondria Triggers Neuroinflammation and Neurodegeneration. Nat. Metab. 2023, 5, 445–465. [Google Scholar] [CrossRef] [PubMed]
- Dhopeshwarkar, G.A.; Subramanian, C.; Mead, J.F. Rapid Uptke of [1-14C] Acetate by the Adult Rat Brain 15 Seconds after Carotid Injection. Biochim. Et Biophys. Acta (BBA)/Lipids Lipid Metab. 1971, 248, 41–47. [Google Scholar] [CrossRef]
- Gnaedinger, J.M.; Miller, J.C.; Latker, C.H.; Rapoport, S.I. Cerebral Metabolism of Plasma [14C]Palmitate in Awake, Adult Rat: Subcellular Localization. Neurochem. Res. 1988, 13, 21–29. [Google Scholar] [CrossRef]
- Panov, A.; Orynbayeva, Z.; Vavilin, V.; Lyakhovich, V. Fatty Acids in Energy Metabolism of the Central Nervous System. Biomed. Res. Int. 2014, 2014, 472459. [Google Scholar] [CrossRef]
- Edmond, J.; Robbins, R.A.; Bergstrom, J.D.; Cole, R.A.; de Vellis, J. Capacity for Substrate Utilization in Oxidative Metabolism by Neurons, Astrocytes, and Oligodendrocytes from Developing Brain in Primary Culture. J. Neurosci. Res. 1987, 18, 551–561. [Google Scholar] [CrossRef]
- Ouellet, M.; Emond, V.; Chen, C.T.; Julien, C.; Bourasset, F.; Oddo, S.; LaFerla, F.; Bazinet, R.P.; Calon, F. Diffusion of Docosahexaenoic and Eicosapentaenoic Acids through the Blood-Brain Barrier: An in Situ Cerebral Perfusion Study. Neurochem. Int. 2009, 55, 476–482. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and Function of the Blood-Brain Barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Fitscher, B.A.; Riedel, H.D.; Young, K.C.; Stremmel, W. Tissue Distribution and CDNA Cloning of a Human Fatty Acid Transport Protein (HsFATP4). Biochim. Biophys. Acta 1998, 1443, 381–385. [Google Scholar] [CrossRef]
- Mitchell, R.W.; On, N.H.; Del Bigio, M.R.; Miller, D.W.; Hatch, G.M. Fatty Acid Transport Protein Expression in Human Brain and Potential Role in Fatty Acid Transport across Human Brain Microvessel Endothelial Cells. J. Neurochem. 2011, 117, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.N.; Ma, D.; Shui, G.; Wong, P.; Cazenave-Gassiot, A.; Zhang, X.; Wenk, M.R.; Goh, E.L.K.; Silver, D.L. Mfsd2a Is a Transporter for the Essential Omega-3 Fatty Acid Docosahexaenoic Acid. Nature 2014, 509, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Cater, R.J.; Chua, G.L.; Erramilli, S.K.; Keener, J.E.; Choy, B.C.; Tokarz, P.; Chin, C.F.; Quek, D.Q.Y.; Kloss, B.; Pepe, J.G.; et al. Structural Basis of Ω−3 Fatty Acid Transport across the Blood-Brain Barrier. Nature 2021, 595, 315. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.K.T.; Schwartz, G.J.; Rossetti, L. Hypothalamic Sensing of Fatty Acids. Nat. Neurosci. 2005, 8, 579–584. [Google Scholar] [CrossRef]
- Ebert, D.; Haller, R.G.; Walton, M.E. Energy Contribution of Octanoate to Intact Rat Brain Metabolism Measured by 13C Nuclear Magnetic Resonance Spectroscopy. J. Neurosci. 2003, 23, 5928–5935. [Google Scholar] [CrossRef]
- Xie, Z.; Jones, A.; Deeney, J.T.; Hur, S.K.; Bankaitis, V.A. Inborn Errors of Long-Chain Fatty Acid β-Oxidation Link Neural Stem Cell Self-Renewal to Autism. Cell Rep. 2016, 14, 991–999. [Google Scholar] [CrossRef]
- Knobloch, M.; Pilz, G.A.; Ghesquière, B.; Kovacs, W.J.; Wegleiter, T.; Moore, D.L.; Hruzova, M.; Zamboni, N.; Carmeliet, P.; Jessberger, S. A Fatty Acid Oxidation-Dependent Metabolic Shift Regulates Adult Neural Stem Cell Activity. Cell Rep. 2017, 20, 2144–2155. [Google Scholar] [CrossRef]
- Schönfeld, P.; Reiser, G. Why Does Brain Metabolism Not Favor Burning of Fatty Acids to Provide Energy-Reflections on Disadvantages of the Use of Free Fatty Acids as Fuel for Brain. J. Cereb. Blood Flow Metab. 2013, 33, 1493–1499. [Google Scholar] [CrossRef]
- Bird, M.I.; Munday, L.A.; Saggerson, E.D.; Clark, J.B. Carnitine Acyltransferase Activities in Rat Brain Mitochondria. Bimodal Distribution, Kinetic Constants, Regulation by Malonyl-CoA and Developmental Pattern. Biochem. J. 1985, 226, 323–330. [Google Scholar] [CrossRef]
- Schönfeld, P.; Reiser, G. How the Brain Fights Fatty Acids’ Toxicity. Neurochem. Int. 2021, 148, 105050. [Google Scholar] [CrossRef]
- Jones, L.L.; McDonald, D.A.; Borum, P.R. Acylcarnitines: Role in Brain. Prog. Lipid Res. 2010, 49, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Amemiya, F. Brain and Liver Pathology in a Patient with Carnitine Deficiency. Brain Dev. 1990, 12, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Jernberg, J.N.; Bowman, C.E.; Wolfgang, M.J.; Scafidi, S. Developmental Regulation and Localization of Carnitine Palmitoyltransferases (CPTs) in Rat Brain. J. Neurochem. 2017, 142, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Fecher, C.; Trovò, L.; Müller, S.A.; Snaidero, N.; Wettmarshausen, J.; Heink, S.; Ortiz, O.; Wagner, I.; Kühn, R.; Hartmann, J.; et al. Cell-Type-Specific Profiling of Brain Mitochondria Reveals Functional and Molecular Diversity. Nat. Neurosci. 2019, 22, 1731–1742. [Google Scholar] [CrossRef]
- Eraso-Pichot, A.; Brasó-Vives, M.; Golbano, A.; Menacho, C.; Claro, E.; Galea, E.; Masgrau, R. GSEA of Mouse and Human Mitochondriomes Reveals Fatty Acid Oxidation in Astrocytes. Glia 2018, 66, 1724–1735. [Google Scholar] [CrossRef]
- Sayre, N.L.; Sifuentes, M.; Holstein, D.; Cheng, S.Y.; Zhu, X.; Lechleiter, J.D. Stimulation of Astrocyte Fatty Acid Oxidation by Thyroid Hormone Is Protective against Ischemic Stroke-Induced Damage. J. Cereb. Blood Flow Metab. 2017, 37, 514–527. [Google Scholar] [CrossRef]
- Fitzner, D.; Bader, J.M.; Penkert, H.; Bergner, C.G.; Su, M.; Weil, M.T.; Surma, M.A.; Mann, M.; Klose, C.; Simons, M. Cell-Type- and Brain-Region-Resolved Mouse Brain Lipidome. Cell Rep. 2020, 32, 108132. [Google Scholar] [CrossRef]
- Uzor, N.E.; McCullough, L.D.; Tsvetkov, A.S. Peroxisomal Dysfunction in Neurological Diseases and Brain Aging. Front. Cell Neurosci. 2020, 14, 44. [Google Scholar] [CrossRef]
- Islinger, M.; Voelkl, A.; Fahimi, H.D.; Schrader, M. The Peroxisome: An Update on Mysteries 2.0. Histochem. Cell Biol. 2018, 150, 443–471. [Google Scholar] [CrossRef]
- Deb, R.; Joshi, N.; Nagotu, S. Peroxisomes of the Brain: Distribution, Functions, and Associated Diseases. Neurotox. Res. 2021, 39, 986–1006. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Poll-The, B.T. Role of Peroxisomes in Human Lipid Metabolism and Its Importance for Neurological Development. Neurosci. Lett. 2017, 637, 11–17. [Google Scholar] [CrossRef]
- Wanders, R.J.A. Metabolic Functions of Peroxisomes in Health and Disease. Biochimie 2014, 98, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Takahata, S. Role of Peroxisomal Fatty Acyl-CoA β-Oxidation in Phospholipid Biosynthesis. Arch. Biochem. Biophys. 1991, 284, 326–331. [Google Scholar] [CrossRef]
- Schutgens, R.B.H.; Wanders, R.J.A.; Heymans, H.S.A.; Schram, A.W.; Tager, J.M.; Schrakamp, G.; van den Bosch, H. Zellweger Syndrome: Biochemical Procedures in Diagnosis, Prevention and Treatment. J. Inherit. Metab. Dis. 1987, 10, 33–45. [Google Scholar] [CrossRef]
- Su, X.Q.; Wang, J.; Sinclair, A.J. Plasmalogens and Alzheimer’s Disease: A Review. Lipids Health Dis. 2019, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Dorninger, F.; Forss-Petter, S.; Kunze, M. Peroxisomes in Brain Development and Function. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 934–955. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, P.; Reiser, G. Brain Lipotoxicity of Phytanic Acid and Very Long-Chain Fatty Acids. Harmful Cellular/Mitochondrial Activities in Refsum Disease and X-Linked Adrenoleukodystrophy. Aging Dis. 2016, 7, 136–149. [Google Scholar] [CrossRef]
- Li, S.; Sheng, Z.H. Oligodendrocyte-Derived Transcellular Signaling Regulates Axonal Energy Metabolism. Curr. Opin. Neurobiol. 2023, 80, 102722. [Google Scholar] [CrossRef]
- López-Muguruza, E.; Matute, C. Alterations of Oligodendrocyte and Myelin Energy Metabolism in Multiple Sclerosis. Int. J. Mol. Sci. 2023, 24, 2912. [Google Scholar] [CrossRef]
- Asadollahi, E.; Trevisiol, A.; Saab, A.S.; Looser, Z.J.; Dibaj, P.; Kusch, K.; Ruhwedel, T.; Möbius, W.; Jahn, O.; Baes, M.; et al. Myelin Lipids as Nervous System Energy Reserves. bioRxiv 2022. [Google Scholar] [CrossRef]
- Flowers, A.; Bell-Temin, H.; Jalloh, A.; Stevens, S.M.; Bickford, P.C. Proteomic Anaysis of Aged Microglia: Shifts in Transcription, Bioenergetics, and Nutrient Response. J. Neuroinflamm. 2017, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xia, X.; Wang, Y.; Zheng, J.C. Mitochondrial Dysfunction in Microglia: A Novel Perspective for Pathogenesis of Alzheimer’s Disease. J. Neuroinflamm. 2022, 19. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yuan, Z.; Yang, S.; Zhu, Y.; Xue, M.; Zhang, J.; Leng, L. Brain Energy Metabolism: Astrocytes in Neurodegenerative Diseases. CNS Neurosci. Ther. 2023, 29, 24–36 . [Google Scholar] [CrossRef] [PubMed]
- Bouzier-Sore, A.K.; Pellerin, L. Unraveling the Complex Metabolic Nature of Astrocytes. Front. Cell Neurosci. 2013, 7, 179. [Google Scholar] [CrossRef]
- Drulis-Fajdasz, D.; Gizak, A.; Wójtowicz, T.; Wiśniewski, J.R.; Rakus, D. Aging-Associated Changes in Hippocampal Glycogen Metabolism in Mice. Evidence for and against Astrocyte-to-Neuron Lactate Shuttle. Glia 2018, 66, 1481–1495. [Google Scholar] [CrossRef]
- Guzmán, M.; Blázquez, C. Is There an Astrocyte- Neuron Ketone Body Shuttle? Trends Endocrinol. Metab. 2001, 12, 169–173. [Google Scholar] [CrossRef]
- Takahashi, S. Metabolic Compartmentalization between Astroglia and Neurons in Physiological and Pathophysiological Conditions of the Neurovascular Unit. Neuropathology 2020, 40, 121–137. [Google Scholar] [CrossRef]
- Thevenet, J.; De Marchi, U.; Domingo, J.S.; Christinat, N.; Bultot, L.; Lefebvre, G.; Sakamoto, K.; Descombes, P.; Masoodi, M.; Wiederkehr, A. Medium-Chain Fatty Acids Inhibit Mitochondrial Metabolism in Astrocytes Promoting Astrocyte-Neuron Lactate and Ketone Body Shuttle Systems. FASEB J. 2016, 30, 1913–1926. [Google Scholar] [CrossRef]
- Blázquez, C.; Woods, A.; De Ceballos, M.L.; Carling, D.; Guzmán, M. The AMP-Activated Protein Kinase Is Involved in the Regulation of Ketone Body Production by Astrocytes. J. Neurochem. 1999, 73, 1674–1682. [Google Scholar] [CrossRef]
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 1995, 229, 558–565. [Google Scholar] [CrossRef]
- Westhaus, A.; Blumrich, E.M.; Dringen, R. The Antidiabetic Drug Metformin Stimulates Glycolytic Lactate Production in Cultured Primary Rat Astrocytes. Neurochem. Res. 2017, 42, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Iizumi, T.; Mashima, K.; Abe, T.; Suzuki, N. Roles and Regulation of Ketogenesis in Cultured Astroglia and Neurons under Hypoxia and Hypoglycemia. ASN Neuro 2014, 6, 1759091414550997. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Ikeshima-Kataoka, H.; Kreft, M.; Vardjan, N.; Zorec, R.; Noda, M. Metabolic Plasticity of Astrocytes and Aging of the Brain. Int. J. Mol. Sci. 2019, 20, 941. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Morales, P.; Pedraza-Chaverri, J.; Tapia, E. Ketone Bodies, Stress Response, and Redox Homeostasis. Redox Biol. 2020, 29, 101395. [Google Scholar] [CrossRef] [PubMed]
- Fortier, M.; Castellano, C.A.; Croteau, E.; Langlois, F.; Bocti, C.; St-Pierre, V.; Vandenberghe, C.; Bernier, M.; Roy, M.; Descoteaux, M.; et al. A Ketogenic Drink Improves Brain Energy and Some Measures of Cognition in Mild Cognitive Impairment. Alzheimer’s Dement. 2019, 15, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Jensen, N.J.; Wodschow, H.Z.; Nilsson, M.; Rungby, J. Effects of Ketone Bodies on Brain Metabolism and Function in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8767. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain Energy Rescue: An Emerging Therapeutic Concept for Neurodegenerative Disorders of Ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Mu, J.; Wang, T.; Li, M.; Guan, T.; Guo, Y.; Zhang, X.; Zhang, G.; Kong, J. Ketogenic Diet Protects Myelin and Axons in Diffuse Axonal Injury. Nutr. Neurosci. 2022, 25, 1534–1547. [Google Scholar] [CrossRef]
- Ioannou, M.S. Current Insights into Fatty Acid Transport in the Brain. J. Membr. Biol. 2020, 253, 1–5. [Google Scholar] [CrossRef]
- Ioannou, M.S.; Jackson, J.; Sheu, S.H.; Chang, C.L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535.e14. [Google Scholar] [CrossRef]
- Rose, J.; Brian, C.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Mitochondrial Metabolism in Astrocytes Regulates Brain Bioenergetics, Neurotransmission and Redox Balance. Front. Neurosci. 2020, 14, 536682. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Wang, X.; Zhang, L.; Fang, Y.; Zheng, Q.; Liu, X.; Yu, W.; Chen, S.; Ying, J.; Hua, F. Lipid Metabolism and Storage in Neuroglia: Role in Brain Development and Neurodegenerative Diseases. Cell Biosci. 2022, 12, 106. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.; Mi, Y.; Shi, X.; Gu, H.; Brinton, R.D.; Yin, F. ApoE4 Impairs Neuron-Astrocyte Coupling of Fatty Acid Metabolism. Cell Rep. 2021, 34, 108572. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein e and Alzheimer Disease: Risk, Mechanisms and Therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Belloy, M.E.; Napolioni, V.; Greicius, M.D. A Quarter Century of APOE and Alzheimer’s Disease: Progress to Date and the Path Forward. Neuron 2019, 101, 820–838. [Google Scholar] [CrossRef]
- Qi, G.; Mi, Y.; Yin, F. Cellular Specificity and Inter-Cellular Coordination in the Brain Bioenergetic System: Implications for Aging and Neurodegeneration. Front. Physiol. 2020, 10, 1531. [Google Scholar] [CrossRef]
- Uddin, M.S.; Kabir, M.T.; Al Mamun, A.; Abdel-Daim, M.M.; Barreto, G.E.; Ashraf, G.M. APOE and Alzheimer’s Disease: Evidence Mounts That Targeting APOE4 May Combat Alzheimer’s Pathogenesis. Mol. Neurobiol. 2019, 56, 2450–2465. [Google Scholar] [CrossRef]
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An Emerging Therapeutic Target for Alzheimer’s Disease. BMC Med. 2019, 17, 64. [Google Scholar] [CrossRef]
- Rubio-Atonal, L.F.; Ioannou, M.S. Astrocytic OxPhos: More than Just Energy Production. Nat. Metab. 2023, 5, 362–363. [Google Scholar] [CrossRef]
- Cabodevilla, A.G.; Sánchez-Caballero, L.; Nintou, E.; Boiadjieva, V.G.; Picatoste, F.; Gubern, A.; Claro, E. Cell Survival during Complete Nutrient Deprivation Depends on Lipid Droplet-Fueled β-Oxidation of Fatty Acids. J. Biol. Chem. 2013, 288, 27777–27788. [Google Scholar] [CrossRef]
- Kreft, M.; Bak, L.K.; Waagepetersen, H.S.; Schousboe, A. Aspects of Astrocyte Energy Metabolism, Amino Acid Neurotransmitter Homoeostasis and Metabolic Compartmentation. ASN Neuro 2012, 4, e00086. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Lee, Y.; Lee, G.H. The Regulation of Glutamic Acid Decarboxylases in GABA Neurotransmission in the Brain. Arch. Pharm. Res. 2019, 42, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Tao, Y.; Li, M.; Che, T.; Qu, J. Protein Acetylation and Deacetylation: An Important Regulatory Modification in Gene Transcription (Review). Exp. Ther. Med. 2020, 20, 2923–2940. [Google Scholar] [CrossRef] [PubMed]
- Labarre, A.; Guitard, E.; Tossing, G.; Forest, A.; Bareke, E.; Labrecque, M.; Tétreault, M.; Ruiz, M.; Alex Parker, J. Fatty Acids Derived from the Probiotic Lacticaseibacillus Rhamnosus HA-114 Suppress Age-Dependent Neurodegeneration. Commun. Biol. 2022, 5, 1–19. [Google Scholar] [CrossRef]
- Pei, L.; Wallace, D.C. Mitochondrial Etiology of Neuropsychiatric Disorders. Biol. Psychiatry 2018, 83, 722–730. [Google Scholar] [CrossRef]
- Brown, A.; Crowe, L.; Andresen, B.S.; Anderson, V.; Boneh, A. Neurodevelopmental Profiles of Children with Very Long Chain Acyl-CoA Dehydrogenase Deficiency Diagnosed by Newborn Screening. Mol. Genet. Metab. 2014, 113, 278–282. [Google Scholar] [CrossRef]
- Strandqvist, A.; Haglind, C.B.; Zetterström, R.H.; Nemeth, A.; von Döbeln, U.; Stenlid, M.H.; Nordenström, A. Neuropsychological Development in Patients with Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase (LCHAD) Deficiency. JIMD Rep. 2016, 28, 75–84. [Google Scholar] [CrossRef]
- Barone, R.; Alaimo, S.; Messina, M.; Pulvirenti, A.; Bastin, J.; Ferro, A.; Frye, R.E.; Rizzo, R.; Fiumara, A.; Meli, C.; et al. A Subset of Patients with Autism Spectrum Disorders Show a Distinctive Metabolic Profile by Dried Blood Spot Analyses. Front. Psychiatry 2018, 9, 636. [Google Scholar] [CrossRef]
- Konttinen, H.; Gureviciene, I.; Oksanen, M.; Grubman, A.; Loppi, S.; Huuskonen, M.T.; Korhonen, P.; Lampinen, R.; Keuters, M.; Belaya, I.; et al. PPARβ/δ-Agonist GW0742 Ameliorates Dysfunction in Fatty Acid Oxidation in PSEN1ΔE9 Astrocytes. Glia 2019, 67, 146–159. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, W.; Devarakonda, T.; Zhou, H.; Wang, X.Y.; Salloum, F.N.; Spiegel, S.; Fang, X. Functional Analysis of Molecular and Pharmacological Modulators of Mitochondrial Fatty Acid Oxidation. Sci. Rep. 2020, 10, 1450. [Google Scholar] [CrossRef]
- Lin, H.; Patel, S.; Affeck, V.S.; Wilson, I.; Turnbull, D.M.; Joshi, A.R.; Maxwell, R.; Stoll, E.A. Fatty Acid Oxidation Is Required for the Respiration and Proliferation of Malignant Glioma Cells. Neuro Oncol. 2017, 19, 43–54. [Google Scholar] [CrossRef]
- Bogie, J.F.J.; Haidar, M.; Kooij, G.; Hendriks, J.J.A. Fatty Acid Metabolism in the Progression and Resolution of CNS Disorders. Adv. Drug Deliv. Rev. 2020, 159, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Toomey, C.E.; Heywood, W.E.; Evans, J.R.; Lachica, J.; Pressey, S.N.; Foti, S.C.; Al Shahrani, M.; D’Sa, K.; Hargreaves, I.P.; Heales, S.; et al. Mitochondrial Dysfunction Is a Key Pathological Driver of Early Stage Parkinson’s. Acta Neuropathol. Commun. 2022, 10, 134. [Google Scholar] [CrossRef]
- Polyzos, A.A.; Lee, D.Y.; Datta, R.; Hauser, M.; Budworth, H.; Holt, A.; Mihalik, S.; Goldschmidt, P.; Frankel, K.; Trego, K.; et al. Metabolic Reprogramming in Astrocytes Distinguishes Region-Specific Neuronal Susceptibility in Huntington Mice. Cell Metab. 2019, 29, 1258–1273.e11. [Google Scholar] [CrossRef]
- Bertapelle, C.; Carillo, M.R.; Cacciola, N.A.; Shidlovskii, Y.V.; Peluso, G.; Digilio, F.A. The Reversible Carnitine Palmitoyltransferase 1 Inhibitor (Teglicar) Ameliorates the Neurodegenerative Phenotype in a Drosophila Huntington’s Disease Model by Acting on the Expression of Carnitine-Related Genes. Molecules 2022, 27, 3125. [Google Scholar] [CrossRef]
- Hofmann, S.; Kedersha, N.; Anderson, P.; Ivanov, P. Molecular Mechanisms of Stress Granule Assembly and Disassembly. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118876. [Google Scholar] [CrossRef]
- Amen, T.; Kaganovich, D. Stress Granules Inhibit Fatty Acid Oxidation by Modulating Mitochondrial Permeability. Cell Rep. 2021, 35, 109237. [Google Scholar] [CrossRef]
- Yin, F. Lipid Metabolism and Alzheimer’s Disease: Clinical Evidence, Mechanistic Link and Therapeutic Promise. FEBS J. 2023, 290, 1420–1453. [Google Scholar] [CrossRef]
- Wehbe, Z.; Tucci, S. Therapeutic Potential of Triheptanoin in Metabolic and Neurodegenerative Diseases. J. Inherit. Metab. Dis. 2020, 43, 385–391. [Google Scholar] [CrossRef]
- Adanyeguh, I.; Rinaldi, D.; Henry, P.; Caillet, S.; Valabregue, R.; Durr, A.; Mochel, F. N04 Anaplerotic Therapy Using Triheptanoin Improves Brain Energy Metabolism In Patients With Huntington Disease. J. Neurol. Neurosurg. Psychiatry 2014, 85, A103. [Google Scholar] [CrossRef]
- Cotto, B.; Natarajaseenivasan, K.; Langford, D. Astrocyte Activation and Altered Metabolism in Normal Aging, Age-Related CNS Diseases, and HAND. J. Neurovirol. 2019, 25, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Laranjeira, A.; Schulz, J.; Dotti, C.G. Genes Related to Fatty Acid β-Oxidation Play a Role in the Functional Decline of the Drosophila Brain with Age. PLoS ONE 2016, 11, e0161143. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szrok-Jurga, S.; Turyn, J.; Hebanowska, A.; Swierczynski, J.; Czumaj, A.; Sledzinski, T.; Stelmanska, E. The Role of Acyl-CoA β-Oxidation in Brain Metabolism and Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 13977. https://doi.org/10.3390/ijms241813977
Szrok-Jurga S, Turyn J, Hebanowska A, Swierczynski J, Czumaj A, Sledzinski T, Stelmanska E. The Role of Acyl-CoA β-Oxidation in Brain Metabolism and Neurodegenerative Diseases. International Journal of Molecular Sciences. 2023; 24(18):13977. https://doi.org/10.3390/ijms241813977
Chicago/Turabian StyleSzrok-Jurga, Sylwia, Jacek Turyn, Areta Hebanowska, Julian Swierczynski, Aleksandra Czumaj, Tomasz Sledzinski, and Ewa Stelmanska. 2023. "The Role of Acyl-CoA β-Oxidation in Brain Metabolism and Neurodegenerative Diseases" International Journal of Molecular Sciences 24, no. 18: 13977. https://doi.org/10.3390/ijms241813977
APA StyleSzrok-Jurga, S., Turyn, J., Hebanowska, A., Swierczynski, J., Czumaj, A., Sledzinski, T., & Stelmanska, E. (2023). The Role of Acyl-CoA β-Oxidation in Brain Metabolism and Neurodegenerative Diseases. International Journal of Molecular Sciences, 24(18), 13977. https://doi.org/10.3390/ijms241813977