
Unraveling the Impact of Acetylation Patterns in Chitosan Oligomers on Cu2+ Ion Binding: Insights from DFT Calculations



Abstract
:1. Introduction
- i
- Electronic structure of the GlcN monomer (D) and its copper-bound complex (D-Cu = M) were investigated regarding its coordination and assessing the stability of bridge and pendant model.
- ii
- GlcN- and GlcNAc (A)-containing dimers (D-D, A-D, D-A) were studied to analyze the effect of reducing and non-reducing ends on copper binding (M-D, D-M, A-M, M-A).
- iii
- Chitosan trimers (D-D-D, A-D-D, D-D-A, A-D-A) were analyzed to define the combined effect of reducing and non-reducing ends on the metal binding subunit (M-D-D, D-M-D, D-D-M, A-M-D, D-M-A, A-M-A).
- iv
- Chitosan pentamers (D-A-D-D-D, D-D-D-A-D, D-A-D-A-D) were investigated to define the influence of the pattern of acetylation, i.e., of neighboring deacetylated or acetylated units, on copper binding (D-A-M-D-D, D-D-M-A-D, D-A-M-A-D) without the effects of reducing and non-reducing ends.
2. Results and Discussion
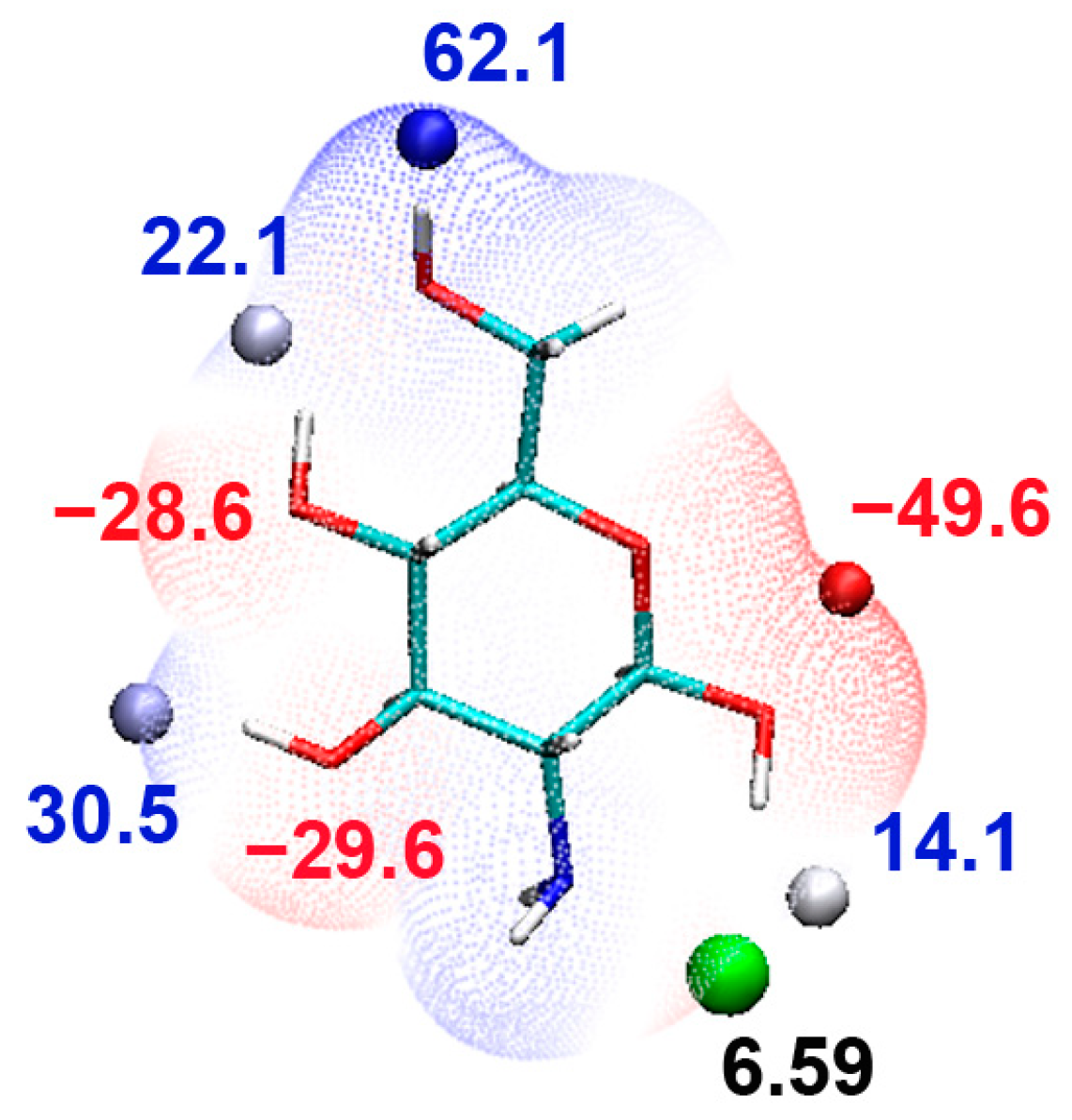
2.1. Monomer
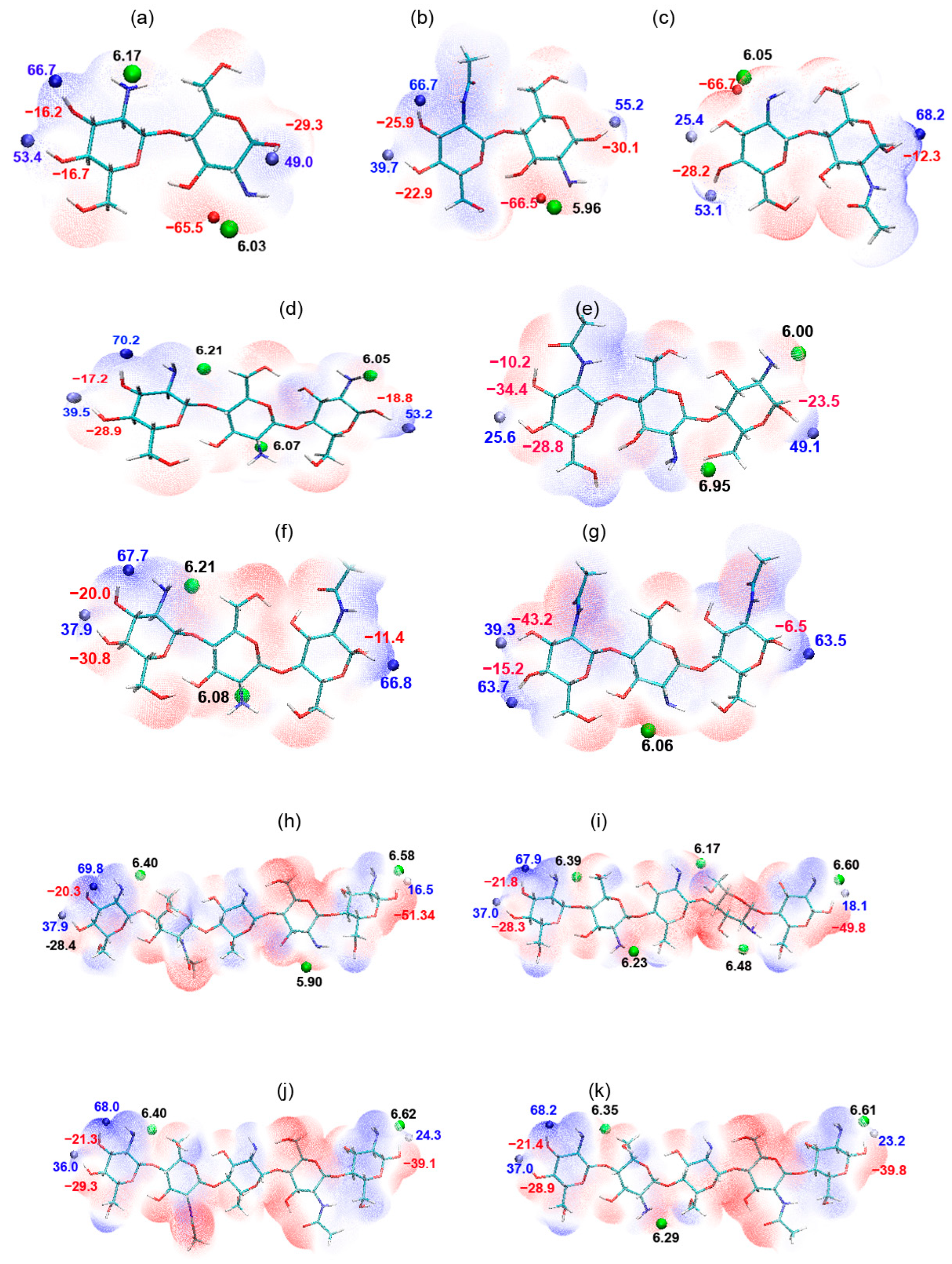
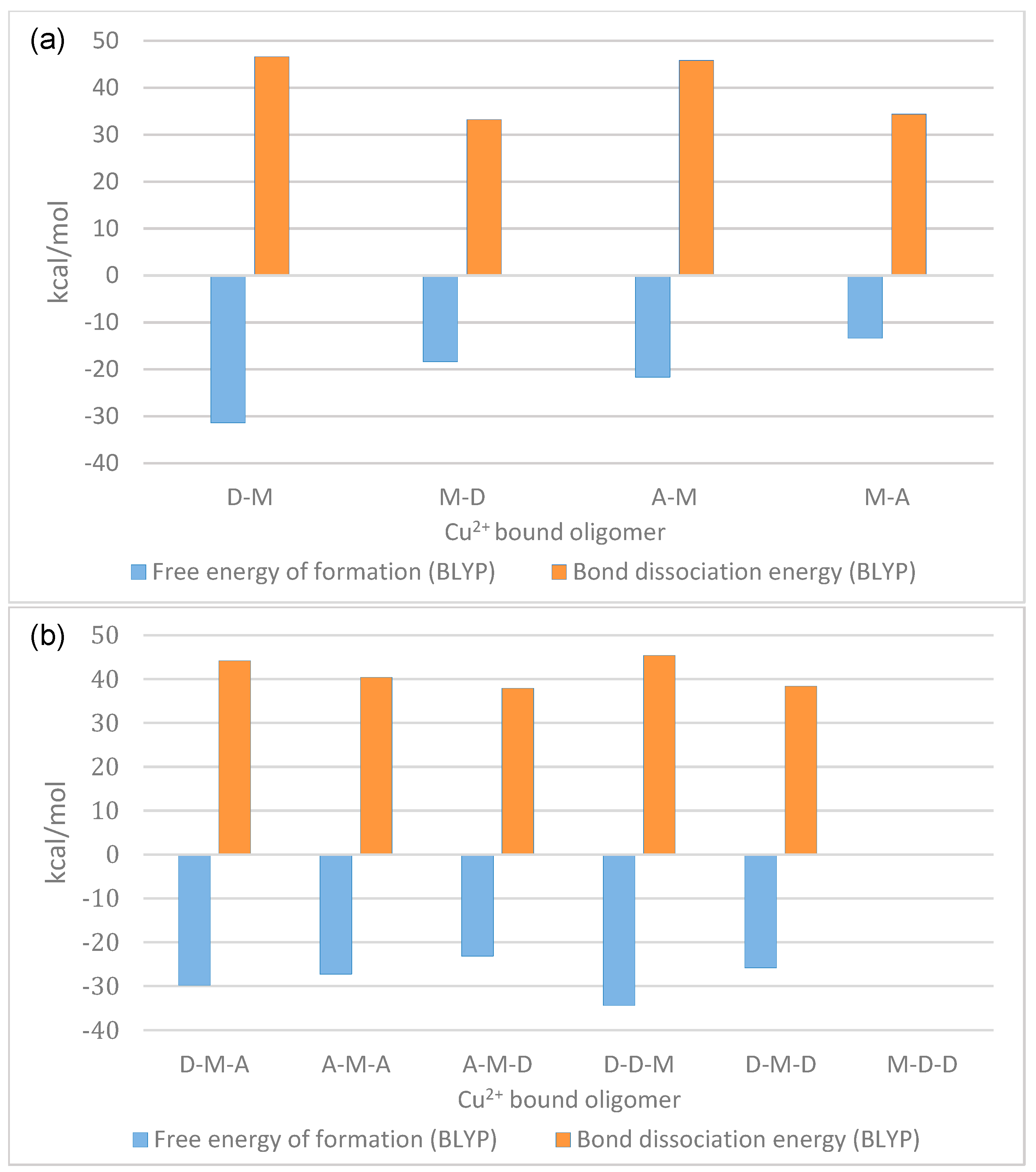
2.2. Dimers
2.3. Trimers
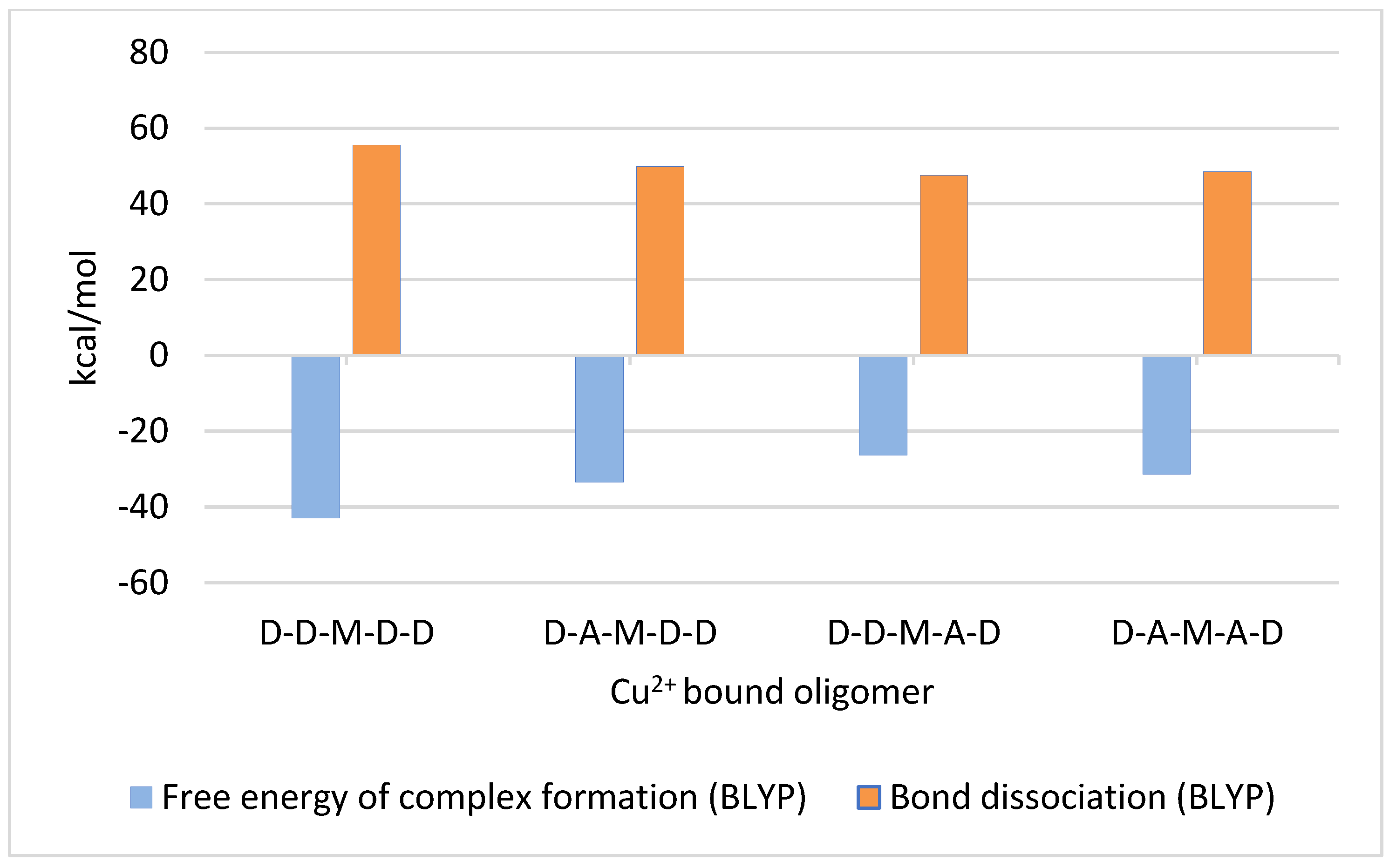
2.4. Pentamer
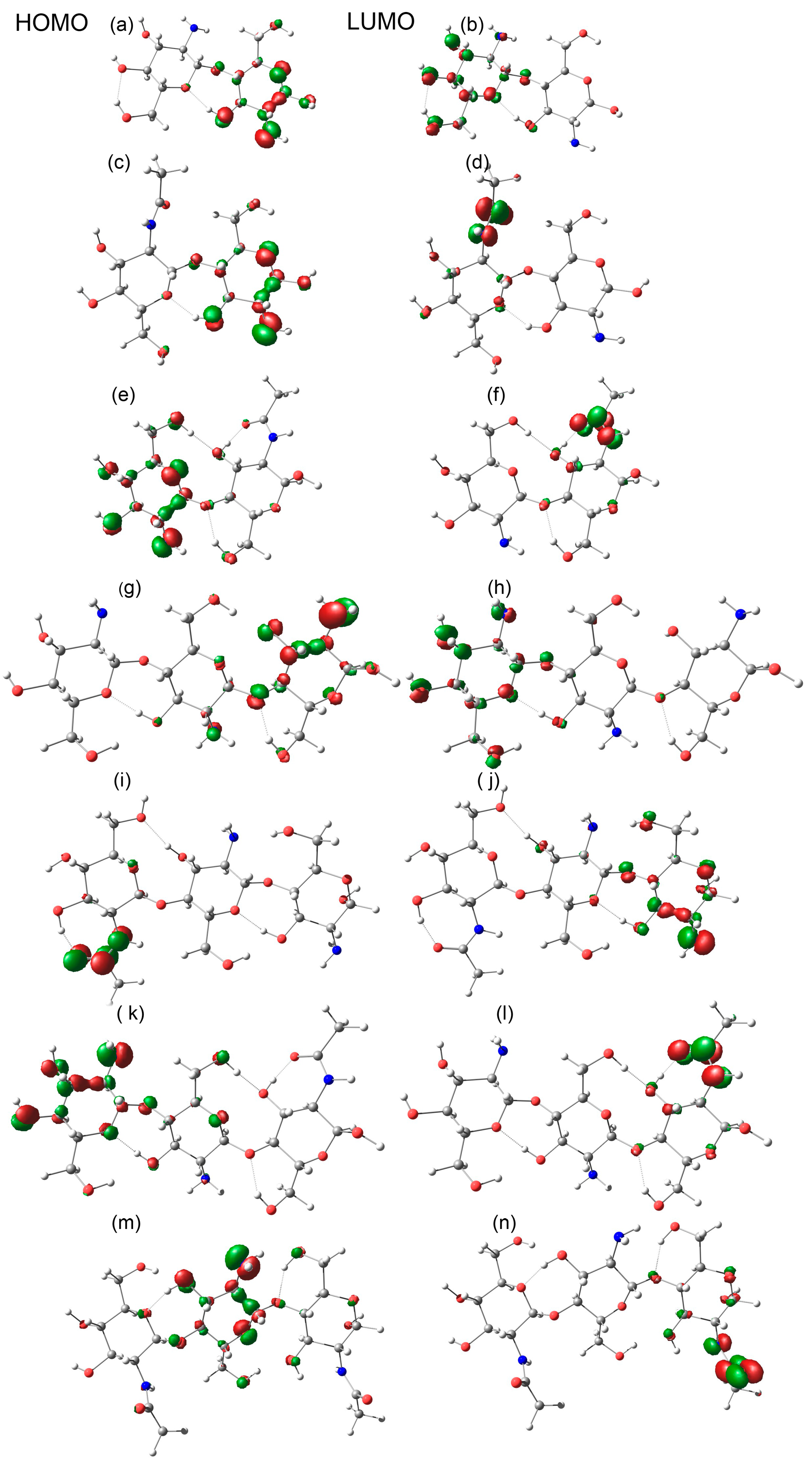
2.5. Energy Gap
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lamichhane, J.R.; Osdaghi, E.; Behlau, F.; Köhl, J.; Jones, J.B.; Aubertot, J.N. Thirteen decades of antimicrobial copper compounds applied in agriculture. A review. Agron. Sustain. Dev. 2018, 38, 28. [Google Scholar] [CrossRef]
- Sorenson, J.R.J. Antiarthritic, Antiulcer, and Analgesic Activities of Copper Complexes. In Trace Elements in Clinical Medicine; Springer: Tokyo, Japan, 1990; pp. 261–268. [Google Scholar] [CrossRef]
- Dutta, P.K.; Ravikumar, M.N.V.; Dutta, J. Chitin and chitosan for versatile applications. J. Macromol. Sci. Polym. Rev. 2002, 42, 307–354. [Google Scholar] [CrossRef]
- Brunel, F.; El Gueddari, N.E.; Moerschbacher, B.M. Complexation of copper(II) with chitosan nanogels: Toward control of microbial growth. Carbohydr. Polym. 2013, 92, 1348–1356. [Google Scholar] [CrossRef] [PubMed]
- Wattjes, J.; Sreekumar, S.; Richter, C.; Cord-Landwehr, S.; Singh, R.; El Gueddari, N.E.; Moerschbacher, B.M. Patterns matter part 1: Chitosan polymers with non-random patterns of acetylation. React. Funct. Polym. 2020, 151, 104583. [Google Scholar] [CrossRef]
- Domard, A. pH and c.d. measurements on a fully deacetylated chitosan: Application to CuII-polymer interactions. Int. J. Biol. Macromol. 1987, 9, 98–104. [Google Scholar] [CrossRef]
- Lü, R.; Cao, Z.; Shen, G. Comparative study on interaction between copper (II) and chitin/chitosan by density functional calculation. J. Mol. Struct. THEOCHEM 2008, 860, 80–85. [Google Scholar] [CrossRef]
- Terreux, R.; Domard, M.; Viton, C.; Domard, A. Interactions study between the copper II ion and constitutive elements of chitosan structure by DFT calculation. Biomacromolecules 2006, 7, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kurth, S.; Marques, M.A.L.; Gross, E.K.U. Density-Functional Theory. In Encyclopedia of Condensed Matter Physics; Elsevier Inc.: Amsterdam, The Netherlands, 2005; pp. 395–402. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Filippi, C.; Gonze, X.; Umrigar, C.J. Generalized gradient approximations to density functional theory: Comparison with exact results. Theor. Comput. Chem. 1996, 4, 295–326. [Google Scholar] [CrossRef]
- Fabiano, E.; Trevisanutto, P.E.; Terentjevs, A.; Constantin, L.A. Generalized gradient approximation correlation energy functionals based on the uniform electron gas with gap model. J. Chem. Theory Comput. 2014, 10, 2016–2026. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.P.; Roos, B.O.; Ryde, U. Performance of density functionals for first row transition metal systems. J. Chem. Phys. 2007, 126, 014103. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Zapata, J.C.; McKemmish, L.K. Computation of Dipole Moments: A Recommendation on the Choice of the Basis Set and the Level of Theory. J. Phys. Chem. A 2020, 124, 7538–7548. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Schaftenaar, G.; Noordik, J.H. Molden: A pre- and post-processing program for molecular and electronic structures. J. Comput. Aided. Mol. Des. 2000, 14, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Huang, C.C.; Ferrin, T.E. Software extensions to UCSF chimera for interactive visualization of large molecular assemblies. Structure 2005, 13, 473–482. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligomer | HOMO (eV) | LUMO (eV) | Bandgap (eV) |
|---|---|---|---|
| D | −5.324 | 0.498 | 5.823 |
| DD | −5.040 | 0.339 | 5.387 |
| DA | −5.049 | −0.688 | 4.355 |
| AD | −5.039 | −0.376 | 4.662 |
| DDD | −5.173 | 0.413 | 5.587 |
| ADD | −5.193 | −0.621 | 4.572 |
| DDA | −5.210 | −0.685 | 4.473 |
| ADA | −5.103 | −0.439 | 4.663 |
| DDDD | −5.229 | 0.4111 | 5.641 |
| DDDDD | −4.986 | 0.415 | 5.401 |
| DADAD | −4.898 | −0.294 | 4.682 |
| DDDAD | −5.185 | −0.527 | 4.658 |
| DADDD | −5.202 | −0.519 | 4.603 |
| DDDDDD | −4.974 | 0.482 | 5.457 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, R.; Smiatek, J.; Moerschbacher, B.M. Unraveling the Impact of Acetylation Patterns in Chitosan Oligomers on Cu2+ Ion Binding: Insights from DFT Calculations. Int. J. Mol. Sci. 2023, 24, 13792. https://doi.org/10.3390/ijms241813792
Singh R, Smiatek J, Moerschbacher BM. Unraveling the Impact of Acetylation Patterns in Chitosan Oligomers on Cu2+ Ion Binding: Insights from DFT Calculations. International Journal of Molecular Sciences. 2023; 24(18):13792. https://doi.org/10.3390/ijms241813792
Chicago/Turabian StyleSingh, Ratna, Jens Smiatek, and Bruno M. Moerschbacher. 2023. "Unraveling the Impact of Acetylation Patterns in Chitosan Oligomers on Cu2+ Ion Binding: Insights from DFT Calculations" International Journal of Molecular Sciences 24, no. 18: 13792. https://doi.org/10.3390/ijms241813792
APA StyleSingh, R., Smiatek, J., & Moerschbacher, B. M. (2023). Unraveling the Impact of Acetylation Patterns in Chitosan Oligomers on Cu2+ Ion Binding: Insights from DFT Calculations. International Journal of Molecular Sciences, 24(18), 13792. https://doi.org/10.3390/ijms241813792