
Crystal Structures of Bacterial Pectin Methylesterases Pme8A and PmeC2 from Rumen Butyrivibrio

 , , ,
, , , 
Abstract
:1. Introduction
2. Results and Discussion
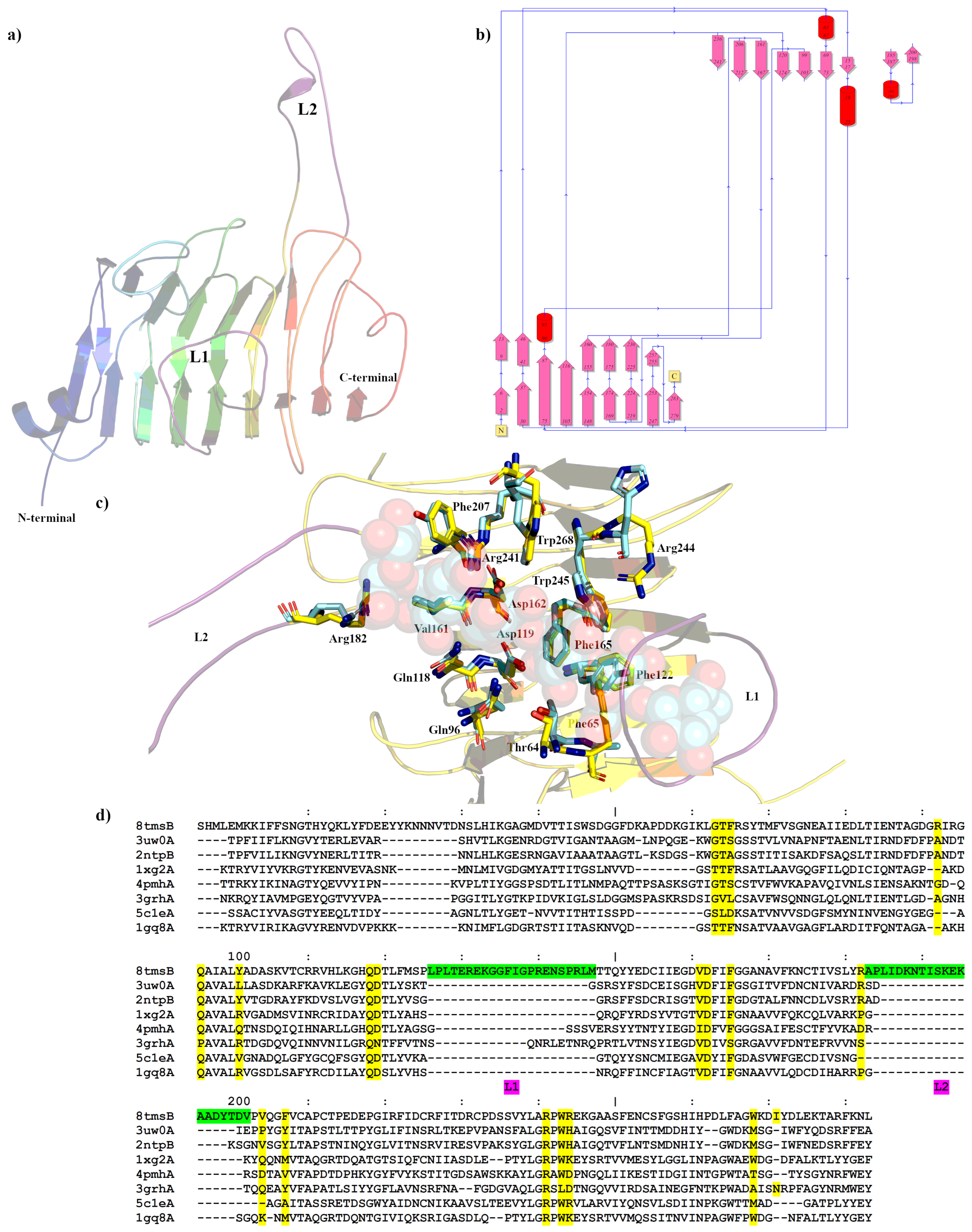
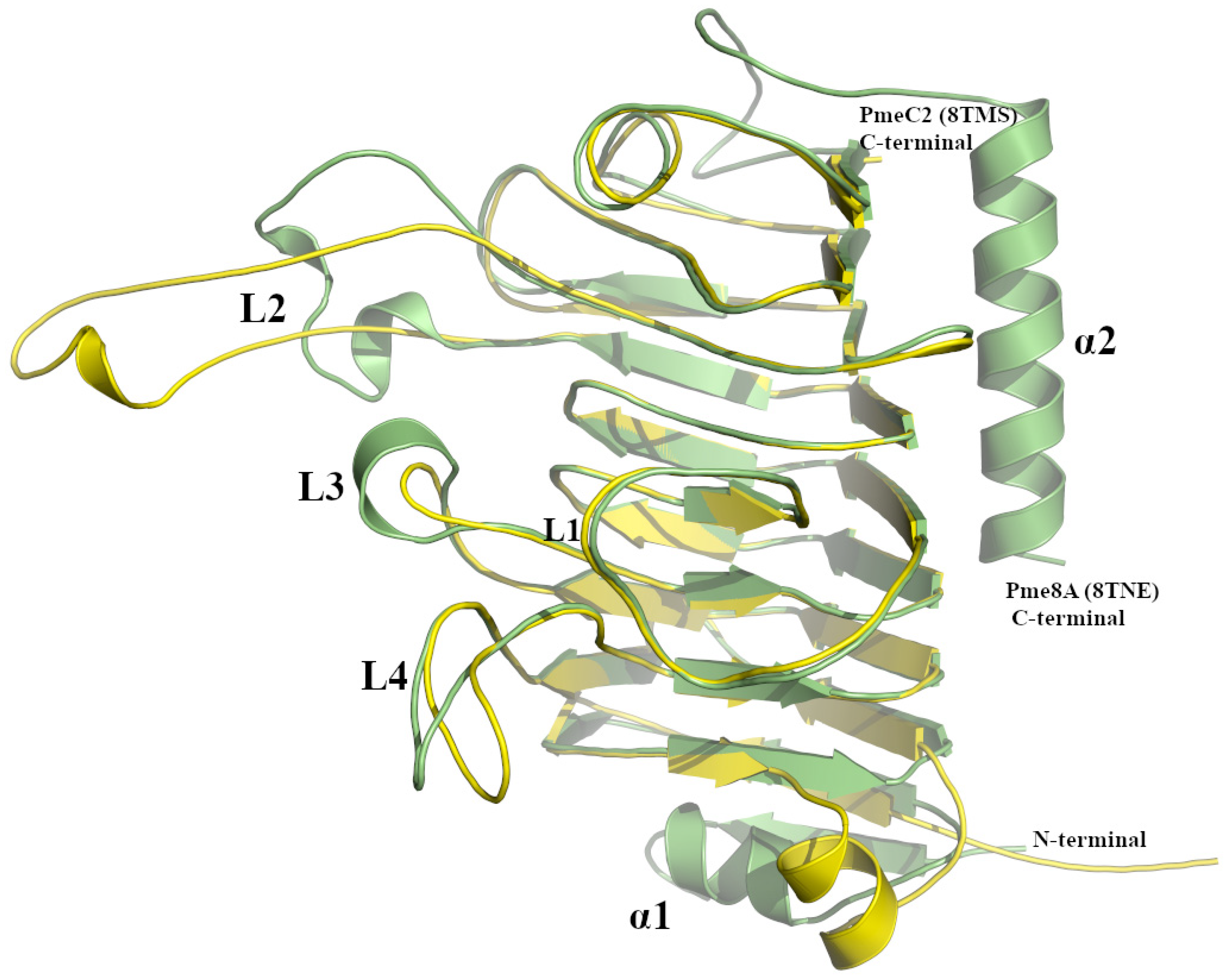
2.1. Analysis of the Pme8A Structure from Butyrivibrio proteoclasticus B316T
2.2. Analysis of the PmeC2 Structure from Butyrivibrio fibrisolvens D1T
2.3. PME Activity in Butyrivibrio Pme8A and PmeC2
3. Materials and Methods
3.1. Molecular Modelling
3.2. Protein Expression and Purification
3.3. Crystallization
3.4. Data Collection and Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morgavi, D.P.; Forano, E.; Martin, C.; Newbold, C.J. Microbial ecosystem and methanogenesis in ruminants. Animal 2010, 4, 1024–1036. [Google Scholar] [CrossRef]
- Czerkawski, J. Methane production in ruminants and its significance. World Rev. Nutr. Diet. 1969, 11, 240–282. [Google Scholar]
- Janssen, P.H. Influence of hydrogen on rumen methane formation and fermentation balances through microbial growth kinetics and fermentation thermodynamics. Anim. Feed Sci. Technol. 2010, 160, 1–22. [Google Scholar] [CrossRef]
- Kelly, W.J.; Leahy, S.C.; Kamke, J.; Soni, P.; Koike, S.; Mackie, R.; Seshadri, R.; Cook, G.M.; Morales, S.E.; Greening, C.; et al. Occurrence and expression of genes encoding methyl-compound production in rumen bacteria. Anim. Microbiome 2019, 1, 1–13. [Google Scholar] [CrossRef]
- Kelly, W.J.; Mackie, R.I.; Attwood, G.T.; Janssen, P.H.; McAllister, T.A.; Leahy, S.C. Hydrogen and formate production and utilisation in the rumen and the human colon. Anim. Microbiome 2022, 4, 22. [Google Scholar] [CrossRef]
- Leahy, S.C.; Janssen, P.H.; Attwood, G.T.; Mackie, R.I.; McAllister, T.A.; Kelly, W.J. Electron flow: Key to mitigating ruminant methanogenesis. Trends Microbiol. 2022, 30, 209–212. [Google Scholar] [CrossRef]
- Bailey, R. Pasture quality and ruminant nutrition: I. Carbohydrate composition of ryegrass varieties grown as sheep pastures. N. Z. J. Agric. Res. 1964, 7, 497–507. [Google Scholar] [CrossRef]
- Vogel, J. Unique aspects of the grass cell wall. Curr. Opin. Plant Biol. 2008, 11, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, D.J. Growth of the plant cell wall. Nat. Rev. Mol. Cell Biol. 2005, 6, 850–861. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, M.C. Structure and properties of pectin gels in plant cell walls. Plant Cell Environ. 1984, 7, 153–164. [Google Scholar]
- Ralet, M.-C.; Lerouge, P.; Quéméner, B. Mass spectrometry for pectin structure analysis. Carbohydr. Res. 2009, 344, 1798–1807. [Google Scholar] [CrossRef] [PubMed]
- Carpita, N.C. Structure and biogenesis of the cell walls of grasses. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996, 47, 445–476. [Google Scholar] [CrossRef] [PubMed]
- Micheli, F. Pectin methylesterases: Cell wall enzymes with important roles in plant physiology. Trends Plant Sci. 2001, 6, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Pelloux, J.; Rusterucci, C.; Mellerowicz, E.J. New insights into pectin methylesterase structure and function. Trends Plant Sci. 2007, 12, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Seedorf, H.; Kittelmann, S.; Janssen, P.H. Few highly abundant operational taxonomic units dominate within rumen methanogenic archaeal species in New Zealand sheep and cattle. Appl. Environ. Microbiol. 2015, 81, 986–995. [Google Scholar] [CrossRef]
- Blackburn, T.; Hobson, P. Further studies on the isolation of proteolytic bacteria from the sheep rumen. Microbiology 1962, 29, 69–81. [Google Scholar] [CrossRef]
- Maia, M.R.; Chaudhary, L.C.; Figueres, L.; Wallace, R.J. Metabolism of polyunsaturated fatty acids and their toxicity to the microflora of the rumen. Antonie Van Leeuwenhoek 2007, 91, 303–314. [Google Scholar] [CrossRef]
- McKain, N.; Shingfield, K.J.; Wallace, R.J. Metabolism of conjugated linoleic acids and 18: 1 fatty acids by ruminal bacteria: Products and mechanisms. Microbiology 2010, 156, 579–588. [Google Scholar] [CrossRef]
- Seshadri, R.; Leahy, S.C.; Attwood, G.T.; Teh, K.H.; Lambie, S.C.; Cookson, A.L.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Hadjithomas, M.; Varghese, N.J.; et al. Cultivation and sequencing of rumen microbiome members from the Hungate1000 Collection. Nat. Biotechnol. 2018, 36, 359–367. [Google Scholar] [CrossRef]
- Sengupta, K.; Hivarkar, S.S.; Palevich, N.; Chaudhary, P.P.; Dhakephalkar, P.K.; Dagar, S.S. Genomic architecture of three newly isolated unclassified Butyrivibrio species elucidate their potential role in the rumen ecosystem. Genomics 2022, 114, 110281. [Google Scholar] [CrossRef]
- Kelly, W.J.; Leahy, S.C.; Altermann, E.; Yeoman, C.J.; Dunne, J.C.; Kong, Z.; Pacheco, D.M.; Li, D.; Noel, S.J.; Moon, C.D.; et al. The glycobiome of the rumen bacterium Butyrivibrio proteoclasticus B316T highlights adaptation to a polysaccharide-rich environment. PLoS ONE 2010, 5, e11942. [Google Scholar] [CrossRef] [PubMed]
- Palevich, N.; Kelly, W.J.; Ganesh, S.; Rakonjac, J.; Attwood, G.T. Butyrivibrio hungatei MB2003 competes effectively for soluble sugars released by Butyrivibrio proteoclasticus B316T during growth on xylan or pectin. Appl. Environ. Microbiol. 2019, 85, e02056-18. [Google Scholar] [CrossRef] [PubMed]
- Marounek, M.; Dušková, D. Metabolism of pectin in rumen bacteria Butyrivibrio fibrisolvens and Prevotella ruminicola. Lett. Appl. Microbiol. 1999, 29, 429–433. [Google Scholar] [CrossRef]
- Gradel, C.M.; Dehority, B. Fermentation of isolated pectin and pectin from intact forages by pure cultures of rumen bacteria. Appl. Microbiol. 1972, 23, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Dehority, B. Pectin-fermenting bacteria isolated from the bovine rumen. J. Bacteriol. 1969, 99, 189–196. [Google Scholar] [CrossRef]
- Hespell, R.B.; O’Bryan-Shah, P. Esterase activities in Butyrivibrio fibrisolvens strains. Appl. Environ. Microbiol. 1988, 54, 1917–1922. [Google Scholar] [CrossRef]
- Wojciechowicz, M.; Heinrichova, K.; Ziolecki, A. An exopectate lyase of Butyrivibrio fibrisolvens from the bovine rumen. Microbiology 1982, 128, 2661–2665. [Google Scholar] [CrossRef]
- Kasperowicz, A. Comparison of utilization of pectins from various sources by pure cultures of pectinolytic rumen bacteria and mixed cultures of rumen microorganisms. Acta Microbiol. Pol. 1994, 43, 47–56. [Google Scholar]
- Moon, C.D.; Pacheco, D.M.; Kelly, W.J.; Leahy, S.C.; Li, D.; Kopecny, J.; Attwood, G.T. Reclassification of Clostridium proteoclasticum as Butyrivibrio proteoclasticus comb. nov., a butyrate-producing ruminal bacterium. Int. J. Syst. Evol. Microbiol. 2008, 58, 2041–2045. [Google Scholar] [CrossRef]
- Dunne, J.C.; Li, D.; Kelly, W.J.; Leahy, S.C.; Bond, J.J.; Attwood, G.T.; Jordan, T.W. Extracellular polysaccharide-degrading proteome of Butyrivibrio proteoclasticus. J. Proteome Res. 2012, 11, 131–142. [Google Scholar] [CrossRef]
- Dunne, J.C.; Kelly, W.J.; Leahy, S.C.; Li, D.; Bond, J.J.; Peng, L.; Attwood, G.T.; Jordan, T.W. The cytosolic oligosaccharide-degrading proteome of Butyrivibrio proteoclasticus. Proteomes 2015, 3, 347–368. [Google Scholar] [CrossRef] [PubMed]
- Palevich, N.; Kelly, W.J.; Leahy, S.C.; Denman, S.; Altermann, E.; Rakonjac, J.; Attwood, G.T. Comparative genomics of rumen Butyrivibrio spp. uncovers a continuum of polysaccharide-degrading capabilities. Appl. Environ. Microbiol. 2019, 86, e01993-19. [Google Scholar] [CrossRef] [PubMed]
- Pidcock, S.E.; Skvortsov, T.; Santos, F.G.; Courtney, S.J.; Sui-Ting, K.; Creevey, C.J.; Huws, S.A. Phylogenetic systematics of Butyrivibrio and Pseudobutyrivibrio genomes illustrate vast taxonomic diversity, open genomes and an abundance of carbohydrate-active enzyme family isoforms. Microb. Genom. 2021, 7, 000638. [Google Scholar] [CrossRef]
- D’Avino, R.; Camardella, L.; Christensen, T.M.; Giovane, A.; Servillo, L. Tomato pectin methylesterase: Modeling, fluorescence, and inhibitor interaction studies-comparison with the bacterial (Erwinia chrysanthemi) enzyme. Proteins 2003, 53, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Giovane, A.; Servillo, L.; Balestrieri, C.; Raiola, A.; D’Avino, R.; Tamburrini, M.; Ciardiello, M.; Camardella, L. Pectin methylesterase inhibitor. Biochim. Biophys. Acta BBA-Proteins Proteom. 2004, 1696, 245–252. [Google Scholar] [CrossRef]
- Lionetti, V.; Raiola, A.; Camardella, L.; Giovane, A.; Obel, N.; Pauly, M.; Favaron, F.; Cervone, F.; Bellincampi, D. Overexpression of pectin methylesterase inhibitors in Arabidopsis restricts fungal infection by Botrytis cinerea. Plant Physiol. 2007, 143, 1871–1880. [Google Scholar] [CrossRef]
- Jenkins, J.; Mayans, O.; Smith, D.; Worboys, K.; Pickersgill, R.W. Three-dimensional structure of Erwinia chrysanthemi pectin methylesterase reveals a novel esterase active site. J. Mol. Biol. 2001, 305, 951–960. [Google Scholar] [CrossRef]
- Jenkins, J.; Pickersgill, R. The architecture of parallel β-helices and related folds. Prog. Biophys. Mol. Biol. 2001, 77, 111–175. [Google Scholar] [CrossRef]
- Holm, L.; Kääriäinen, S.; Wilton, C.; Plewczynski, D. Using Dali for Structural Comparison of Proteins. Curr. Protoc. Bioinform. 2006, 14, 5.5.1–5.5.24. [Google Scholar] [CrossRef]
- Fries, M.; Ihrig, J.; Brocklehurst, K.; Shevchik, V.E.; Pickersgill, R.W. Molecular basis of the activity of the phytopathogen pectin methylesterase. EMBO J. 2007, 26, 3879–3887. [Google Scholar] [CrossRef]
- Di Matteo, A.; Giovane, A.; Raiola, A.; Camardella, L.; Bonivento, D.; De Lorenzo, G.; Cervone, F.; Bellincampi, D.; Tsernoglou, D. Structural basis for the interaction between pectin methylesterase and a specific inhibitor protein. Plant Cell 2005, 17, 849–858. [Google Scholar] [CrossRef]
- Johansson, K.; El-Ahmad, M.; Friemann, R.; Jornvall, H.; Markovic, O.; Eklund, H. Crystal structure of plant pectin methylesterase. FEBS Lett. 2002, 514, 243–249. [Google Scholar] [CrossRef]
- Teller, D.C.; Behnke, C.A.; Pappan, K.; Shen, Z.; Reese, J.C.; Reeck, G.R.; Stenkamp, R.E. The structure of rice weevil pectin methylesterase. Acta Crystallogr. Sect. F Biol. Crystallogr. Commun. 2014, 70, 1480–1484. [Google Scholar] [CrossRef] [PubMed]
- Boraston, A.B.; Abbott, D.W. Structure of a pectin methylesterase from Yersinia enterocolitica. Acta Crystallogr. Sect. F Biol. Crystallogr. Commun. 2012, 68, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Kent, L.M.; Loo, T.S.; Melton, L.D.; Mercadante, D.; Williams, M.A.; Jameson, G.B. Structure and properties of a non-processive, salt-requiring, and acidophilic pectin methylesterase from Aspergillus niger provide insights into the key determinants of processivity control. J. Biol. Chem. 2016, 291, 1289–1306. [Google Scholar] [CrossRef] [PubMed]
- Eklof, J.M.; Tan, T.C.; Divne, C.; Brumer, H. The crystal structure of the outer membrane lipoprotein YbhC from Escherichia coli sheds new light on the phylogeny of carbohydrate esterase family 8. Proteins 2009, 76, 1029–1036. [Google Scholar] [CrossRef]
- Guillerm, J.; Frere, J.M.; Meersman, F.; Matagne, A. The Right-Handed Parallel beta-Helix Topology of Erwinia chrysanthemi Pectin Methylesterase Is Intimately Associated with Both Sequential Folding and Resistance to High Pressure. Biomolecules 2021, 11, 1083. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM-A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Cardozo, T.; Totrov, M.; Abagyan, R. Homology modeling by the ICM method. Proteins Struct. Funct. Bioinform. 1995, 23, 403–414. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 133–144. [Google Scholar] [CrossRef]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.; Teplyakov, A. MOLREP: An Automated Program for Molecular Replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| B. proteoclasticus B316T Pme8A (8TNE) | B. fibrisolvens D1T PmeC2 (8TMS) | |
|---|---|---|
| Space group | C 1 2 1 | P1 |
| Unit cell parameters: | ||
| a, b, c (Å) | 228.80, 49.27, 100.59 | 48.64, 76.28, 96.78 |
| α, β, γ (°) | 90.00, 101.03, 90.00 | 98.36, 104.16, 90.05 |
| Data collection statistics | ||
| Wavelength (Å) | 0.95374 | 0.95372 |
| Temperature (K) | 100 | 100 |
| Resolution Range (Å) | 48.76–2.30 | 47.13–2.30 |
| No. of observed ref. * | 327,785 (19,428) | 206,403 (16,208) |
| No. of unique ref. * | 48,330 (3213) | 56,000 (4465) |
| Rsym a | 0.098 (1.167) | 0.119 (1.121) |
| Rpim b | 0.061 (0.751) | 0.119 (1.121) |
| Completeness (%) * | 97.3 (71.0) | 95.1 (92.3) |
| Multiplicity * | 6.8 (6.0) | 3.7 (3.6) |
| I/σ(I) * | 8.8 (1.2) | 5.4 (0.6) |
| CC1/2 * | 0.997 (0.693) | 0.994 (0.349) |
| Refinement statistics | ||
| Resolution range (Å) | 48.17–2.30 | 47.170–2.305 |
| All reflections used | 49,652 | 58,852 |
| Size Rfree set (%) | 5 | 5 |
| All reflections (Rfree) | 2459 | 2934 |
| Rcryst (%) | 20.01 | 20.45 |
| Rfree (%) | 24.95 | 24.01 |
| Matthews coefficient (Å3 Da−1) | 2.48 | 2.57 |
| Solvent content (%) | 50 | 51.7 |
| RMSD ** | ||
| Rms Bond Length (Å) | 0.0069 | 0.0066 |
| Rms Bond Angle (°) | 1.3925 | 1.2985 |
| Ramachandran plot | ||
| Residues in favoured regions (%) | 96.8 | 96.2 |
| Residues in allowed regions (%) | 3.2 | 3.8 |
| Average B factors (Å2) | ||
| Protein | ||
| Chain A | 59.616 | 39.231 |
| Chain B | 66.726 | 42.158 |
| Chain C | 82.988 | 48.131 |
| Chain D | 48.853 | |
| Water (HOH) | 54.260 | 36.499 |
| Cl− | - | 56.625 |
| Organism | Class | PDB Monomer | Z-Score a | RMSD b | Lali c | %id d | |
|---|---|---|---|---|---|---|---|
| 8A | Solanum lycopersicum Gene Names: PME1.9 EC: 3.1.1.11 | Pectinesterase | 1xg2-A | 37.9 | 2.0 | 288 | 27 |
| Dickeya dadantii 3937 Gene Names: pmeA, pme, Dda3937_03374 EC: 3.1.1.11 | Pectinesterase | 2ntp-B | 37.5 | 2.2 | 292 | 28 | |
| Daucus carota EC: 3.1.1.11 | Pectinesterase | 1gq8-A | 35.8 | 1.9 | 287 | 28 | |
| Yersinia enterocolitica subsp. enterocolitica 8081 Gene Names: YE0549 EC: 3.1.1.11 | Pectinesterase | 3uw0-A | 35.1 | 2.2 | 280 | 28 | |
| Aspergillus niger ATCC 1015 Gene Names: ASPNIDRAFT_214857 EC: 3.1.1.11 | Pectinesterase | 5c1e-A | 34.8 | 2.0 | 273 | 27 | |
| C2 | Yersinia enterocolitica subsp. enterocolitica 8081 Gene Names: YE0549 EC: 3.1.1.11 | Pectinesterase | 3uw0-A | 29.8 | 2.0 | 232 | 31 |
| Dickeya dadantii 3937 Gene Names: pmeA, pme, Dda3937_03374 EC: 3.1.1.11 | Pectinesterase | 2ntp-B | 29.7 | 2.2 | 232 | 32 | |
| Solanum lycopersicum Gene Names: PME1.9 EC: 3.1.1.11 | Pectinesterase | 1xg2-A | 28.7 | 1.9 | 224 | 31 | |
| Sitophilus oryzae Gene Names: CE8-1 EC: 3.1.1.11 | Pectinesterase | 4pmh-A | 28.1 | 2.0 | 238 | 26 | |
| Escherichia coli K-12 Gene Names: b0772, JW0755, ybhC EC: 3.1.2 | Acyl-coa thioester hydrolase ybgc | 3grh-A | 28.0 | 2.3 | 242 | 21 | |
| Aspergillus niger ATCC 1015 Gene Names: ASPNIDRAFT_214857 EC: 3.1.1.11 | Pectinesterase | 5c1e-A | 27.4 | 1.6 | 213 | 30 | |
| Daucus carota EC: 3.1.1.11 | Pectinesterase | 1gq8-A | 27.1 | 1.7 | 223 | 26 |
| Class Summary | Nominal Pectin Binding Residues | |||||
|---|---|---|---|---|---|---|
| Subsite +4 | Subsite +3 | Subsite +2 | Subsite +1 | Subsite −1 | Subsite −2 | |
| PME (2NTP) Dickeya dadantii 3937 EC: 3.1.1.11 Pectin methylesterase 366 amino acids | Asn226 Val227 | Tyr230 Arg219 Val198 | Arg267 Gln177 | Gln177 Asp178 Asp199 Met306 | Trp269 Thr272 * Thr109 Gln153 Asp178 Phe202 | Thr109 Ala110 Tyr181 |
| Pme8A (8TNE) Butyrivibrio proteoclasticus B316T Carbohydrate Esterase Family 8 Pectin methylesterase EC: 3.1.1.11 341 amino acids | Phe216 Ile217 | Tyr220 Arg202 Val181 | Arg255 Gln138 | Gln138 Asp139 Asp182 Trp282 | Trp257 Arg258 * Thr84 Gln116 Asp139 Phe185 | Thr84 Phe85 Phe142 |
| PmeC2 (8TMS) Butyrivibrio fibrisolvens D1T Carbohydrate Esterase Family 8 Pectin methylesterase EC: 3.1.1.11 283 amino acids | Pro203 Val204 | Phe207 Arg182 Val161 | Arg241 Gln118 | Gln118 Asp119 Asp162 Trp268 | Trp243 Arg244 * Thr64 Gln96 Asp119 Phe165 | Thr64 Phe65 Phe122 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carbone, V.; Reilly, K.; Sang, C.; Schofield, L.R.; Ronimus, R.S.; Kelly, W.J.; Attwood, G.T.; Palevich, N. Crystal Structures of Bacterial Pectin Methylesterases Pme8A and PmeC2 from Rumen Butyrivibrio. Int. J. Mol. Sci. 2023, 24, 13738. https://doi.org/10.3390/ijms241813738
Carbone V, Reilly K, Sang C, Schofield LR, Ronimus RS, Kelly WJ, Attwood GT, Palevich N. Crystal Structures of Bacterial Pectin Methylesterases Pme8A and PmeC2 from Rumen Butyrivibrio. International Journal of Molecular Sciences. 2023; 24(18):13738. https://doi.org/10.3390/ijms241813738
Chicago/Turabian StyleCarbone, Vincenzo, Kerri Reilly, Carrie Sang, Linley R. Schofield, Ron S. Ronimus, William J. Kelly, Graeme T. Attwood, and Nikola Palevich. 2023. "Crystal Structures of Bacterial Pectin Methylesterases Pme8A and PmeC2 from Rumen Butyrivibrio" International Journal of Molecular Sciences 24, no. 18: 13738. https://doi.org/10.3390/ijms241813738
APA StyleCarbone, V., Reilly, K., Sang, C., Schofield, L. R., Ronimus, R. S., Kelly, W. J., Attwood, G. T., & Palevich, N. (2023). Crystal Structures of Bacterial Pectin Methylesterases Pme8A and PmeC2 from Rumen Butyrivibrio. International Journal of Molecular Sciences, 24(18), 13738. https://doi.org/10.3390/ijms241813738