


RNA-Seq and Comparative Transcriptomic Analyses of Asian Soybean Rust Resistant and Susceptible Soybean Genotypes Provide Insights into Identifying Disease Resistance Genes

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
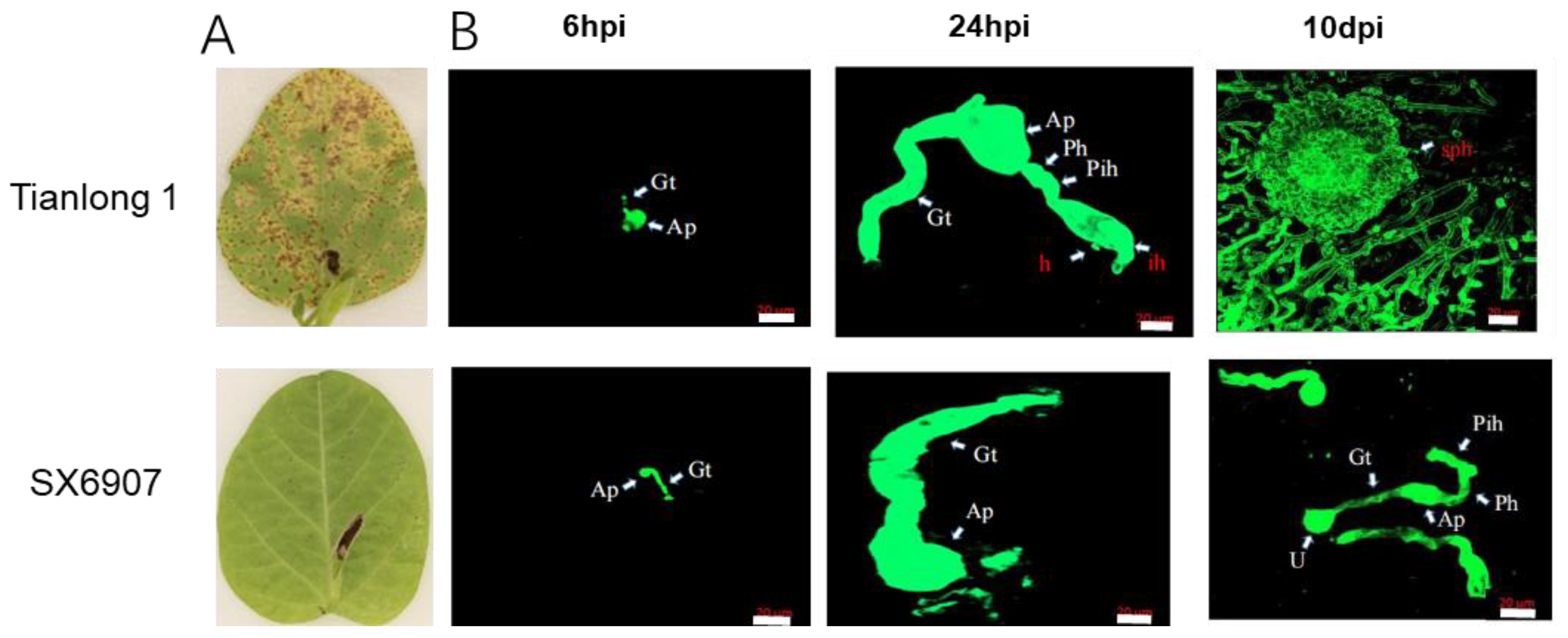
2.1. The Phenotypic Response of Soybean Accessions to P. pachyrhizi
2.2. Analysis of RNA-Seq Data
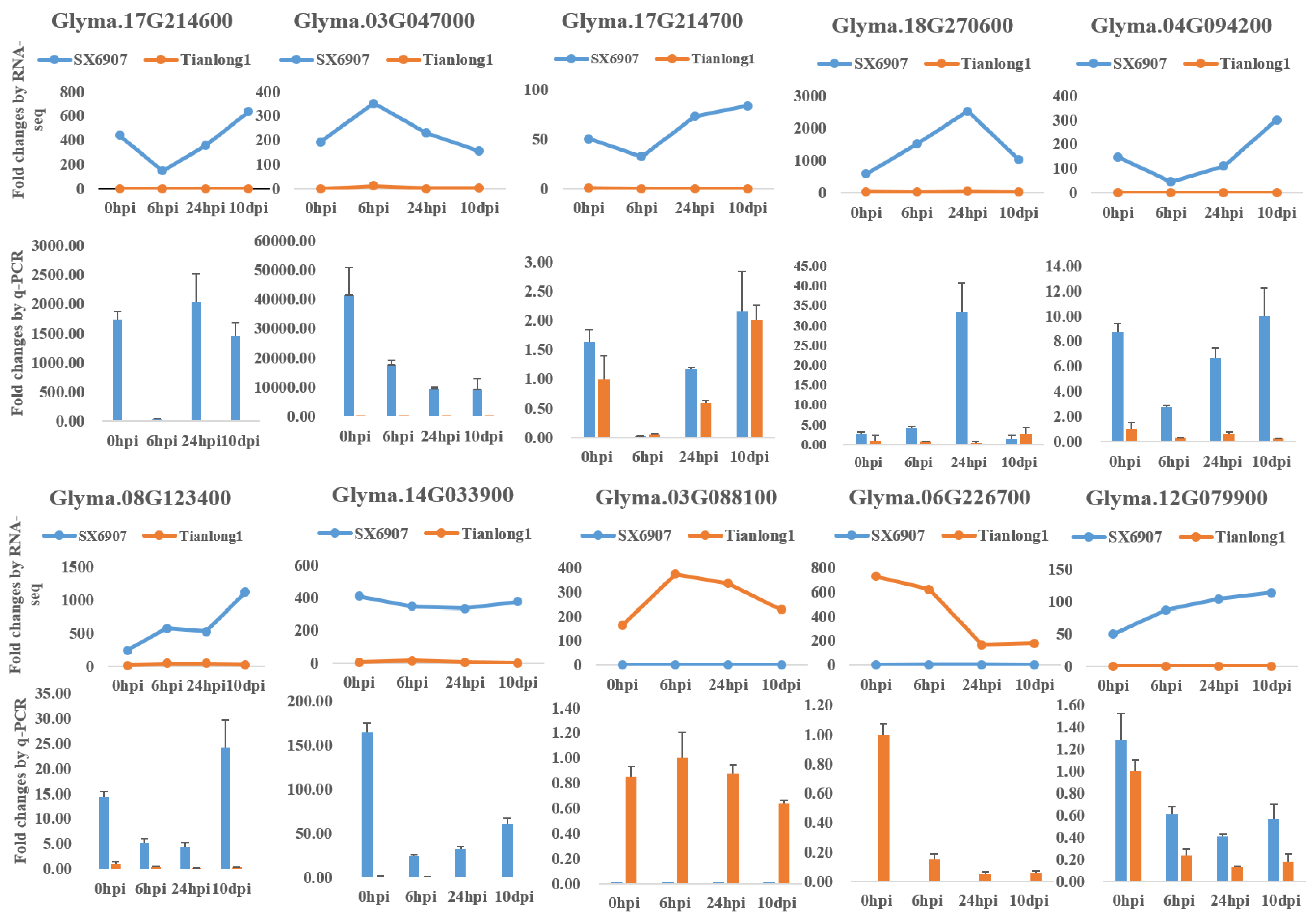
2.3. Validation of Selected Differentially Expressed Genes (DEGs) Using qRT-PCR
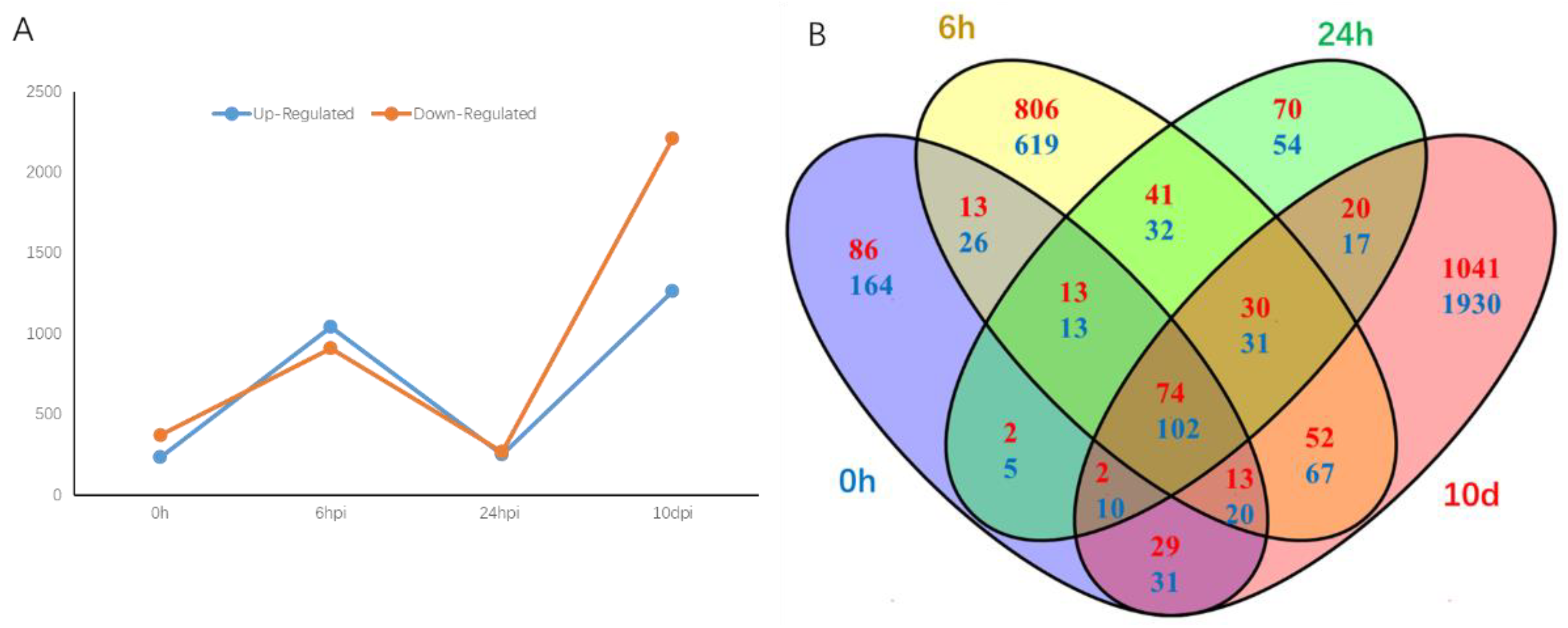
2.4. Comparative Analysis of Tianlong1 and SX6907 Transcriptomes
2.5. Gene Ontology (GO)-Based Analysis of the DEGs
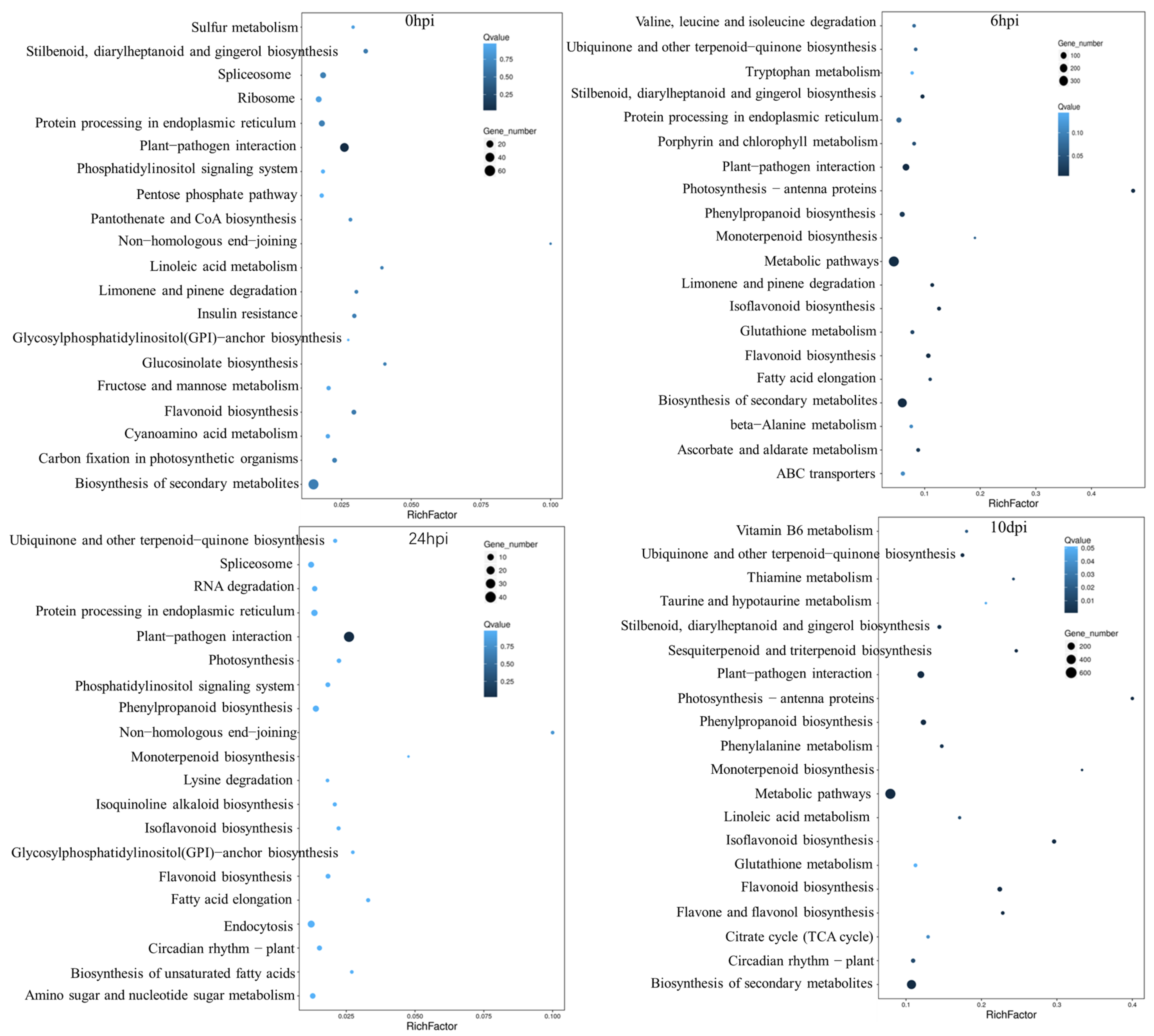
2.6. KEGG Pathway Analysis of the DEGs
2.7. Identification of Transcription Factors
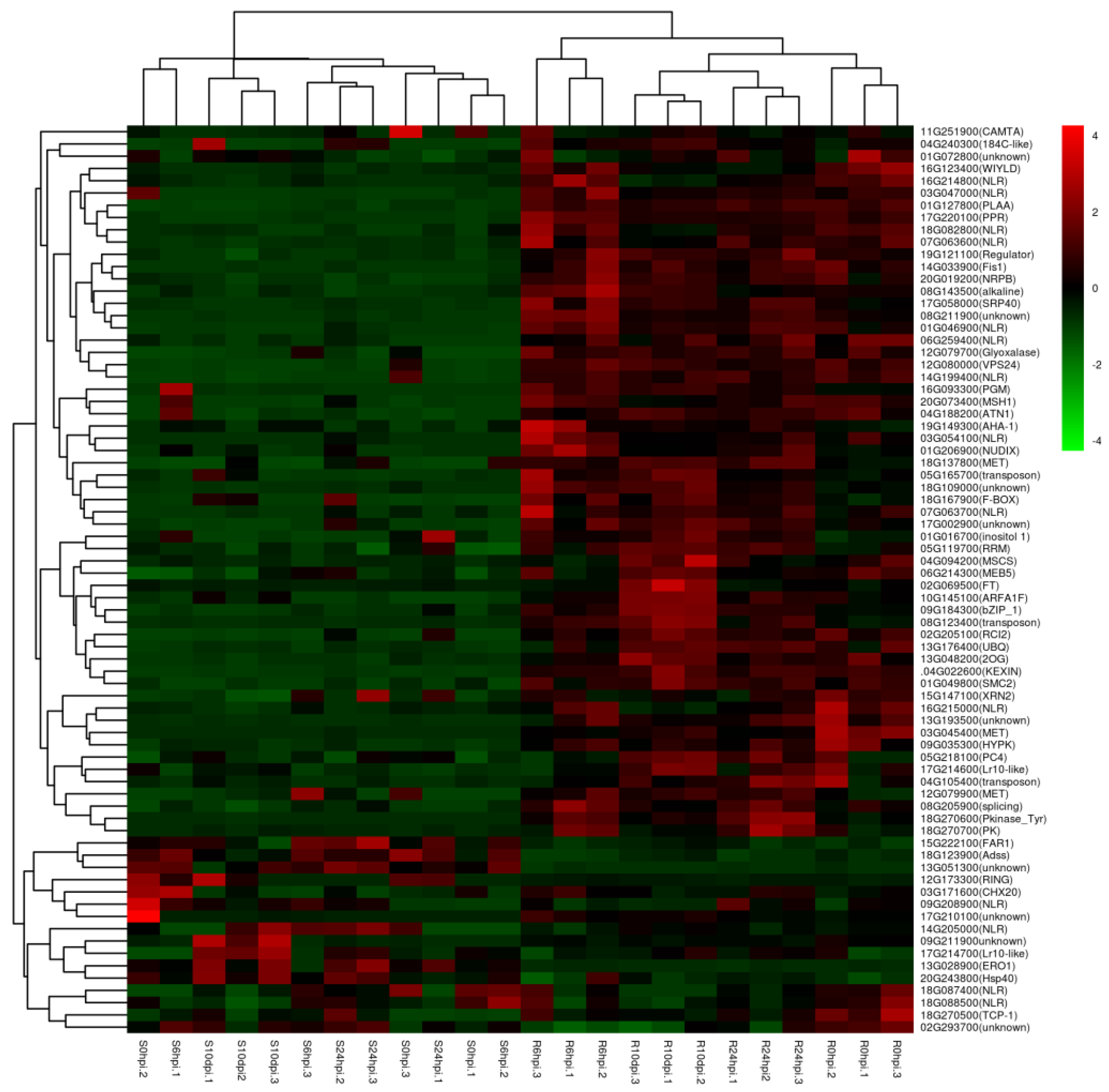
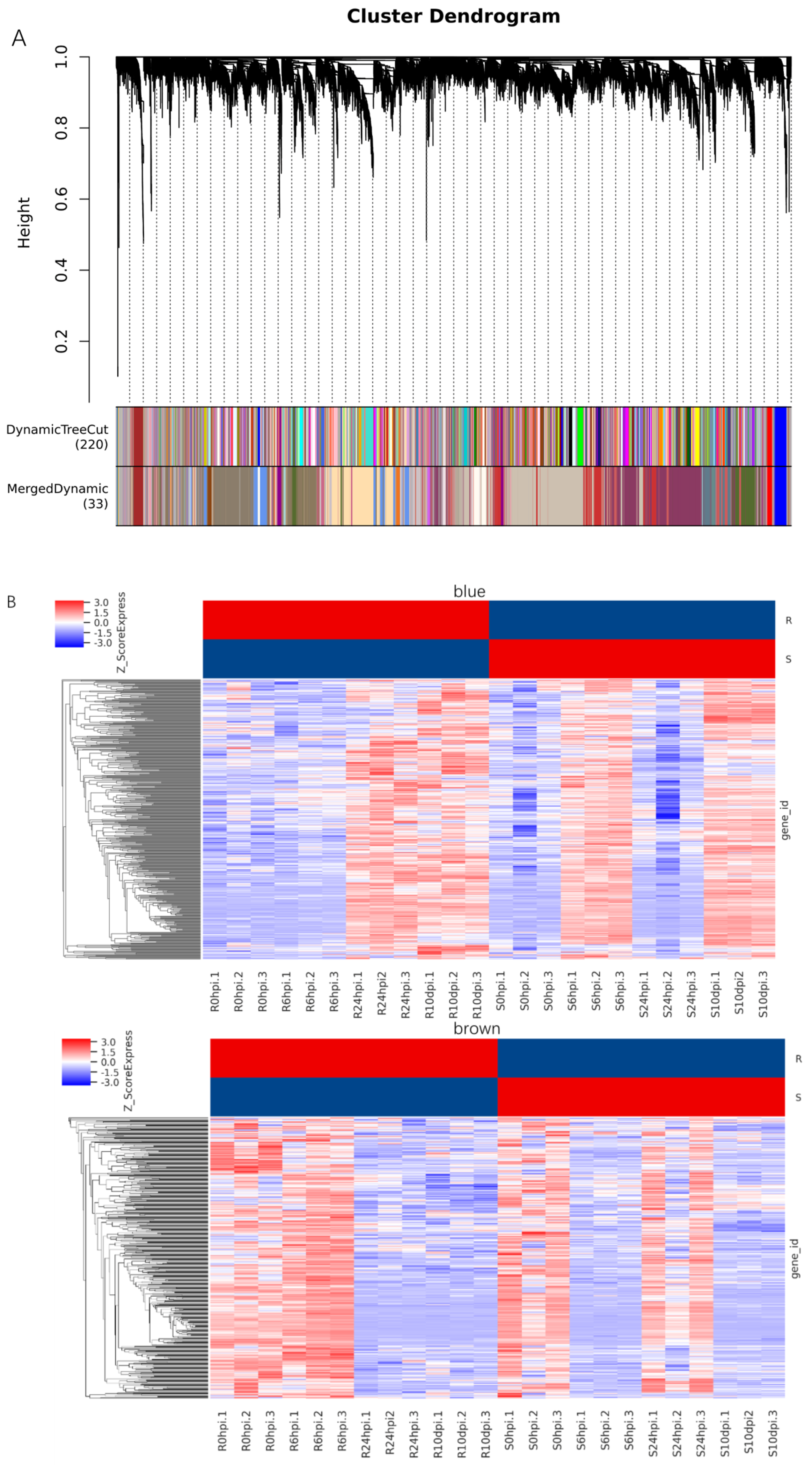
2.8. Co-Expression Network Analysis of Soybean Resistance to P. pachyrhizi
3. Discussion
3.1. Ca2+ Signaling Pathway
3.2. MAPK Signaling Pathway
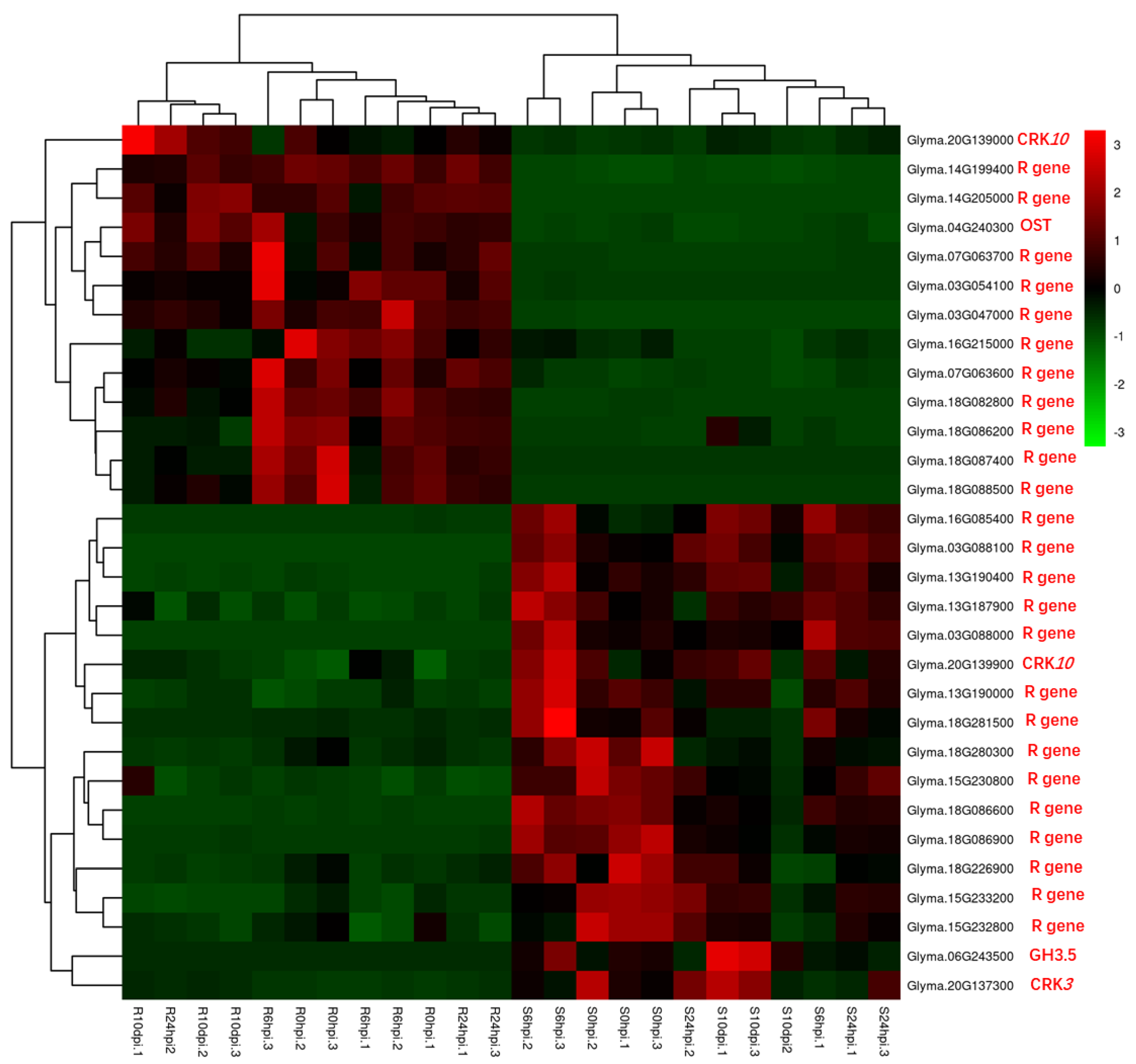
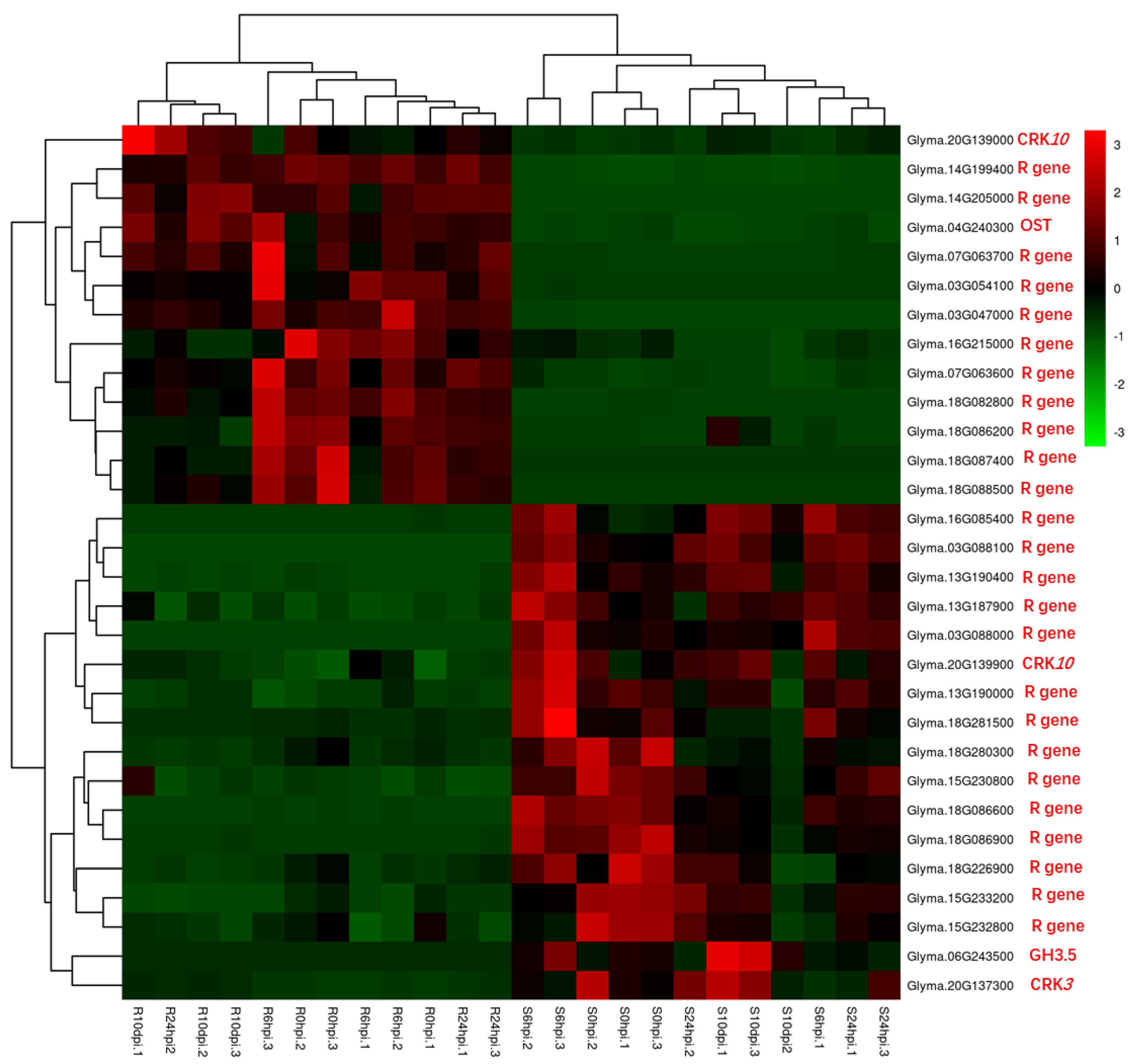
3.3. NLR Genes
3.4. Flavonoid Biosynthesis and Disease Resistance
4. Materials and Methods
4.1. Plant Genotypes, Inoculations, and Experimental Conditions
4.2. Observation by Laser Confocal Microscopy
4.3. RNA Extraction
4.4. Library Preparation and Illumina Hiseq Sequencing
4.5. Reads Quality Control and Mapping
4.6. Differential Expression Analysis and Functional Enrichment
4.7. WGCNA Construct
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hossain, M.Z.; Ishiga, Y.; Yamanaka, N.; Ogiso-Tanaka, E.; Yamaoka, Y. Soybean leaves transcriptomic data dissects the phenylpropanoid pathway genes as a defence response against Phakopsora pachyrhizi. Plant Physiol. Biochem. 2018, 132, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Slaminko, T.L.; Miles, M.R.; Frederick, R.D.; Bonde, M.R.; Hartman, G.L. New legume hosts of Phakopsora pachyrhizi based on greenhouse evaluations. Plant Dis. 2008, 92, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, H.; Yamanaka, N.; Soares, R.M.; Ivancovich, A.; Kato, M. Pathogenic variation of south American Phakopsora pachyrhizi populations isolated from soybeans from 2010 to 2015. Jpn. Agric. Res. Q. Jarq 2017, 51, 221–232. [Google Scholar] [CrossRef]
- Godoy, C.; de Freitas Bueno, A.; Gazziero, D. Brazilian soybean pest management and threats to its sustainability. Outlooks Pest Manag. 2015, 26, 113–117. [Google Scholar] [CrossRef]
- Vuong, T.D.; Walker, D.R.; Nguyen, B.T.; Nguyen, T.T.; Dinh, H.X.; Hyten, D.L.; Cregan, P.B.; Sleper, D.A.; Lee, J.D.; Shannon, J.G.; et al. Molecular characterization of resistance to soybean rust (Phakopsora pachyrhizi Syd. & Syd.) in soybean cultivar DT 2000 (PI 635999). PLoS ONE 2016, 11, e0164493. [Google Scholar] [CrossRef]
- Shan, Z.H.; Chen, H.F.; Zhao, S.; Yang, Z.L.; Yang, Y.Y.; Zhang, C.J.; Chen, L.M.; Yuan, S.L.; Chen, S.L.; Qiu, D.Z.; et al. Screening and evaluation for soybean resistance to Phakopsora pachyrhyzi in Glycine soja. Oil Crop Sci. 2016, 000, 52–59. [Google Scholar]
- Shan, Z.H.; Liu, Y.; Ba, H.P.; Shan, L.M.; Chen, H.F.; Sha, A.H.; Qiu, D.Z.; Yang, Z.L.; Chen, S.L.; Zhou, X.A. New soybean germplasm resistance to Phakopsora pachyrhizi Syd. Chin. J. Oil Crop Sci. 2012, 34, 188–192. (In Chinese) [Google Scholar]
- Tan, Y.J.; Shan, Z.H.; Shen, M.Z.; Yu, Z.L.; Chang, R.Z.; Sun, J.Y.; Luo, Y.; Xiao, S.S. Evaluation of soybean germplasm of China for resistance to soybean rust. Soybean Sci. 1997, 16, 205–209. (In Chinese) [Google Scholar]
- Tan, Y.J.; Sun, Y.L.; Shan, Z.H. Study on inheritance of resistance to rust in soybean. Soybean Sci. 1991, 10, 6. (In Chinese) [Google Scholar]
- Meyer, J.D.; Silva, D.C.; Yang, C.; Pedley, K.F.; Zhang, C.; van de Mortel, M.; Hill, J.H.; Shoemaker, R.C.; Abdelnoor, R.V.; Whitham, S.A.; et al. Identification and analyses of candidate genes for rpp4-mediated resistance to Asian soybean rust in soybean. Plant Physiol. 2009, 150, 295–307. [Google Scholar] [CrossRef]
- Miles, M.R.; Morel, W.; Ray, J.D.; Smith, J.R.; Frederick, R.D.; Hartman, G.L. Adult plant evaluation of soybean accessions for resistance to Phakopsora pachyrhizi in the field and greenhouse in Paraguay. Plant Dis. 2008, 92, 96–105. [Google Scholar] [CrossRef]
- Paul, C.; Hartman, G.L.; Marois, J.J.; Wright, D.L.; Walker, D.R. First report of Phakopsora pachyrhizi adapting to soybean genotypes with Rpp1 or Rpp6 rust resistance genes in field plots in the United States. Plant Dis. 2013, 97, 1379. [Google Scholar] [CrossRef]
- Luo, Y.; Shan, Z.H.; Zhou, X.A. Evaluation of soybean rust resistant germplasm in Fujian. Chin. J. Oil Crop Sci. 2006, 28, 4. (In Chinese) [Google Scholar] [CrossRef]
- Hyten, D.L.; Hartman, G.L.; Nelson, R.L.; Frederick, R.D.; Concibido, V.C.; Narvel, J.M.; Cregan, P.B. Map location of the Rpp1 locus that confers resistance to soybean rust in soybean. Crop Sci. 2007, 47, 837–840. [Google Scholar] [CrossRef]
- Yu, N.; Kim, M.; King, Z.R.; Harris, D.K.; Buck, J.W.; Li, Z.; Diers, B.W. Fine mapping of the Asian soybean rust resistance gene Rpp2 from soybean PI 230970. Theor Appl Genet. 2015, 128, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Davidl, H.; Jamesr, S.; Reidd, F.; Markl, T.; Qijian, S.; Perryb, C. Bulked segregant analysis using the goldengate assay to locate the Rpp3 locus that confers resistance to soybean rust in soybean. Crop Sci. 2009, 49, 265–271. [Google Scholar] [CrossRef]
- Silva, D.C.; Yamanaka, N.; Brogin, R.L.; Arias, C.A.; Nepomuceno, A.L.; Di Mauro, A.O.; Pereira, S.S.; Nogueira, L.M.; Passianotto, A.L.; Abdelnoor, R.V. Molecular mapping of two loci that confer resistance to Asian rust in soybean. Theor. Appl. Genet. 2008, 117, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, M.D.; Harris, D.K.; Ha, B.K.; Hyten, D.L.; Cregan, P.B.; Frederick, R.D.; Boerma, H.R.; Pedley, K.F. Identification of a second Asian soybean rust resistance gene in Hyuuga soybean. Phytopathology 2011, 101, 535–543. [Google Scholar] [CrossRef]
- Li, S.; Smith, J.R.; Ray, J.D.; Frederick, R.D. Identification of a new soybean rust resistance gene in PI 567102B. Theor Appl Genet. 2012, 125, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Childs, S.P.; King, Z.R.; Walker, D.R.; Harris, D.K.; Pedley, K.F.; Buck, J.W.; Boerma, H.R.; Li, Z. Discovery of a seventh Rpp soybean rust resistance locus in soybean accession PI 605823. Theor Appl Genet. 2018, 131, 27–41. [Google Scholar] [CrossRef]
- Chen, H.; Zhao, S.; Yang, Z.; Sha, A.; Wan, Q.; Zhang, C.; Chen, L.; Yuan, S.; Qiu, D.; Chen, S.; et al. Genetic analysis and molecular mapping of resistance gene to Phakopsora pachyrhizi in soybean germplasm SX6907. Theor. Appl. Genet. 2015, 128, 733–743. [Google Scholar] [CrossRef]
- Dodds, P.N.; Rathjen, J.P. Plant immunity: Towards an integrated view of plant-pathogen interactions. Nat. Rev. Genet. 2010, 11, 539–548. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Naveed, Z.A.; Ali, G.S. Comparative transcriptome analysis between a resistant and a susceptible wild tomato accession in response to Phytophthora parasitica. Int. J. Mol. Sci. 2018, 19, 3735. [Google Scholar] [CrossRef]
- Jain, S.; Chittem, K.; Brueggeman, R.; Osorno, J.M.; Richards, J.; Nelson, B.D., Jr. Comparative transcriptome analysis of resistant and susceptible common bean genotypes in response to soybean cyst nematode infection. PLoS ONE 2016, 11, e0159338. [Google Scholar] [CrossRef]
- Matić, S.; Bagnaresi, P.; Biselli, C.; Orru, L.; Amaral Carneiro, G.; Siciliano, I.; Valé, G.; Gullino, M.L.; Spadaro, D. Comparative transcriptome profiling of resistant and susceptible rice genotypes in response to the seedborne pathogen Fusarium fujikuroi. BMC Genom. 2016, 17, 608. [Google Scholar] [CrossRef] [PubMed]
- Panthee, D.R.; Marois, J.J.; Wright, D.L.; Narváez, D.; Yuan, J.S.; Stewart, C.N., Jr. Differential expression of genes in soybean in response to the causal agent of Asian soybean rust (Phakopsora pachyrhizi Sydow) is soybean growth stage-specific. Theor. Appl. Genet. 2009, 118, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, A.; Hosseini, P.; Li, S.; Alkharouf, N.W.; Matthews, B.F. Analysis of Phakopsora pachyrhizi transcript abundance in critical pathways at four time-points during infection of a susceptible soybean cultivar using deep sequencing. BMC Genom. 2013, 14, 614. [Google Scholar] [CrossRef] [PubMed]
- Amorim, L.L.B.; da Fonseca Dos Santos, R.; Neto, J.P.B.; Guida-Santos, M.; Crovella, S.; Benko-Iseppon, A.M. Transcription factors involved in plant resistance to pathogens. Curr. Protein Pept. Sci. 2017, 18, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Köster, P.; DeFalco, T.A.; Zipfel, C. Ca2+ signals in plant immunity. EMBO J. 2022, 41, e110741. [Google Scholar] [CrossRef]
- Tian, W.; Hou, C.; Ren, Z.; Wang, C.; Zhao, F.; Dahlbeck, D.; Hu, S.; Zhang, L.; Niu, Q.; Li, L.; et al. A calmodulin-gated calcium channel links pathogen patterns to plant immunity. Nature 2019, 572, 131–135. [Google Scholar] [CrossRef]
- Clough, S.J.; Fengler, K.A.; Yu, I.C.; Lippok, B.; Smith, R.K., Jr.; Bent, A.F. The Arabidopsis dnd1 “defense, no death” gene encodes a mutated cyclic nucleotide-gated ion channel. Proc. Natl. Acad. Sci. USA 2000, 97, 9323–9328. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.W.; Lee, D.H.; Hwang, B.K. The pepper calmodulin gene CaCaM1 is involved in reactive oxygen species and nitric oxide generation required for cell death and the defense response. Mol. Plant Microbe Interact. 2009, 22, 1389–1400. [Google Scholar] [CrossRef]
- Vadassery, J.; Reichelt, M.; Hause, B.; Gershenzon, J.; Boland, W.; Mithöfer, A. CML42-mediated calcium signaling coordinates responses to Spodoptera herbivory and abiotic stresses in Arabidopsis. Plant Physiol. 2012, 159, 1159–1175. [Google Scholar] [CrossRef]
- Leba, L.J.; Cheval, C.; Ortiz-Martín, I.; Ranty, B.; Beuzón, C.R.; Galaud, J.P.; Aldon, D. CML9, an Arabidopsis calmodulin-like protein, contributes to plant innate immunity through a flagellin-dependent signalling pathway. Plant J. 2012, 71, 976–989. [Google Scholar] [CrossRef]
- Boudsocq, M.; Sheen, J. CDPKs in immune and stress signaling. Trends Plant Sci. 2013, 18, 30–40. [Google Scholar] [CrossRef]
- Zipfel, C. Pattern-recognition receptors in plant innate immunity. Curr. Opin. Immunol. 2008, 20, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Thulasi Devendrakumar, K.; Li, X.; Zhang, Y. MAP kinase signalling: Interplays between plant PAMP- and effector-triggered immunity. Cell Mol. Life Sci. 2018, 75, 2981–2989. [Google Scholar] [CrossRef]
- Gómez-Gómez, L.; Boller, T. FLS2: An LRR receptor-like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis. Mol. Cell. 2000, 5, 1003–1011. [Google Scholar] [CrossRef]
- Chinchilla, D.; Shan, L.; He, P.; de Vries, S.; Kemmerling, B. One for all: The receptor-associated kinase BAK1. Trends Plant Sci. 2009, 14, 535–541. [Google Scholar] [CrossRef]
- Kong, Q.; Qu, N.; Gao, M.; Zhang, Z.; Ding, X.; Yang, F.; Li, Y.; Dong, O.X.; Chen, S.; Li, X.; et al. The MEKK1-MKK1/MKK2-MPK4 kinase cascade negatively regulates immunity mediated by a mitogen-activated protein kinase kinase kinase in Arabidopsis. Plant Cell. 2012, 24, 2225–2236. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Wang, H.; Jang, J.C.; Xiao, T.; He, H.; Jiang, D.; Tang, X. OsWRKY80-OsWRKY4 module as a positive regulatory circuit in rice resistance against Rhizoctonia solani. Rice 2016, 9, 63. [Google Scholar] [CrossRef]
- Boller, T.; He, S.Y. Innate immunity in plants: An arms race between pattern recognition receptors in plants and effectors in microbial pathogens. Science 2009, 324, 742–744. [Google Scholar] [CrossRef] [PubMed]
- Dou, D.; Zhou, J.M. Phytopathogen effectors subverting host immunity: Different foes, similar battleground. Cell Host Microbe 2012, 12, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.; Vance, R.E.; Dangl, J.L. Intracellular innate immune surveillance devices in plants and animals. Science 2016, 354, aaf6395. [Google Scholar] [CrossRef]
- Kim, K.S.; Unfried, J.R.; Hyten, D.L.; Frederick, R.D.; Hartman, G.L.; Nelson, R.L.; Song, Q.; Diers, B.W. Molecular mapping of soybean rust resistance in soybean accession PI 561356 and SNP haplotype analysis of the Rpp1 region in diverse germplasm. Theor. Appl. Genet. 2012, 125, 1339–1352. [Google Scholar] [CrossRef]
- Monteros, M.J.; Ha, B.K.; Phillips, D.V.; Boerma, H.R. SNP assay to detect the ‘Hyuuga’ red-brown lesion resistance gene for Asian soybean rust. Theor. Appl. Genet. 2010, 121, 1023–1032. [Google Scholar] [CrossRef]
- Wang, L.; Guo, D.; Zhao, G.; Wang, J.; Zhang, S.; Wang, C.; Guo, X. Group IIc WRKY transcription factors regulate cotton resistance to Fusarium oxysporum by promoting GhMKK2-mediated flavonoid biosynthesis. New Phytol. 2022, 236, 249–265. [Google Scholar] [CrossRef]
- Gamir, J.; Minchev, Z.; Berrio, E.; García, J.M.; De Lorenzo, G.; Pozo, M.J. Roots drive oligogalacturonide-induced systemic immunity in tomato. Plant Cell Environ. 2021, 44, 275–289. [Google Scholar] [CrossRef]
- Subramanian, S.; Graham, M.Y.; Yu, O.; Graham, T.L. RNA interference of soybean isoflavone synthase genes leads to silencing in tissues distal to the transformation site and to enhanced susceptibility to Phytophthora sojae. Plant Physiol. 2005, 137, 1345–1353. [Google Scholar] [CrossRef]
- Tian, J.; Chen, M.C.; Zhang, J.; Li, K.T.; Song, T.T.; Zhang, X.; Yao, Y.C. Characteristics of dihydroflavonol 4-reductase gene promoters from different leaf colored Malus crabapple cultivars. Hortic. Res. 2017, 4, 17070. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wu, P.; Cao, Y.; Yang, B.; Liu, L.; Chen, J.; Zhuo, R.; Yao, X. Overexpression of dihydroflavonol 4-reductase (CoDFR) boosts flavonoid production involved in the anthracnose resistance. Front. Plant Sci. 2022, 13, 1038467. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Li, N.; Dong, L.; Zhang, D.; Fan, S.; Jiang, L.; Wang, X.; Xu, P.; Zhang, S. Overexpression of soybean isoflavone reductase (GmIFR) enhances resistance to Phytophthora sojae in soybean. Front. Plant Sci. 2015, 6, 1024. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Cho, K.S.; Kim, S.G.; Kang, S.Y.; Kang, K.Y. A rice isoflavone reductase-like gene, OsIRL, is induced by rice blast fungal elicitor. Mol. Cells 2003, 16, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Meyermans, H.; Morreel, K.; Lapierre, C.; Pollet, B.; De Bruyn, A.; Busson, R.; Herdewijn, P.; Devreese, B.; Van Beeumen, J.; Marita, J.M.; et al. Modifications in lignin and accumulation of phenolic glucosides in poplar xylem upon down-regulation of caffeoyl-coenzyme A O-methyltransferase, an enzyme involved in lignin biosynthesis. J. Biol. Chem. 2000, 275, 36899–36909. [Google Scholar] [CrossRef]
- Hegde, N.; Joshi, S.; Soni, N.; Kushalappa, A.C. The caffeoyl-CoA O-methyltransferase gene SNP replacement in Russet Burbank potato variety enhances late blight resistance through cell wall reinforcement. Plant Cell Rep. 2021, 40, 237–254. [Google Scholar] [CrossRef]
- Yang, Q.; He, Y.; Kabahuma, M.; Chaya, T.; Kelly, A.; Borrego, E.; Bian, Y.; El Kasmi, F.; Yang, L.; Teixeira, P.; et al. A gene encoding maize caffeoyl-CoA O-methyltransferase confers quantitative resistance to multiple pathogens. Nat. Genet. 2017, 49, 1364–1372. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Sample Name | Clean Reads (M) | Clean Bases (G) | Mapped Reads (%) | Uniquely Mapped (%) | No. of Mapped Genes |
|---|---|---|---|---|---|---|
| 1 | R-0 hpi (Rep 1) | 39.0659 | 5.8599 | 93.15 | 89.68 | 39,658 |
| R-0 hpi (Rep 2) | 40.9507 | 6.1426 | 92.38 | 90.30 | 39,736 | |
| R-0 hpi (Rep 3) | 39.2121 | 5.8818 | 92.11 | 89.14 | 38,620 | |
| 2 | R-6 hpi (Rep 1) | 40.6590 | 5.9497 | 90.14 | 87.11 | 37,646 |
| R-6 hpi (Rep 2) | 37.8276 | 5.6741 | 70.75 | 69.25 | 37,054 | |
| R-6 hpi (Rep 3) | 39.6647 | 6.0988 | 91.19 | 88.99 | 37,396 | |
| 3 | R-24 hpi (Rep 1) | 38.8942 | 5.7379 | 92.74 | 90.35 | 38,848 |
| R-24 hpi (Rep 2) | 40.3832 | 6.0575 | 92.91 | 90.82 | 38,635 | |
| R-24 hpi (Rep 3) | 38.2529 | 5.8341 | 78.34 | 76.65 | 38,307 | |
| 4 | R-10 dpi (Rep 1) | 41.4718 | 5.7205 | 80.99 | 79.11 | 38,309 |
| R-10 dpi (Rep 2) | 40.0860 | 6.0129 | 82.90 | 80.90 | 37,731 | |
| R-10 dpi (Rep 3) | 40.0860 | 6.2208 | 92.61 | 90.39 | 38,463 | |
| 5 | S-0 hpi (Rep 1) | 45.1133 | 6.7670 | 91.74 | 88.53 | 37,033 |
| S-0 hpi (Rep 2) | 43.1935 | 6.4790 | 91.68 | 89.38 | 38,407 | |
| S-0 hpi (Rep 3) | 41.1932 | 6.1790 | 91.16 | 87.72 | 35,172 | |
| 6 | S-6 hpi (Rep 1) | 42.2855 | 6.2377 | 91.19 | 88.56 | 37,847 |
| S-6 hpi (Rep 2) | 43.3958 | 6.5094 | 91.02 | 88.53 | 40,092 | |
| S-6 hpi (Rep 3) | 41.5849 | 6.3428 | 90.30 | 88.08 | 38,527 | |
| 7 | S-24 hpi (Rep 1) | 47.2346 | 6.6490 | 92.85 | 90.16 | 39,249 |
| S-24 hpi (Rep 2) | 37.8453 | 5.6768 | 92.56 | 89.60 | 31,962 | |
| S-24 hpi (Rep 3) | 44.3266 | 7.0852 | 91.56 | 89.30 | 38,696 | |
| 8 | S-10 dpi (Rep 1) | 38.9951 | 6.4775 | 86.10 | 83.94 | 37,657 |
| S-10 dpi (Rep 2) | 39.0004 | 5.8501 | 73.21 | 56.29 | 35,421 | |
| S-10 dpi (Rep 3) | 43.1835 | 5.8493 | 80.69 | 78.55 | 40,234 |
| Gene ID | Description | Point-in-Time | |
|---|---|---|---|
| Glyma.18G087400, Glyma.03G087800, Glyma.03G047000, Glyma.03G054100, Glyma.14G205000, Glyma.18G082800, Glyma.14G199400, Glyma.18G088500, Glyma.07G063600, Glyma.09G208900, Glyma.07G063700, Glyma.01G046900, Glyma.06G259400, Glyma.16G214800, Glyma.16G215000 | LRR and NB-ARC domain-containing disease-resistant proteins | 6 hpi; 24 hpi; 10 dpi | upregulated |
| Glyma.17G214600; Glyma.17G214700 | Rust resistance kinase Lr10 isoform | 6 hpi; 24 hpi; 10 dpi | upregulated |
| Glyma.16G214000, Glyma.16G210600, Glyma.03G047700, Glyma.13G190000, Glyma.16G214500, Glyma.13G187900, Glyma.15G233200, Glyma.18G280300, Glyma.06G259800, Glyma.18G281500, Glyma.18G086400, Glyma.13G190400, Glyma.16G085400, Glyma.18G086900, Glyma.18G086600, Glyma.03G088000, Glyma.03G088100 | LRR and NB-ARC domain-containing disease-resistant proteins | 6 hpi; 24 hpi; 10 dpi | downregulated |
| Glyma.12G233000, Glyma.18G198800, Glyma.18G268000, Glyma.16G185100, Glyma.09G107600, Glyma.13G188800, Glyma.06G226700 | LRR receptor protein kinase | 6 hpi; 24 hpi; 10 dpi | downregulated |
| Glyma.13G033500, Glyma.13G033400 | Rust resistance kinase lr10-related protein | 6 hpi; 24 hpi; 10 dpi | downregulated |
| Glyma.05G127400, Glyma.04G220200 | Programmed cell death protein | 6 hpi; 24 hpi; 10 dpi | downregulated |
| Glyma.16G211500, Glyma.15G187300 | TMV resistance protein N isoform | 6 hpi; 24 hpi; 10 dpi | downregulated |
| GO Term (Biological Process) | Cluster Frequency | p-Value |
|---|---|---|
| 0 hpi | ||
| defense response | 58 out of 392 genes, 14.8% | 2.96 × 10−21 |
| response to stress | 72 out of 392 genes, 18.4% | 2.18 ×10−7 |
| response to stimulus | 100 out of 392 genes, 25.5% | 1.2 ×10−3 |
| 6 hpi | ||
| defense response | 106 out of 1202 genes, 8.8% | 8.55 × 10−21 |
| photosynthesis, light harvesting in photosystem I | 17 out of 1202 genes, 1.4% | 3.97 × 10−12 |
| photosynthesis, light harvesting | 19 out of 1202 genes, 1.6% | 3.80 × 10−10 |
| protein-chromophore linkage | 19 out of 1202 genes, 1.6% | 6.04 × 10−9 |
| secondary metabolic process | 55 out of 1202 genes, 4.6% | 1.85 × 10−7 |
| response to stress | 163 out of 1202 genes, 13.6% | 1.41 × 10−6 |
| photosynthesis, light reaction | 20 out of 1202 genes, 1.7% | 7.40 × 10−6 |
| response to stimulus | 270 out of 1202 genes, 22.5% | 1.05 × 10−5 |
| flavonoid metabolic process | 34 out of 1202 genes, 2.8% | 4.57 × 10−5 |
| flavonoid biosynthetic process | 33 out of 1202 genes, 2.7% | 1.12 × 10−3 |
| 24 hpi | ||
| defense response | 47 out of 320 genes, 14.7% | 7.71 × 10−17 |
| response to stress | 66 out of 320 genes, 20.6% | 6.07 × 10−9 |
| response to stimulus | 86 out of 320 genes, 26.9% | 5.62 × 10−3 |
| 10 dpi | ||
| defense response | 172 out of 2179 genes, 7.9% | 7.67 × 10−30 |
| secondary metabolic process | 108 out of 2179 genes, 5.0% | 1.40 × 10−19 |
| multi-organism process | 66 out of 2179 genes, 3.0% | 2.21 × 10−12 |
| protein phosphorylation | 307 out of 2179 genes, 14.1% | 4.79 × 10−12 |
| secondary metabolite biosynthetic process | 75 out of 2179 genes, 3.4% | 5.15 × 10−12 |
| oxidation-reduction process | 374 out of 2179 genes, 17.2% | 4.85 × 10−11 |
| cell recognition | 42 out of 2179 genes, 1.9% | 3.47 × 10−10 |
| pollen–pistil interaction | 42 out of 2179 genes, 1.9% | 3.47 × 10−10 |
| recognition of pollen | 42 out of 2179 genes, 1.9% | 3.47 × 10−10 |
| response to biotic stimulus | 42 out of 2179 genes, 1.9% | 1.10 × 10−9 |
| Pathways | 6 hpi | 24 hpi | 10 dpi |
|---|---|---|---|
| Plant–pathogen interaction | 115 | 45 | 78 |
| Plant hormone signal transduction | 53 | 10 | 21 |
| Flavonoid biosynthesis | 29 | 5 | 14 |
| Phenylpropanoid biosynthesis | 47 | 11 | 21 |
| Galactose metabolism | 11 | 4 | 8 |
| Photosynthesis-antenna proteins | 19 | 0 | 2 |
| Ubiquitin-mediated proteolysis | 15 | 3 | 7 |
| ABC transporters | 23 | 2 | 7 |
| Brassinosteroid biosynthesis | 1 | 1 | 2 |
| Proteasome | 2 | 0 | 2 |
| Phenylalanine metabolism | 9 | 1 | 21 |
| Protein processing in endoplasmic reticulum | 48 | 12 | 17 |
| Flavone and flavonol biosynthesis | 7 | 1 | 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, Q.; Yang, H.; Chen, S.; Qu, Y.; Zhang, C.; Chen, L.; Cao, D.; Yuan, S.; Guo, W.; Yang, Z.; et al. RNA-Seq and Comparative Transcriptomic Analyses of Asian Soybean Rust Resistant and Susceptible Soybean Genotypes Provide Insights into Identifying Disease Resistance Genes. Int. J. Mol. Sci. 2023, 24, 13450. https://doi.org/10.3390/ijms241713450
Hao Q, Yang H, Chen S, Qu Y, Zhang C, Chen L, Cao D, Yuan S, Guo W, Yang Z, et al. RNA-Seq and Comparative Transcriptomic Analyses of Asian Soybean Rust Resistant and Susceptible Soybean Genotypes Provide Insights into Identifying Disease Resistance Genes. International Journal of Molecular Sciences. 2023; 24(17):13450. https://doi.org/10.3390/ijms241713450
Chicago/Turabian StyleHao, Qingnan, Hongli Yang, Shuilian Chen, Yanhui Qu, Chanjuan Zhang, Limiao Chen, Dong Cao, Songli Yuan, Wei Guo, Zhonglu Yang, and et al. 2023. "RNA-Seq and Comparative Transcriptomic Analyses of Asian Soybean Rust Resistant and Susceptible Soybean Genotypes Provide Insights into Identifying Disease Resistance Genes" International Journal of Molecular Sciences 24, no. 17: 13450. https://doi.org/10.3390/ijms241713450
APA StyleHao, Q., Yang, H., Chen, S., Qu, Y., Zhang, C., Chen, L., Cao, D., Yuan, S., Guo, W., Yang, Z., Huang, Y., Shan, Z., Chen, H., & Zhou, X. (2023). RNA-Seq and Comparative Transcriptomic Analyses of Asian Soybean Rust Resistant and Susceptible Soybean Genotypes Provide Insights into Identifying Disease Resistance Genes. International Journal of Molecular Sciences, 24(17), 13450. https://doi.org/10.3390/ijms241713450