
Potential Association of Cytochrome P450 Copy Number Alteration in Tumour with Chemotherapy Resistance in Lung Adenocarcinoma Patients


 , and
, and
Abstract
:
1. Introduction
2. Results
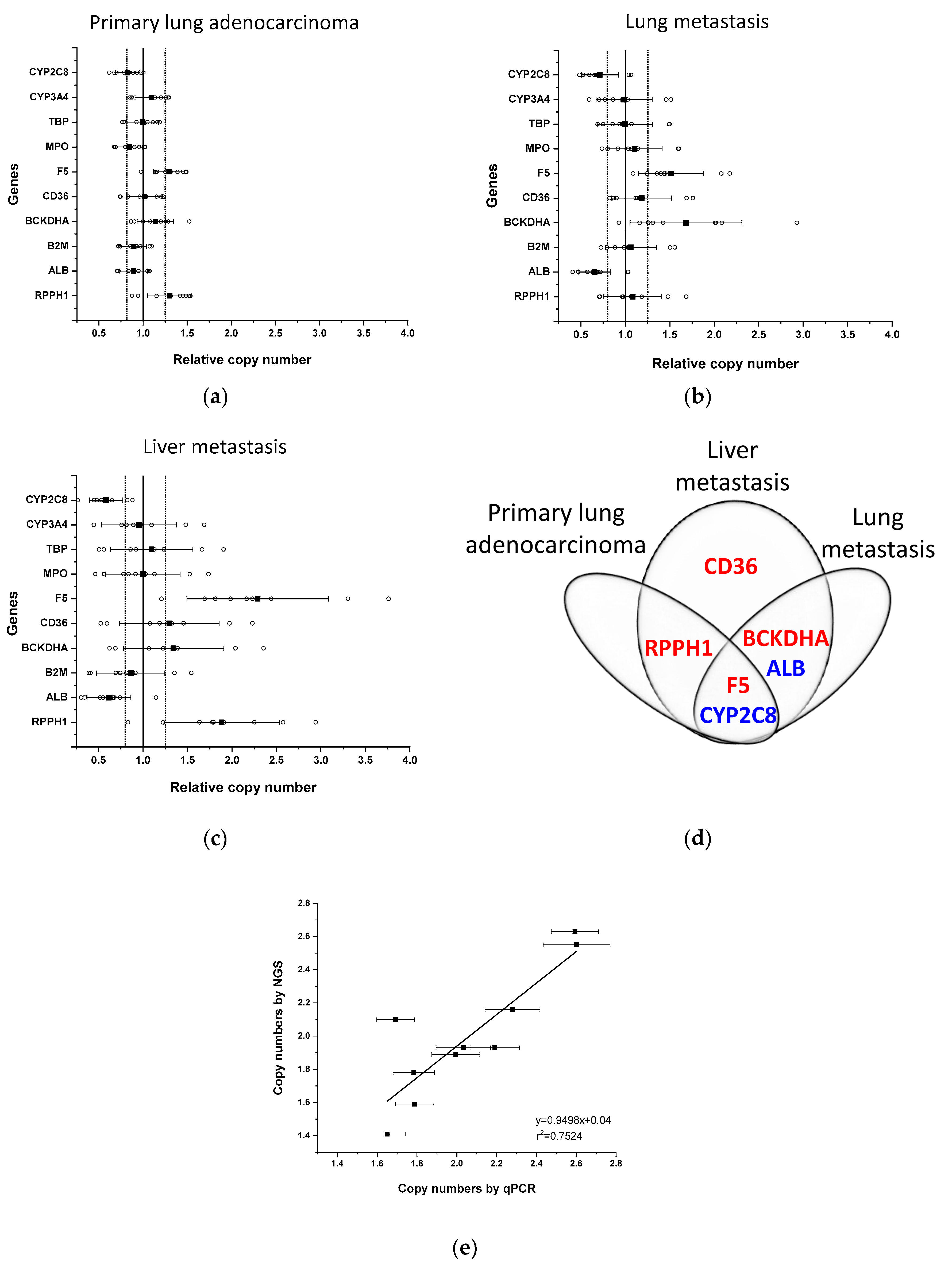
2.1. Copy Number Alterations in Tumour Determined by qPCR-Based Method
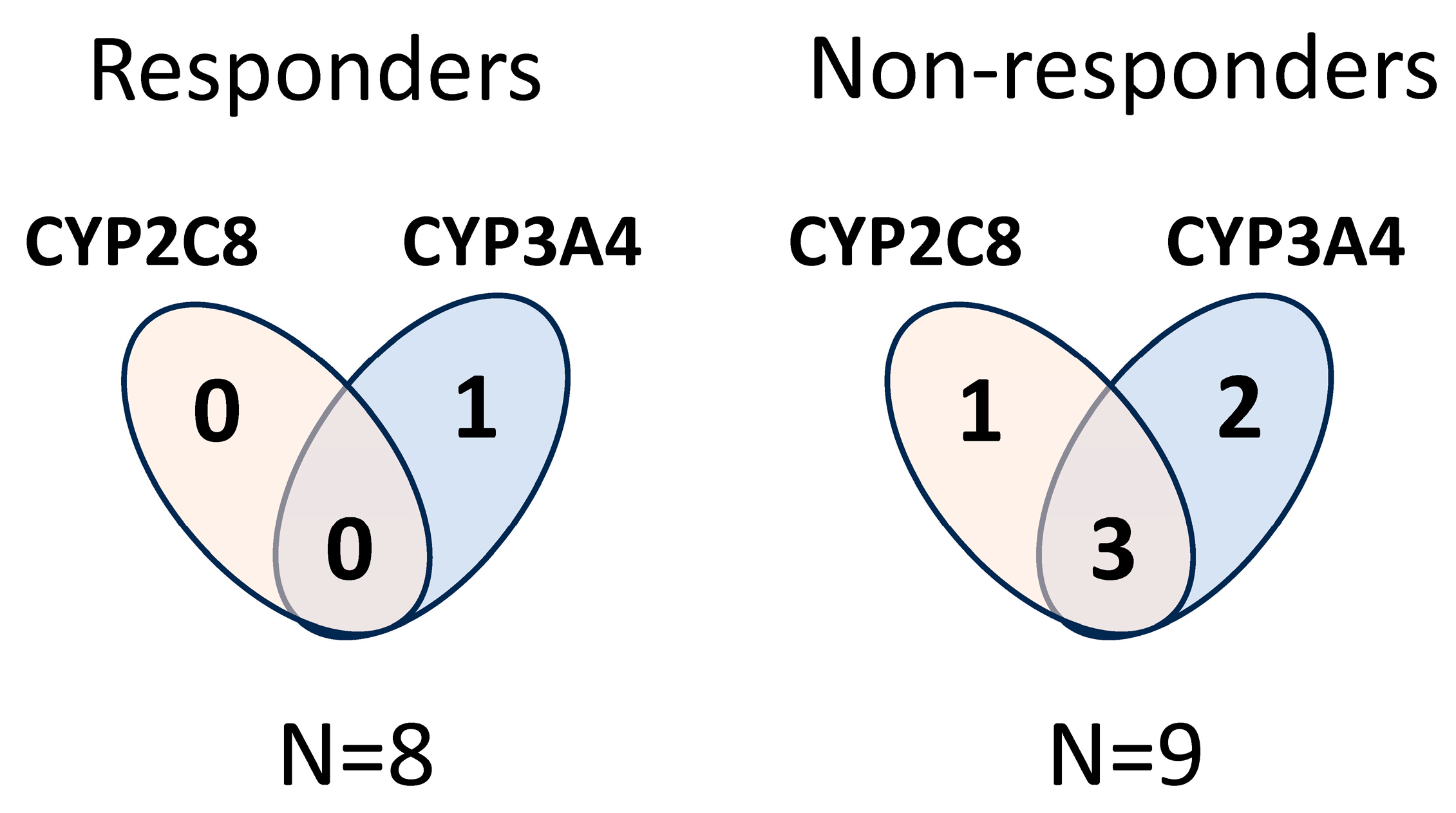
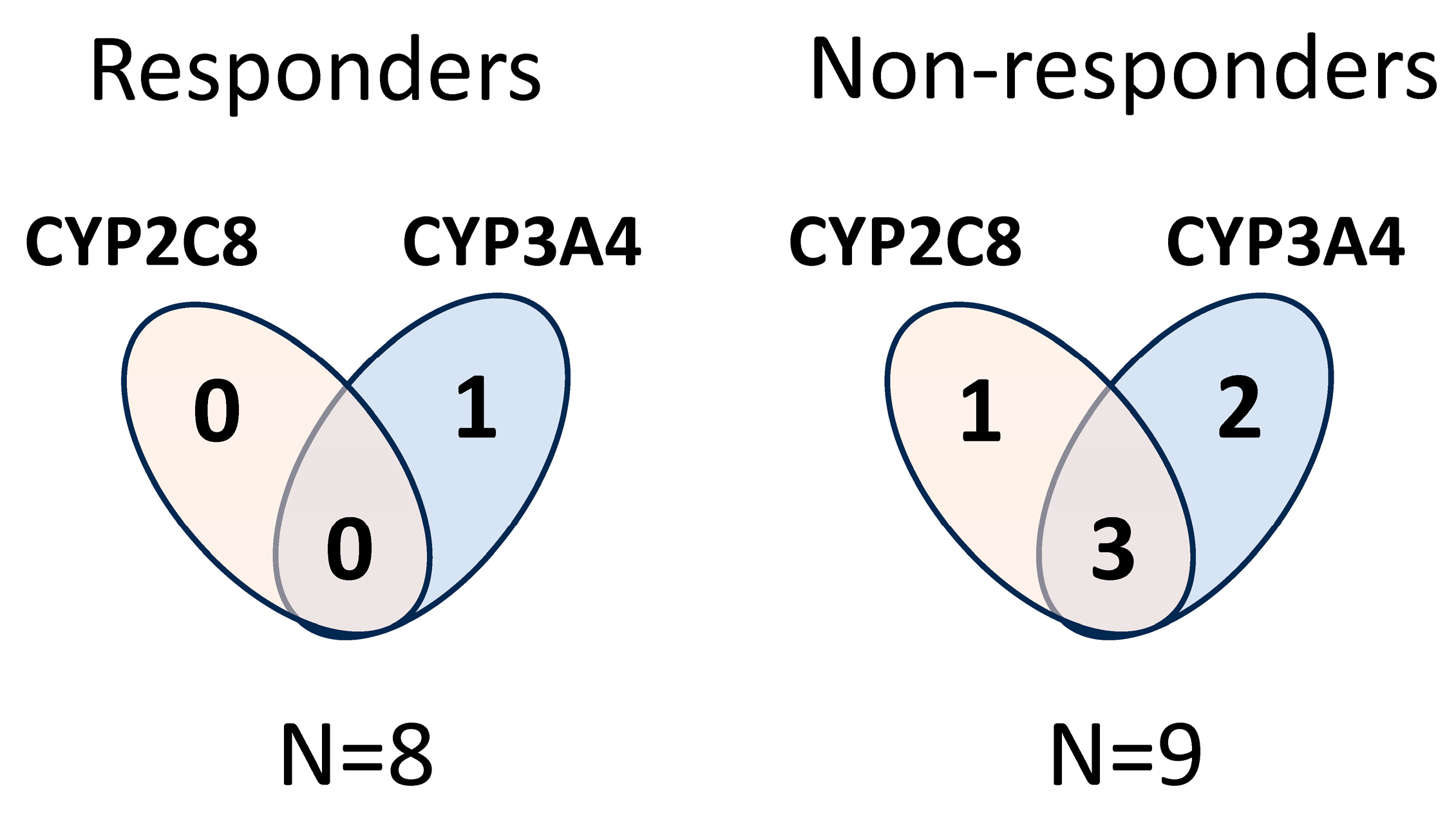
2.2. CYP Haplotype Distribution and Copy Number Alterations in Lung Adenocarcinoma Samples
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. CYP Genotyping
4.3. Copy Number Analyses Using High-Throughput qPCR
4.4. Whole Genome Sequencing
4.5. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef]
- Kaur, G.; Gupta, S.K.; Singh, P.; Ali, V.; Kumar, V.; Verma, M. Drug-metabolizing enzymes: Role in drug resistance in cancer. Clin. Transl. Oncol. 2020, 22, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Bruckmueller, H.; Cascorbi, I. ABCB1, ABCG2, ABCC1, ABCC2, and ABCC3 drug transporter polymorphisms and their impact on drug bioavailability: What is our current understanding? Expert Opin. Drug Metab. Toxicol. 2021, 17, 369–396. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Mittal, B.; Tulsyan, S.; Kumar, S.; Mittal, R.D.; Agarwal, G. Cytochrome P450 in cancer susceptibility and treatment. In Advances in Clinical Chemistry; Makowski, G.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 77–139. [Google Scholar] [CrossRef]
- Rodriguez-Antona, C.; Ingelman-Sundberg, M. Cytochrome P450 pharmacogenetics and cancer. Oncogene 2006, 25, 1679–1691. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Antona, C.; Gomez, A.; Karlgren, M.; Sim, S.C.; Ingelman-Sundberg, M. Molecular genetics and epigenetics of the cytochrome P450 gene family and its relevance for cancer risk and treatment. Hum. Genet. 2010, 127, 1–17. [Google Scholar] [CrossRef]
- van Eijk, M.; Boosman, R.J.; Schinkel, A.H.; Huitema, A.D.R.; Beijnen, J.H. Cytochrome P450 3A4, 3A5, and 2C8 expression in breast, prostate, lung, endometrial, and ovarian tumors: Relevance for resistance to taxanes. Cancer Chemother. Pharmacol. 2019, 84, 487–499. [Google Scholar] [CrossRef]
- Rochat, B. Role of cytochrome P450 activity in the fate of anticancer agents and in drug resistance. Clin. Pharmacokinet. 2005, 44, 349–366. [Google Scholar] [CrossRef]
- Turner, A.P.; Alam, C.; Bendayan, R. Efflux transporters in cancer resistance: Molecular and functional characterization of P-glycoprotein. In Cancer Sensitizing Agents for Chemotherapy; Sosnik, A., Bendayan, R., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 1–30. [Google Scholar] [CrossRef]
- Hofman, J.; Vagiannis, D.; Chen, S.; Guo, L. Roles of CYP3A4, CYP3A5 and CYP2C8 drug-metabolizing enzymes in cellular cytostatic resistance. Chem. Biol. Interact. 2021, 340, 109448. [Google Scholar] [CrossRef]
- Lavrov, A.V.; Ustaeva, O.A.; Adilgereeva, E.P.; Smirnikhina, S.A.; Chelysheva, E.Y.; Shukhov, O.A.; Shatokhin, Y.V.; Mordanov, S.V.; Turkina, A.G.; Kutsev, S.I. Copy number variation analysis in cytochromes and glutathione S-transferases may predict efficacy of tyrosine kinase inhibitors in chronic myeloid leukemia. PLoS ONE 2017, 12, e0182901. [Google Scholar] [CrossRef] [PubMed]
- Sneha, S.; Baker, S.C.; Green, A.; Storr, S.; Aiyappa, R.; Martin, S.; Pors, K. Intratumoural cytochrome P450 expression in breast cancer: Impact on standard of care treatment and new efforts to develop tumour-selective therapies. Biomedicines 2021, 9, 290. [Google Scholar] [CrossRef] [PubMed]
- Coutelier, M.; Holtgrewe, M.; Jäger, M.; Flöttman, R.; Mensah, M.A.; Spielmann, M.; Krawitz, P.; Horn, D.; Beule, D.; Mundlos, S. Combining callers improves the detection of copy number variants from whole-genome sequencing. Eur. J. Hum. Genet. 2022, 30, 178–186. [Google Scholar] [CrossRef]
- Luo, F. A systematic evaluation of copy number alterations detection methods on real SNP array and deep sequencing data. BMC Bioinform. 2019, 20, 692. [Google Scholar] [CrossRef] [PubMed]
- Balagué-Dobón, L.; Cáceres, A.; González, J.R. Fully exploiting SNP arrays: A systematic review on the tools to extract underlying genomic structure. Brief. Bioinform. 2022, 23, bbac043. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chung, W.K. Quantitative analysis of copy number variants based on real-time LightCycler PCR. Curr. Protoc. Hum. Genet. 2014, 80, 7.21.1–7.21.8. [Google Scholar] [CrossRef]
- Seguin, L.; Durandy, M.; Feral, C.C. Lung adenocarcinoma tumor origin: A guide for personalized medicine. Cancers. 2022, 14, 1759. [Google Scholar] [CrossRef]
- Barbuti, A.; Chen, Z.-S. Paclitaxel through the ages of anticancer therapy: Exploring its role in chemoresistance and radiation therapy. Cancers 2015, 7, 2360–2371. [Google Scholar] [CrossRef]
- Cresteil, T.; Monsarrat, B.; Dubois, J.; Sonnier, M.; Alvinerie, P.; Gueritte, F. Regioselective metabolism of taxoids by human CYP3A4 and 2C8: Structure-activity relationship. Drug Metab. Dispos. 2002, 30, 438–445. [Google Scholar] [CrossRef]
- Sparreboom, A.; Huizing, M.T.; Boesen, J.J.B.; Nooijen, W.J.; van Tellingen, O.; Beijnen, J.H. Isolation, purification, and biological activity of mono- and dihydroxylated paclitaxel metabolites from human feces. Cancer Chemother. Pharmacol. 1995, 36, 299–304. [Google Scholar] [CrossRef]
- Dai, D.; Zeldin, D.C.; Blaisdell, J.A.; Chanas, B.; Coulter, S.J.; Ghanayem, B.I.; Goldstein, J.A. Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics 2001, 11, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Liu, D.; Wang, H.; Zhu, J.; Chen, C. Functional characterization of five CYP2C8 variants and prediction of CYP2C8 genotype-dependent effects on in vitro and in vivo drug–drug interactions. Xenobiotica 2010, 40, 467–475. [Google Scholar] [CrossRef]
- Henningsson, A.; Marsh, S.; Loos, W.J.; Karlsson, M.O.; Garsa, A.; Mross, K.; Mielke, S.; Viganò, L.; Locatelli, A.; Verweij, J.; et al. Association of CYP2C8, CYP3A4, CYP3A5, and ABCB1 polymorphisms with the pharmacokinetics of paclitaxel. Clin. Cancer Res. 2005, 11, 8097–8104. [Google Scholar] [CrossRef]
- Leskelä, S.; Jara, C.; Leandro-García, L.J.; Martínez, A.; García-Donas, J.; Hernando, S.; Hurtado, A.; Vicario, J.C.C.; Montero-Conde, C.; Landa, I.; et al. Polymorphisms in cytochromes P450 2C8 and 3A5 are associated with paclitaxel neurotoxicity. Pharmacogenomics J. 2011, 11, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Marsh, S.; Somlo, G.; Li, X.; Frankel, P.; King, C.R.; Shannon, W.D.; McLeod, H.L.; Synold, T.W. Pharmacogenetic analysis of paclitaxel transport and metabolism genes in breast cancer. Pharmacogenomics J. 2007, 7, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Gréen, H.; Söderkvist, P.; Rosenberg, P.; Mirghani, R.A.; Rymark, P.; Lundqvist, E.Å.; Peterson, C. Pharmacogenetic studies of paclitaxel in the treatment of qvarian cancer. Basic Clin. Pharmacol. Toxicol. 2009, 104, 130–137. [Google Scholar] [CrossRef]
- Van Loo, P.; Nordgard, S.H.; Lingjærde, O.C.; Russnes, H.G.; Rye, I.H.; Sun, W.; Weigman, V.J.; Marynen, P.; Zetterberg, A.; Naume, B.; et al. Allele-specific copy number analysis of tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 16910–16915. [Google Scholar] [CrossRef]
- Floyd, M.D.; Gervasini, G.; Masica, A.L.; Mayo, G.; George, A.L.; Bhat, K.; Kim, R.B.; Wilkinson, G.R. Genotype-phenotype associations for common CYP3A4 and CYP3A5 variants in the basal and induced metabolism of midazolam in European- and African-American men and women. Pharmacogenetics 2003, 13, 595–606. [Google Scholar] [CrossRef]
- Zhou, Y.; Ingelman-Sundberg, M.; Lauschke, V.M.V. Worldwide distribution of cytochrome P450 alleles: A meta-analysis of population-scale sequencing projects. Clin. Pharmacol. Ther. 2017, 102, 688–700. [Google Scholar] [CrossRef]
- Joerger, M.; Huitema, A.D.R.; Richel, D.J.; Dittrich, C.; Pavlidis, N.; Briasoulis, E.; Vermorken, J.B.; Strocchi, E.; Martoni, A.; Sorio, R.; et al. Population pharmacokinetics and pharmacodynamics of paclitaxel and carboplatin in ovarian cancer patients: A study by the European Organization for Research and Treatment of Cancer-Pharmacology and Molecular Mechanisms Group and New Drug Development Group. Clin. Cancer Res. 2007, 13, 6410–6418. [Google Scholar] [CrossRef]
- Mielke, S.; Sparreboom, A.; Behringer, D.; Mross, K. Paclitaxel pharmacokinetics and response to chemotherapy in patients with advanced cancer treated with a weekly regimen. Anticancer Res. 2005, 25, 4423–4427. [Google Scholar] [PubMed]
- Bergmann, T.K.; Brasch-Andersen, C.; Gréen, H.; Mirza, M.; Pedersen, R.S.; Nielsen, F.; Skougaard, K.; Wihl, J.; Keldsen, N.; Damkier, P.; et al. Impact of CYP2C8*3 on paclitaxel clearance: A population pharmacokinetic and pharmacogenomic study in 93 patients with ovarian cancer. Pharmacogenomics J. 2011, 11, 113–120. [Google Scholar] [CrossRef]
- Marcath, L.A.; Kidwell, K.M.; Robinson, A.C.; Vangipuram, K.; Burness, M.L.; Griggs, J.J.; Van Poznak, C.; Schott, A.F.; Hayes, D.F.; Henry, N.L.; et al. Patients carrying CYP2C8*3 have shorter systemic paclitaxel exposure. Pharmacogenomics 2019, 20, 95–104. [Google Scholar] [CrossRef]
- Yu, L.J.; Matias, J.; Scudiero, D.A.; Hite, K.M.; Monks, A.; Sausville, E.A.; Waxman, D.J. P450 enzyme expression patterns in the NCI human tumor cell line panel. Drug Metab. Dispos. 2001, 29, 304–312. [Google Scholar]
- De Conti, G.; Dias, M.H.; Bernards, R. Fighting drug resistance through the targeting of drug-tolerant persister cells. Cancers 2021, 13, 1118. [Google Scholar] [CrossRef]
- Nallani, S.C.; Goodwin, B.; Buckley, A.R.; Buckley, D.J.; Desai, P.B. Differences in the induction of cytochrome P450 3A4 by taxane anticancer drugs, docetaxel and paclitaxel, assessed employing primary human hepatocytes. Cancer Chemother. Pharmacol. 2004, 54, 219–229. [Google Scholar] [CrossRef]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Lim, Z.-F.; Ma, P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 2019, 12, 134. [Google Scholar] [CrossRef]
- Manikandan, P.; Nagini, S. Cytochrome P450 structure, function and clinical significance: A review. Curr. Drug Targets 2018, 19, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Han, X.; Tan, Q.; Yang, S.; Li, J.; Xu, J.; Hao, X.; Hu, X.; Xing, P.; Liu, Y.; Lin, L.; et al. Comprehensive profiling of gene copy number alterations predicts patient prognosis in resected stages I–III lung adenocarcinoma. Front. Oncol. 2019, 9, 556. [Google Scholar] [CrossRef] [PubMed]
- Qixing, M.; Juqing, X.; Yajing, W.; Gaochao, D.; Wenjie, X.; Run, S.; Anpeng, W.; Lin, X.; Feng, J.; Jun, W. The expression levels of CYP3A4 and CYP3A5 serve as potential prognostic biomarkers in lung adenocarcinoma. Tumor Biol. 2017, 39, 101042831769834. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, W.; Chen, F.; Hu, G.; Li, F.; Li, J.; Xuan, A. Pregnane X receptors regulate CYP2C8 and P-glycoprotein to impact on the resistance of NSCLC cells to Taxol. Cancer Med. 2016, 5, 3564–3571. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, G.; Lv, S.; Wen, X.; Liu, P. miRNA-301b-3p accelerates migration and invasion of high-grade ovarian serous tumor via targeting CPEB3/EGFR axis. J. Cell. Biochem. 2019, 120, 12618–12627. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-H.; Wu, C.-F.; Rajasekaran, N.; Shin, Y.K. Loss of tumor suppressor gene function in human cancer: An overview. Cell. Physiol. Biochem. 2018, 51, 2647–2693. [Google Scholar] [CrossRef]
- Schwartz, L.H.; Litière, S.; de Vries, E.; Ford, R.; Gwyther, S.; Mandrekar, S.; Shankar, L.; Bogaerts, J.; Chen, A.; Dancey, J.; et al. RECIST 1.1—Update and clarification: From the RECIST committee. Eur. J. Cancer 2016, 62, 132–137. [Google Scholar] [CrossRef]
- Amemiya, H.M.; Kundaje, A.; Boyle, A.P. The ENCODE blacklist: Identification of problematic regions of the genome. Sci. Rep. 2019, 9, 9354. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Gene | Primary Lung Adenocarcinoma | Lung Metastasis | Liver Metastasis |
|---|---|---|---|
| RPPH1 | 6 * | 0 | 7 |
| ALB | −3 | −8 | −8 |
| B2M | −3 | 0 | −2 |
| BCKDHA | 3 | 7 | 3 |
| CD36 | −2 | 2 | 3 |
| F5 | 6 | 7 | 8 |
| MPO | −4 | 1 | −1 |
| TBP | −2 | −1 | 0 |
| CYP3A4 | 3 | −1 | −1 |
| CYP2C8 | −5 | −7 | −7 |
| CYP Allele/Genotype | N | Frequency (%) | |
|---|---|---|---|
| Lung Adenocarcinoma Patients | Caucasian Population * | ||
| CYP allele | |||
| CYP2C8 | |||
| *1 | 31 | 91.2 | 79–90.5 |
| *3 | 3 | 8.8 | 6.5–14 |
| *4 | 0 | 0 | 3–7 |
| CYP3A4 | |||
| *1 | 32 | 94.1 | 83–95.5 |
| *1B | 2 | 5.9 | 2–9 |
| *22 | 0 | 0 | 2.5–8 |
| CYP3A5 | |||
| *1 | 3 | 8.8 | 3–12 |
| *3 | 31 | 91.2 | 88–97 |
| CYP genotype | |||
| CYP2C8 | |||
| *1/*1 | 14 | 82.3 | 81.8–85 |
| *1/*3 | 3 | 17.7 | 11.9–18.1 |
| CYP3A4 | |||
| *1/*1 | 15 | 88.2 | 90–90.9 |
| *1/*1B | 2 | 11.8 | 7.7–9 |
| CYP3A5 | |||
| *1/*1 | 0 | 0 | 3.4 |
| *1/*3 | 2 | 11.8 | 11.6–19.71 |
| *3/*3 | 15 | 88.2 | 76.7–87.4 |
| Complete Remission | Progressive Disease/Exit | ||
|---|---|---|---|
| Number of patients | 8 | 10 b | |
| Gender (male/female) | 2/6 | 4/6 | |
| Age (year) a | 64 (49; 74) | 63.5 (22; 72) | |
| Smoking | active | 6 | 4 |
| quit after 1 year | 0 | 2 | |
| never | 2 | 3 | |
| NA c | - | 1 | |
| Primary tumour d | 1 (1a–1b) | - | 2 |
| 2 (2a–2b) | 7 | 7 | |
| 3 (3a–3b) | 1 | - | |
| Lymph nodes d | 0 | 3 | 5 |
| 1 | 2 | - | |
| 2 | 2 | 3 | |
| x | 1 | 1 | |
| Metastasis d | 0 | 2 | 2 |
| x | 6 | 7 | |
| Surgical intervention | right superior lobectomy | 5 | 3 |
| left superior lobectomy | - | 2 | |
| right middle lobectomy | 1 | - | |
| right inferior lobectomy | 2 | 1 | |
| left inferior lobectomy | - | 1 | |
| atypical resection in right superior lobe | - | 1 | |
| atypical resection in left inferior lobe | - | 1 | |
| autopsy: left inferior lobe | - | 1 e | |
| Therapy | paclitaxel+carboplatin | 8 | 9 |
| gefitinib, cisplatin, afatinib | - | 1 e | |
| adjuvant radiotherapy | 1 | 4 |
| Gene | Gene Name | Chromosome Location | Forward Primer | Reverse Primer | Probe |
|---|---|---|---|---|---|
| RPPH1 | Ribonuclease P RNA component H1 | 14q11.2 | TaqMan® Copy Number Reference Assay, Thermo Fisher Scientific Catalog number: 4403326 | ||
| ALB | albumin | 4q13.3 | TCT TCT GTC AAC CCC ACA | ATC TCG ACG AAA CAC ACC | GGC ACA ATG AAG TGG GTA ACC |
| B2M | beta-2-microglobulin | 15q21.1 | GAA TGA GTC CCA TCC CAT CT | CAG GGA AAC TAC TGG TTC AGA | CAG TAT CTC AGC AGG TGC CA |
| BCKDHA | branched-chain alpha-keto acid dehydrogenase | 19q1.2 | GTC GGC AGG TTA CCA CTA | GAA GGG CTG GGA CAT ACA | TAT CTG GGT GCC ATC TCC TC |
| CD36 | cluster of differentiation 36 | 7q21.11 | TGA AGC TGA AAT TGA AG TGG AG | AAC AGG TCT CAG TAC AGC AT | AGT CAG TTG TCC TCG TCG AAA TC |
| F5 | coagulation factor V | 1q24.2 | TGA TCG AGG ATT TCA ACT CG | TCC CAT GAC AGA ACT CCT | AAG GTA AGA ACA CCC CCA CC |
| MPO | myelo-peroxidase | 17p22.1 | ACA CTA GGG TCA GTC CTT G | CAA GCC TGG GTC ATT ATG AC | CAT CTG GAG GAT TCC TGG AAC A |
| TBP | TATA box binding protein | 6q27 | CCT GCT CTG TTT TCA GAT GG | ATA GCT TTG CTT CCC TTT CC | AGT GGC ACT AAC GGT AAT TGT GT |
| CYP2C8 | cytochrome P450 2C8 | 10q23.33 | CGT TTC TCC CTC ACA ACC TTG C | ACT GTT AAG GTC AAT GAC GCA GA | CCT CAA TGC TCC TCT TCC CCA TCC CA |
| CYP3A4 | cytochrome P450 3A4 | 7q22.1 | CAG AGG TAG GTC TAA TTC AGT TCA | AGA TCA CCT TCT ATC ACA CTC C | ATC ACA CCC AGC GTA GGG C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Incze, E.; Mangó, K.; Fekete, F.; Kiss, Á.F.; Póti, Á.; Harkó, T.; Moldvay, J.; Szüts, D.; Monostory, K. Potential Association of Cytochrome P450 Copy Number Alteration in Tumour with Chemotherapy Resistance in Lung Adenocarcinoma Patients. Int. J. Mol. Sci. 2023, 24, 13380. https://doi.org/10.3390/ijms241713380
Incze E, Mangó K, Fekete F, Kiss ÁF, Póti Á, Harkó T, Moldvay J, Szüts D, Monostory K. Potential Association of Cytochrome P450 Copy Number Alteration in Tumour with Chemotherapy Resistance in Lung Adenocarcinoma Patients. International Journal of Molecular Sciences. 2023; 24(17):13380. https://doi.org/10.3390/ijms241713380
Chicago/Turabian StyleIncze, Evelyn, Katalin Mangó, Ferenc Fekete, Ádám Ferenc Kiss, Ádám Póti, Tünde Harkó, Judit Moldvay, Dávid Szüts, and Katalin Monostory. 2023. "Potential Association of Cytochrome P450 Copy Number Alteration in Tumour with Chemotherapy Resistance in Lung Adenocarcinoma Patients" International Journal of Molecular Sciences 24, no. 17: 13380. https://doi.org/10.3390/ijms241713380
APA StyleIncze, E., Mangó, K., Fekete, F., Kiss, Á. F., Póti, Á., Harkó, T., Moldvay, J., Szüts, D., & Monostory, K. (2023). Potential Association of Cytochrome P450 Copy Number Alteration in Tumour with Chemotherapy Resistance in Lung Adenocarcinoma Patients. International Journal of Molecular Sciences, 24(17), 13380. https://doi.org/10.3390/ijms241713380